
Crosstalk between microRNA and Oxidative Stress in Heart Failure: A Systematic Review

 , ,
, ,
Abstract
:1. Introduction
1.1. microRNA
1.2. Oxidative Stress
1.2.1. ROS Origin, Formation, and Function
1.2.2. microRNA and Oxidative Stress in Cardiovascular Diseases
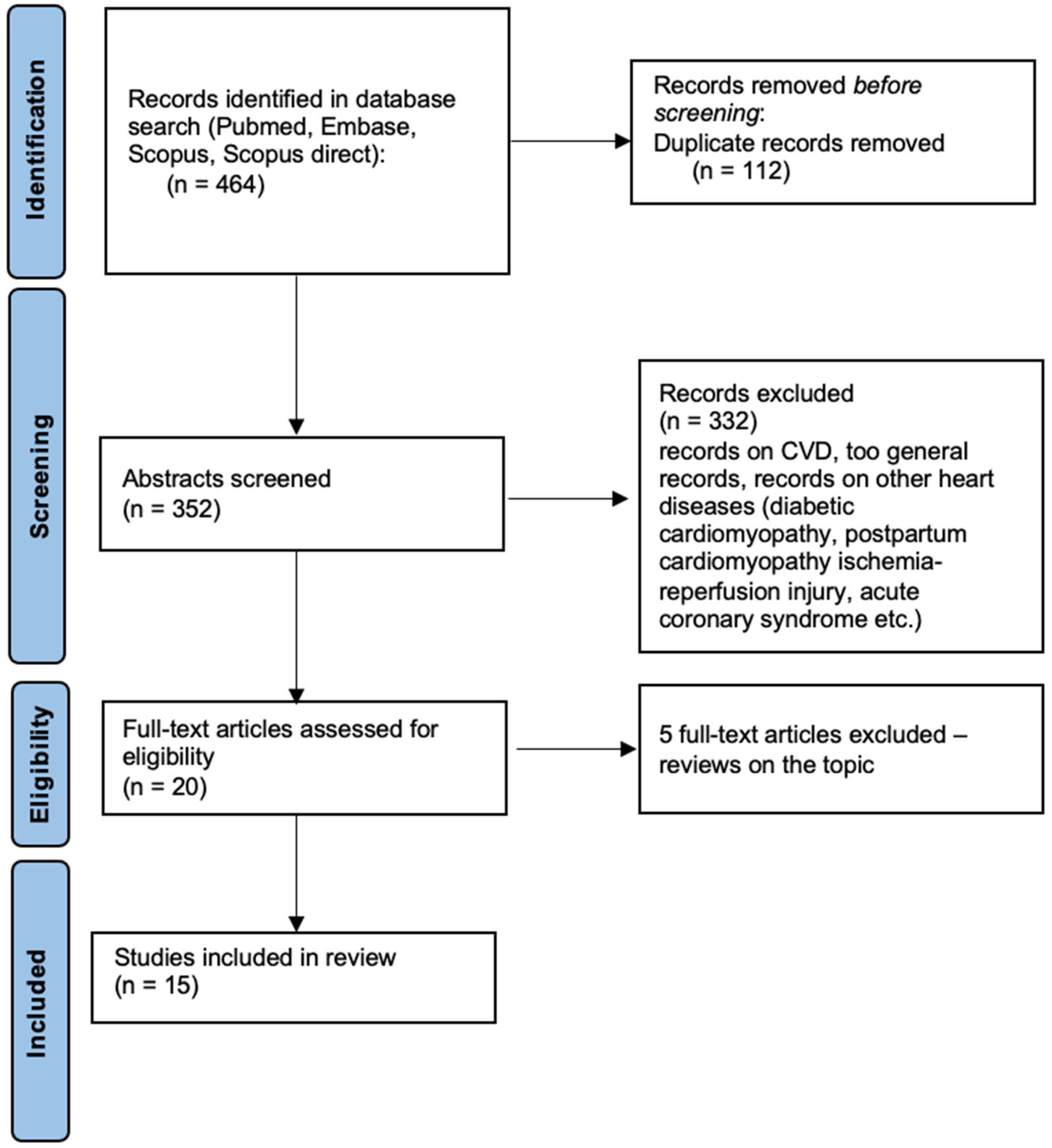
2. Materials and Methods
3. Results
3.1. Mitochondrial Integrity and Function
3.2. Antioxidant Defence
3.3. Iron Overload and Ferroptosis
3.4. Cardiac Hypertrophy and Remodelling
3.5. Apoptosis
3.6. Human Studies
3.7. Therapeutic Potential
3.7.1. miRNA-15b
3.7.2. miRNA-195
3.7.3. miRNA-27a
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Donde, M.J.; Rochussen, A.M.; Kapoor, S.; Taylor, A.I. Targeting non-coding RNA family members with artificial endonuclease XNAzymes. Commun. Biol. 2022, 5, 1010. [Google Scholar] [CrossRef]
- Chipman, L.B.; Pasquinelli, A.E. miRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef]
- Zhou, S.-S.; Jin, J.-P.; Wang, J.-Q.; Zhang, Z.-G.; Freedman, J.H.; Zheng, Y.; Cai, L. miRNAS in cardiovascular diseases: Potential biomarkers, therapeutic targets and challenges. Acta Pharmacol. Sin. 2018, 39, 1073–1084. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Çakmak, H.A.; Demir, M. MicroRNA and Cardiovascular Diseases. Balkan. Med. J. 2020, 37, 60–71. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, A.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, 55–162. [Google Scholar] [CrossRef] [PubMed]
- Klisic, A.; Kavaric, N.; Ninic, A.; Kotur-Stevuljevic, J. Oxidative stress and cardiometabolic biomarkers in patients with non-alcoholic fatty liver disease. Sci. Rep. 2021, 11, 18455. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, I.; Dhar, R.; Singh, S.; Sharma, J.B.; Nag, T.C.; Mridha, A.R.; Jaiswal, P.; Biswas, S.; Karmakar, S. Oxidative stress-induced impairment of trophoblast function causes preeclampsia through the unfolded protein response pathway. Sci. Rep. 2021, 11, 18415. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, R.; Almutairi, N.M.; Al-Muammar, M.N.; Bhat, R.S.; Moubayed, N.; El-Ansary, A. Controlled diabetes amends oxidative stress as mechanism related to severity of diabetic retinopathy. Sci. Rep. 2021, 11, 17670. [Google Scholar] [CrossRef]
- Ghosh, A.; Shcherbik, N. Effects of Oxidative Stress on Protein Translation: Implications for Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 2661. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 16, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 17, 1292. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.-H.; Cheng, W.-H.; Bao, Y.; Ho, Y.-S.; Reddi, A.R.; Holmgren, A.; Arnér, E.S.J. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef] [Green Version]
- Climent, M.; Viggiani, G.; Chen, Y.W.; Coulis, G.; Castaldi, A. MicroRNA and ROS Crosstalk in Cardiac and Pulmonary Diseases. Int. J. Mol. Sci. 2020, 21, 4370. [Google Scholar] [CrossRef]
- Li, M.; Duan, L.; Li, Y.; Liu, B. Long noncoding RNA/circular noncoding RNA–miRNA–mRNA axes in cardiovascular diseases. Life Sci. 2019, 233, 116440. [Google Scholar] [CrossRef]
- Colpaert, R.M.W.; Calore, M. MicroRNAs in Cardiac Diseases. Cells 2019, 8, 737. [Google Scholar] [CrossRef]
- Deng, J.; Zhong, Q. Advanced research on the microRNA mechanism in heart failure. Int. J. Cardiol. 2016, 220, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.Y.; Luo, J.Y.; Wang, L.; Huang, Y. MicroRNAs Regulating Reactive Oxygen Species in Cardiovascular Diseases. Antioxid. Redox Signal. 2018, 29, 1092–1107. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 21, 7. [Google Scholar]
- Wang, X.; Song, C.; Zhou, X.; Han, X.; Li, J.; Wang, Z.; Shang, H.; Liu, Y.; Cao, H. Mitochondria Associated MicroRNA Expression Profiling of Heart Failure. Biomed. Res. Int. 2017, 2017, 4042509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Zhang, Z.; Yin, Q.; Fu, C.; Barszczyk, A.; Zhang, X.; Wang, J.; Yang, D. Cardiac-specific overexpression of miR-122 induces mitochondria-dependent cardiomyocyte apoptosis and promotes heart failure by inhibiting Hand2. J. Cell Mol. Med. 2021, 25, 5326–5334. [Google Scholar] [CrossRef]
- Roy, S.; Banerjee, J.; Gnyawali, S.C.; Khanna, S.; He, G.; Pfeiffer, D.; Zweier, J.L.; Sen, C.K. Suppression of Induced microRNA-15b Prevents Rapid Loss of Cardiac Function in a Dicer Depleted Model of Cardiac Dysfunction. PLoS ONE 2013, 8, e66789. [Google Scholar] [CrossRef]
- Tian, C.; Gao, L.; Zimmerman, M.C.; Zucker, I.H. Myocardial infarction-induced microRNA-enriched exosomes contribute to cardiac Nrf2 dysregulation in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, 928–939. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Hu, G.; Gao, L.; Hackfort, B.T.; Zucker, I.H. Extracellular vesicular MicroRNA-27a* contributes to cardiac hypertrophy in chronic heart failure. J. Mol. Cell Cardiol. 2020, 143, 120–131. [Google Scholar] [CrossRef]
- Shimizu, T.; Taguchi, A.; Higashijima, Y.; Takubo, N.; Kanki, Y.; Urade, Y.; Wada, Y. PERK-mediated suppression of microRNAs by sildenafil improves mitochondrial dysfunction in heart failure. iScience 2020, 23, 101410. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shi, L.; Tong, C.; Liu, Y.; Hou, M. circSnx12 Is Involved in Ferroptosis During Heart Failure by Targeting miR-224-5p. Front. Cardiovasc. Med. 2021, 8, 656093. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Tong, L.; Liang, X.; Zhang, H.; Li, L.; Fan, G.; Wang, Y. Analysis of microRNA Expression Profiles Induced by Yiqifumai Injection in Rats with Chronic Heart Failure. Front. Physiol. 2018, 9, 48. [Google Scholar] [CrossRef]
- Su, Q.; Zhang, P.; Yu, D.; Wu, Z.; Li, D.; Shen, F.; Liao, P.; Yin, G. Upregulation of miR-93 and inhibition of LIMK1 improve ventricular remodeling and alleviate cardiac dysfunction in rats with chronic heart failure by inhibiting RhoA/ROCK signaling pathway activation. Aging 2019, 11, 7570–7586. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Liu, J.; Wei, J.; Yuan, H.; Zhang, T.; Bishopric, N.H. Repression of miR-142 by p300 and MAPK is required for survival signalling via gp130 during adaptive hypertrophy. EMBO Mol. Med. 2012, 4, 617–632. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, W.; Lee, L.; Hong, M.; Lee, M.; Chou, G.; Yu, L.; Sui, Y.; Chou, B. Down-regulated microRNA-195-5p and up-regulated CXCR4 attenuates the heart function injury of heart failure mice via inactivating JAK/STAT pathway. Int. Immunopharmacol. 2020, 82, 106225. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, W.; Bai, X.; Wang, X.; Wang, Y.; Yin, Y. microRNA-454-mediated NEDD4-2/TrkA/cAMP axis in heart failure: Mechanisms and cardioprotective implications. J. Cell Mol. Med. 2021, 25, 5082–5098. [Google Scholar] [CrossRef]
- Ramachandran, S.; Lowenthal, A.; Ritner, C.; Lowenthal, S.; Bernstein, H.S. Plasma microvesicle analysis identifies microRNA 129-5p as a biomarker of heart failure in univentricular heart disease. PLoS ONE 2017, 12, e0183624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, A.; Karami, A.R.B.; Mashtani, V.D.; Sahraei, T.; Tarashoki, Z.B.; Khattavian, E.; Mobarak, S.; Kazerouni, H.M.; Radmanesh, E. Evaluation of Oxidative Stress, Apoptosis, and Expression of MicroRNA-208a and MicroRNA-1 in Cardiovascular Patients. Rep. Biochem. Mol. Biol. 2021, 10, 183–196. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. ESC Scientific Document Group, 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [PubMed]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagan, L.U.; Gomes, M.J.; Martinez, P.F.; Okoshi, M.P. Oxidative Stress and Heart Failure: Mechanisms, Signalling Pathways, and Therapeutics. Oxid. Med. Cell Longev. 2022, 2022, 9829505. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, Y.; Lyu, X. Promoting roles of KLF5 in myocardial infarction in mice involving microRNA-27a suppression and the following GFPT2/TGF-β/Smad2/3 axis activation. Cell Cycle 2021, 20, 874–893. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.C.; Liu, X.M.; Liang, L.R.; Wang, L.F.; Zhong, J.C. Targeting the microRNA-34a as a Novel Therapeutic Strategy for Cardiovascular Diseases. Front. Cardiovasc. Med. 2022, 8, 784044. [Google Scholar] [CrossRef] [PubMed]
- Guglin, M.; Rajagopalan, N.; Anaya, P.; Charnigo, R. Sildenafil in heart failure with reactive pulmonary hypertension (Sildenafil HF) clinical trial (rationale and design). Pulm. Circ. 2016, 6, 161–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.D.; Lachmann, J.; Camuso, J.; Lepore, J.J.; Shin, J.; Martinovic, M.E.; Systrom, D.M.; Bloch, K.D.; Semigran, M.J. Sildenafil improves exercise hemodynamics and oxygen uptake in patients with systolic heart failure. Circulation 2007, 115, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Anstrom, K.; Ilkayeva, O.; Muehlbauer, M.J.; Bain, J.R.; McNulty, S.; Newgard, C.B.; Kraus, W.E.; Hernandez, A.; Felker, G.M.; et al. Sildenafil Treatment in Heart Failure With Preserved Ejection Fraction: Targeted Metabolomic Profiling in the RELAX Trial. JAMA Cardiol. 2017, 2, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.R.; Shah, Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013, 3, 315–330. [Google Scholar] [PubMed] [Green Version]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Lee, C.S.; Auld, J. Heart Failure: A Primer. Crit. Care Nurs. Clin. N. Am. 2015, 27, 413–425. [Google Scholar] [CrossRef]
- Yang, W.H.; Tsai, C.H.; Fong, Y.C.; Huang, Y.L.; Wang, S.J.; Chang, Y.S.; Tang, C.H. Leptin induces oncostatin M production in osteoblasts by downregulating miR-93 through the Akt signaling pathway. Int. J. Mol. Sci. 2014, 15, 15778–15790. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Lin, T.; Chen, L.; Wang, N. MicroRNA-93 elevation after myocardial infarction is cardiac protective. Med. Hypotheses 2017, 106, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Tan, Y.S.; Chen, H.L.; Yan, Y.; Zhai, K.F.; Li, D.P.; Kou, J.P.; Yu, B.Y. Identification of schisandrin as a vascular endothelium protective component in YiQiFuMai Powder Injection using HUVECs binding and HPLC-DAD-Q-TOF-MS/MS analysis. J. Pharmacol. Sci. 2015, 129, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xiao, N.; Zhang, J.; Chen, C.; Wan, Y.; Wang, N.; Yang, J. miR-129-5p improves cardiac function in rats with chronic heart failure through targeting HMGB1. Mamm. Genome 2019, 30, 276–288. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| No. | miRNA | Ref. | Type of Study 1 | Techniqe 2 | Main Conclusion |
|---|---|---|---|---|---|
| 3.1 Mitochondrial integrity and function | |||||
| 1 | miRNA-690 | Wang, X. et al. (2017) [24] | animal | RT qRT-PCR | microRNAs were enriched in mitochondria during heart failure, which establishes a link between microRNA and mitochondrion in heart failure. |
| miRNA-696 | |||||
| miRNA-532-5p | |||||
| miRNA-345-3p | |||||
| 2 | miRNA-122 | Shi, Y. et al. (2021) [25] | animal | qRT-PCR with the miRCute Enhanced Fluorescence Quantitative Assay Kit (Tiangen) | miR-122 causes cardiomyocyte apoptosis by inhibiting Hand2 transcription factor and consequently increasing mitochondrial fission. This mechanism likely contributes to heart failure and modulating this pathway could be therapeutically valuable against heart failure. |
| 3 | miRNA-15b | Roy, S. et al. (2013) [26] | animal/in vivo | qRT-PCR | Suppression of inducible miRNA-15b can prevent rapid loss of cardiac function in an animal adult heart and can be a key approach worthy of therapeutic consideration. |
| 3.2 Antioxidant defence | |||||
| 4 | miRNA-27a | Tian, C. et al. (2018) [27] | animal/in vivo | qRT-PCR | Increased expression of local microRNAs induced by myocardial infarction may contribute to oxidative stress by the inhibition of nuclear factor erythroid 2-related factor 2(NRF2) translation in chronic heart failure. |
| miRNA-28-3p | |||||
| miRNA-34a | |||||
| 5 | miRNA-27a * | Tian, C. et al. (2020) [28] | animal/in vivo | qRT-PCR | Cardiac fibroblast-secretion of miRNA27a *-enriched extracellular vesicles (EV) might act as a paracrine signaling mediator of cardiac hypertrophy and may have a potential to become novel therapeutic target. |
| 6 | miRNA-24-3p | Shimizu, T. et al. (2020) [29] | animal | RNA-sequencing, western blot, ELISA | Protein kinase R–like endoplasmic reticulum (ER) kinase (PERK)-mediated suppression of miRNAs by sildenafil improves cardiac dysfunction in heart failure. |
| 3.3 Iron overload and ferroptosis | |||||
| 7 | miRNA-224-5p | Zheng, H. et al. (2021) [30] | animal | qRT-PCR, Western blot analysis, luciferase reporter assay | Circulatory RNA (circRNA), microRNA(miRNA) and mRNA work in a regulatory network and reveal potential targets for the treatment of heart failure. |
| miRNA-296-3p | |||||
| miRNA-671-5p | |||||
| miRNA-1306-5p | |||||
| miRNA-3082-5p | |||||
| 3.4 Cardiac hypertrophy and remodelling | |||||
| 8 | miRNA-21-3p | Zhao, Y. et al. (2018) [31] | animal | miRNA microarray, bioinforma-tic analysis, qRT-PCR, Western blot | The injection of Yiqifumai (YQFM) has a potential effect which alleviates cardiac hypertrophy and apoptosis in chronic heart failure by miRNA expression regulation. |
| miRNA216-5p | |||||
| miRNA-219a-2-3p | |||||
| miRNA-381-3p | |||||
| miRNA-466c-5p | |||||
| miRNA-542-3p | |||||
| miRNA-702-5p | |||||
| 9 | miRNA-93 | Su, Q. et al. (2019) [32] | animal/in vivo | RT qRT-PCR, ELISA, Western blot | Upregulated miR-93 and downregulated LIM domain kinase 1 (LIMK1) reduce cardiac dysfunction and improve ventricular remodelling in rats with chronic heart failure. |
| 10 | miRNA-142-5p | Sharma, S. et al. (2012) [33] | animal/in vivo | qRT-PCR, Western blot | Downregulation of miR-142 is a critical element of adaptive cardiac muscle hypertrophy in response to hemodynamic stress. |
| miRNA-142-3p | |||||
| 11 | miRNA-195-5p | Shen, Y. et al. (2020) [34] | animal | RT qRT-PCR | Depleting miR-195-5p and up-regulating Chemokine receptor type 4 alleviates cardiac function injury in mice with heart failure and may be a potential candidate marker and therapeutic target for heart failure. |
| For Section 3.4 see also research studies no.: 4 [27] and 5 [28]. | |||||
| 3.5 Apoptosis | |||||
| 12 | miRNA-454 | Wang, Y. et al. (2021) [35] | animal | RT qRT-PCR, Western blot | The cardioprotective role of miR-454 is achieved through activation of the cyclic adenosine 3′5′-monophosphate(cAMP). |
| 3.6 Human studies | |||||
| 13 | miRNA-129-5p | Ramachandran, S. et al. (2017) [36] | human | isolation of mir129-5p from plasma microvesicles, qRT-PCR | miR129-5p was found to be a sensitive and specific biomarker for heart failure in univentricular heart disease in pediatric patients independent of ventricular morphology or stage of palliation. |
| 14 | miRNA-208a | Mohammadi et al. (2021) [37] | human | qRT-PCR | The results on different populations of cardiovascular patients (after myocardial infarction, with arrhythmia, heart failure etc.) showed increases in both oxidative stress, inflammation, apoptotic factors, and in the expression of miR-208a. |
| 3.7 Therapeutic potential | |||||
| 15 | miRNA-15b | Roy, S. et al. (2013) [26] | animal/in vivo | qRT-PCR | Suppression of inducible miRNA-15b can prevent rapid loss of cardiac function in an animal adult heart and can be a key approach worthy of therapeutic consideration. |
| For Section 3.5 see also research studies no.: 5 [28] and 11 [34]. | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimczak-Tomaniak, D.; Haponiuk-Skwarlińska, J.; Kuch, M.; Pączek, L. Crosstalk between microRNA and Oxidative Stress in Heart Failure: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 15013. https://doi.org/10.3390/ijms232315013
Klimczak-Tomaniak D, Haponiuk-Skwarlińska J, Kuch M, Pączek L. Crosstalk between microRNA and Oxidative Stress in Heart Failure: A Systematic Review. International Journal of Molecular Sciences. 2022; 23(23):15013. https://doi.org/10.3390/ijms232315013
Chicago/Turabian StyleKlimczak-Tomaniak, Dominika, Julia Haponiuk-Skwarlińska, Marek Kuch, and Leszek Pączek. 2022. "Crosstalk between microRNA and Oxidative Stress in Heart Failure: A Systematic Review" International Journal of Molecular Sciences 23, no. 23: 15013. https://doi.org/10.3390/ijms232315013
APA StyleKlimczak-Tomaniak, D., Haponiuk-Skwarlińska, J., Kuch, M., & Pączek, L. (2022). Crosstalk between microRNA and Oxidative Stress in Heart Failure: A Systematic Review. International Journal of Molecular Sciences, 23(23), 15013. https://doi.org/10.3390/ijms232315013