
A Central Contribution of TG2 Activity to the Antiproliferative and Pro-Apoptotic Effects of Caffeic Acid in K562 Cells of Human Chronic Myeloid Leukemia
 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
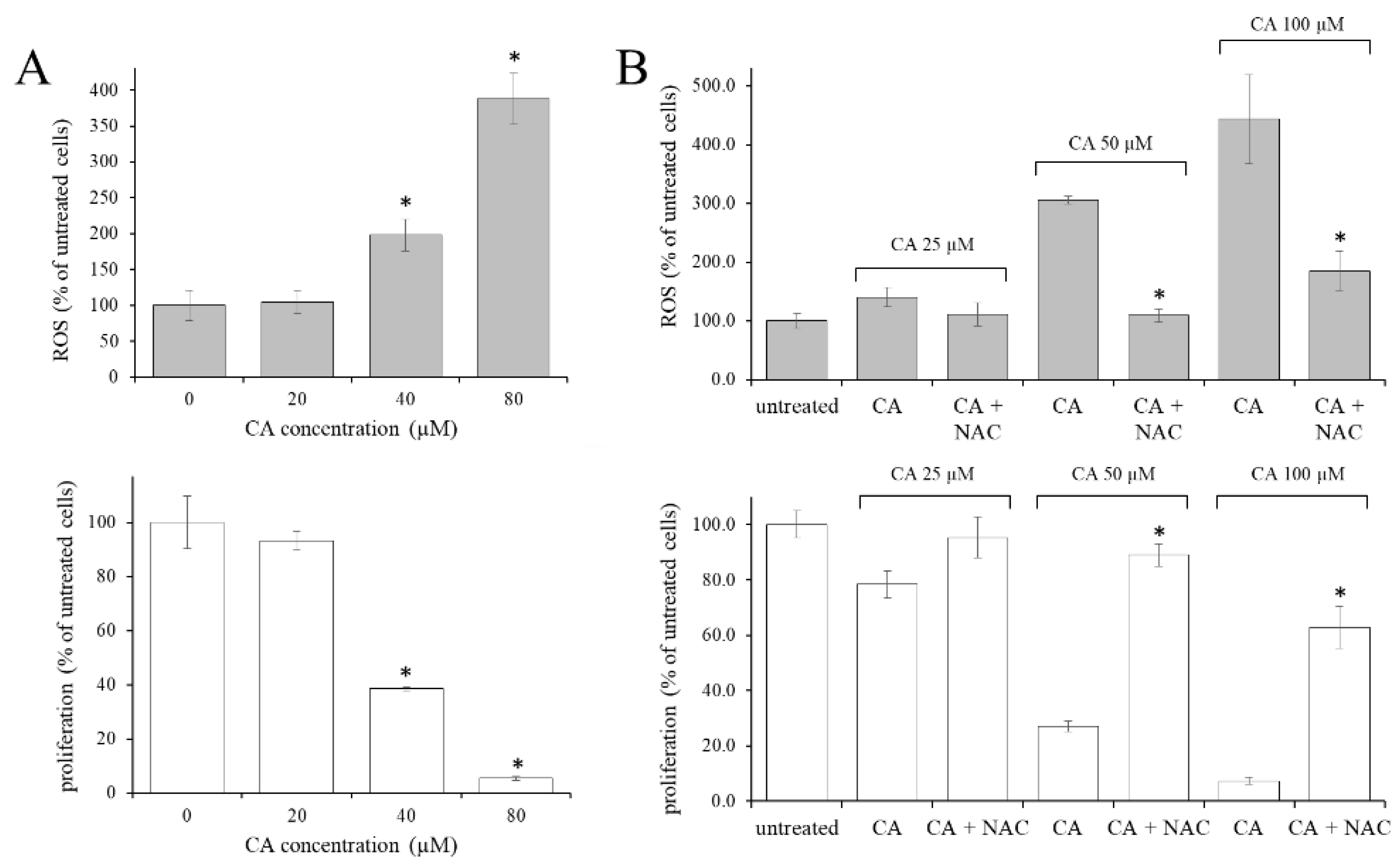
2.1. The Increase in the Intracellular Oxidative State Was Responsible for the Antiproliferative Effect of CA in CML Cells
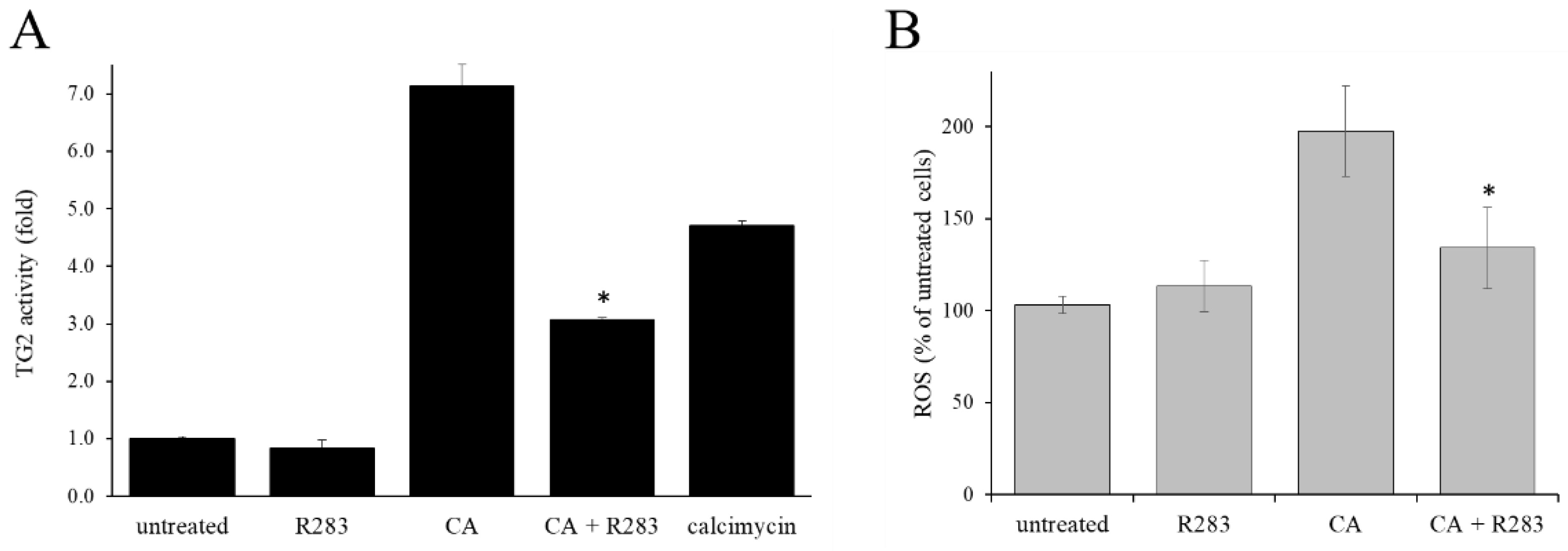
2.2. TG2 Activity Supported the CA-Increased Level of ROS
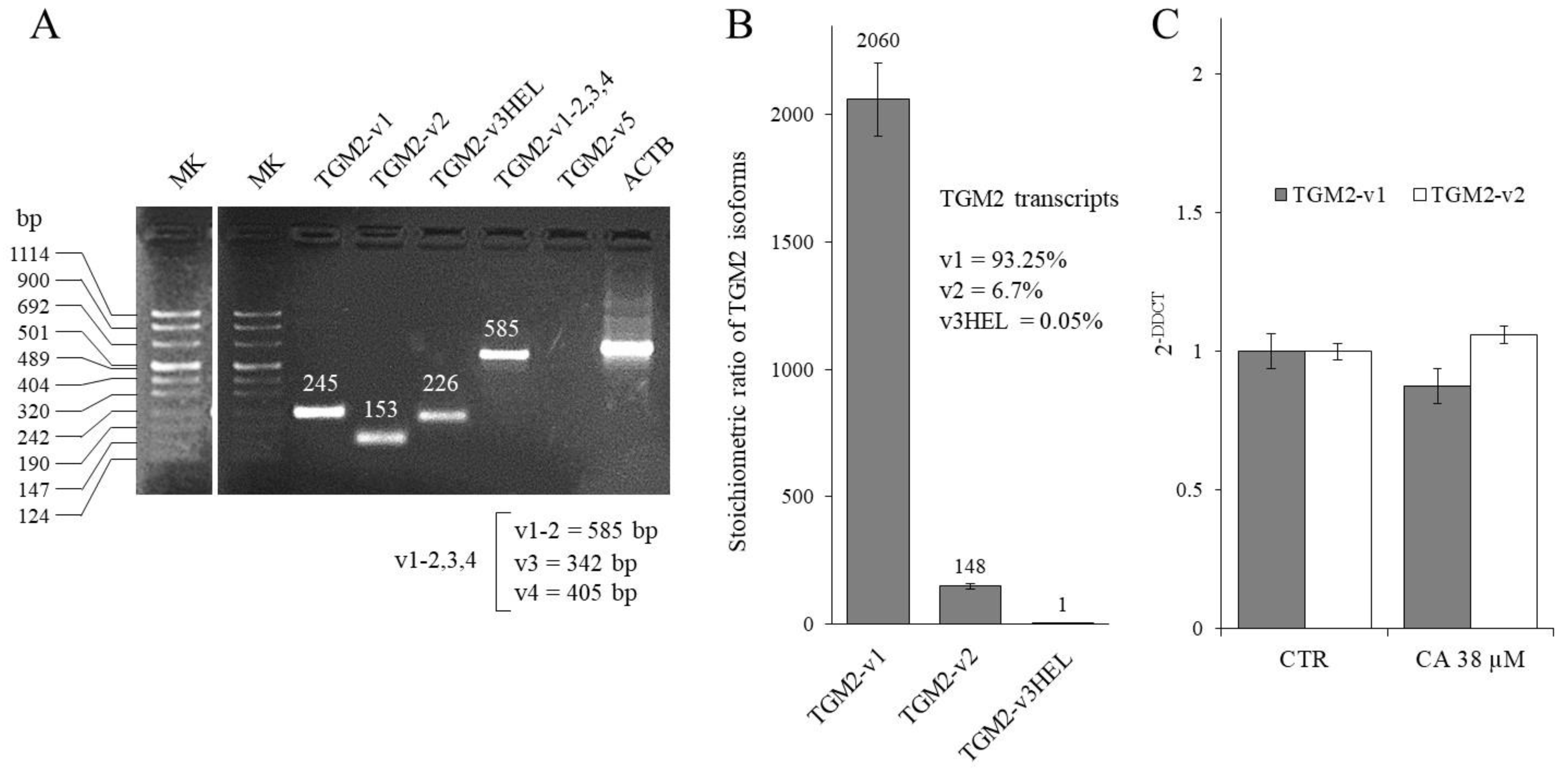
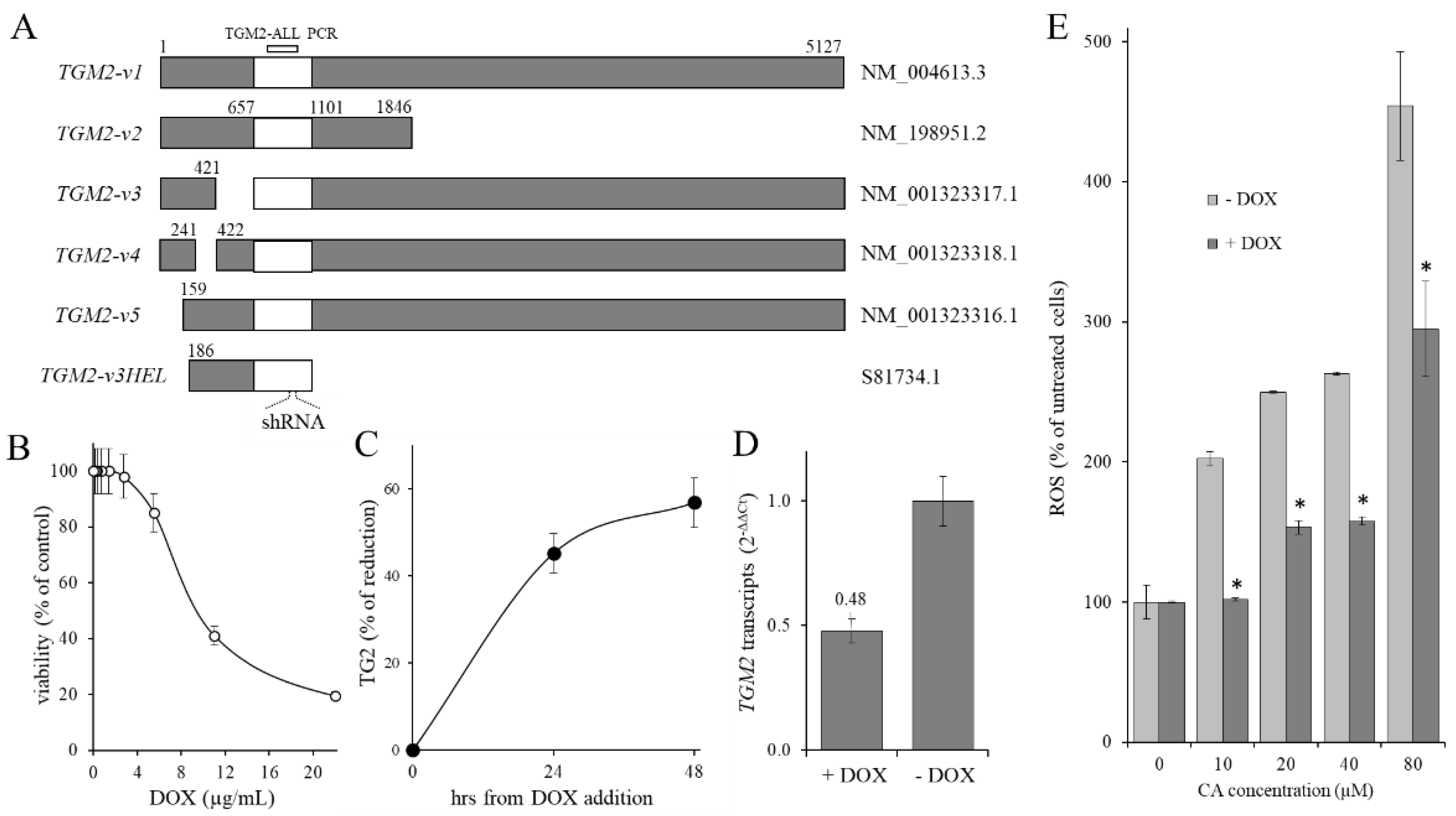
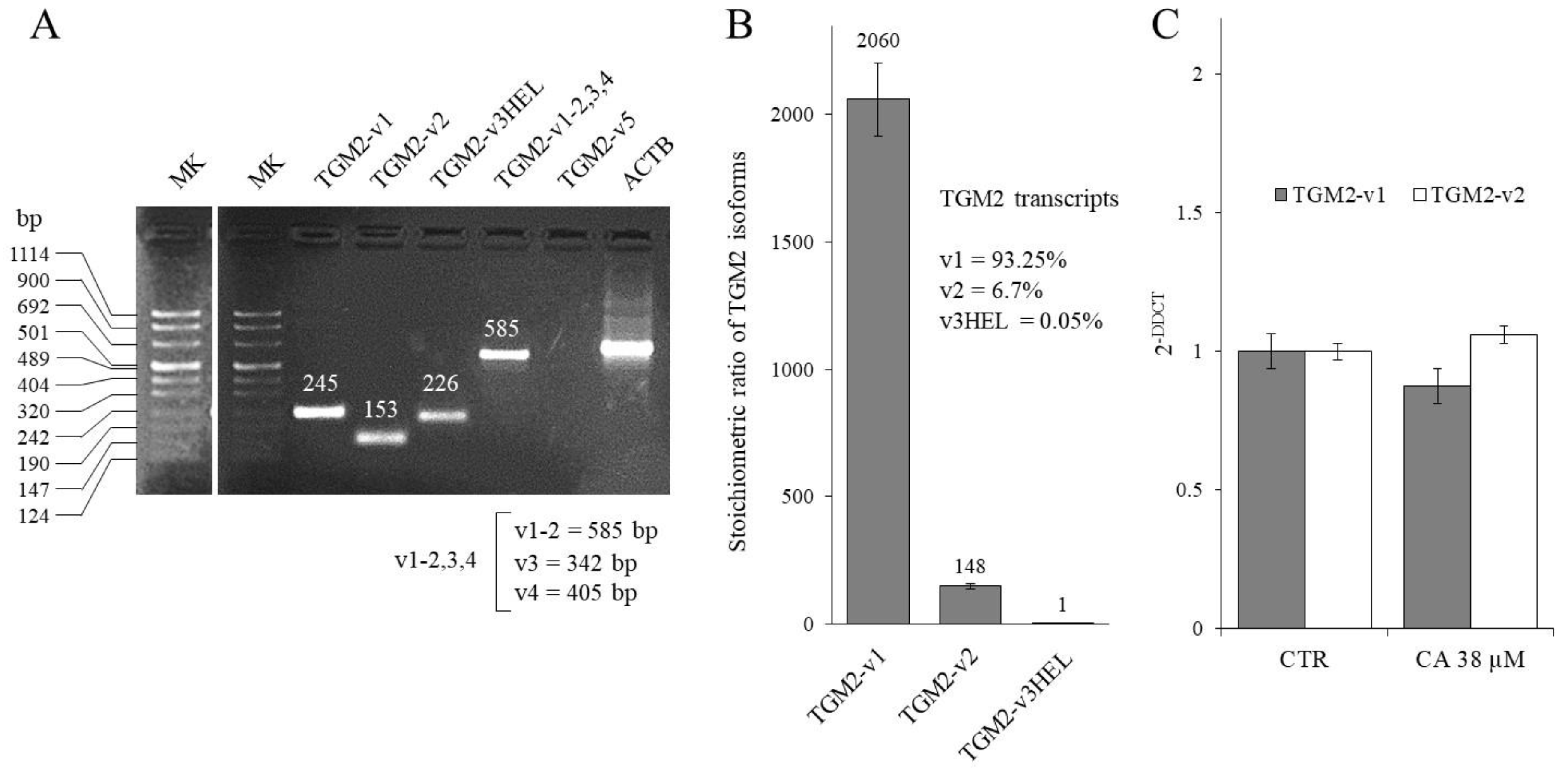
2.3. Effect of CA Treatment on TGM2 Gene Transcription
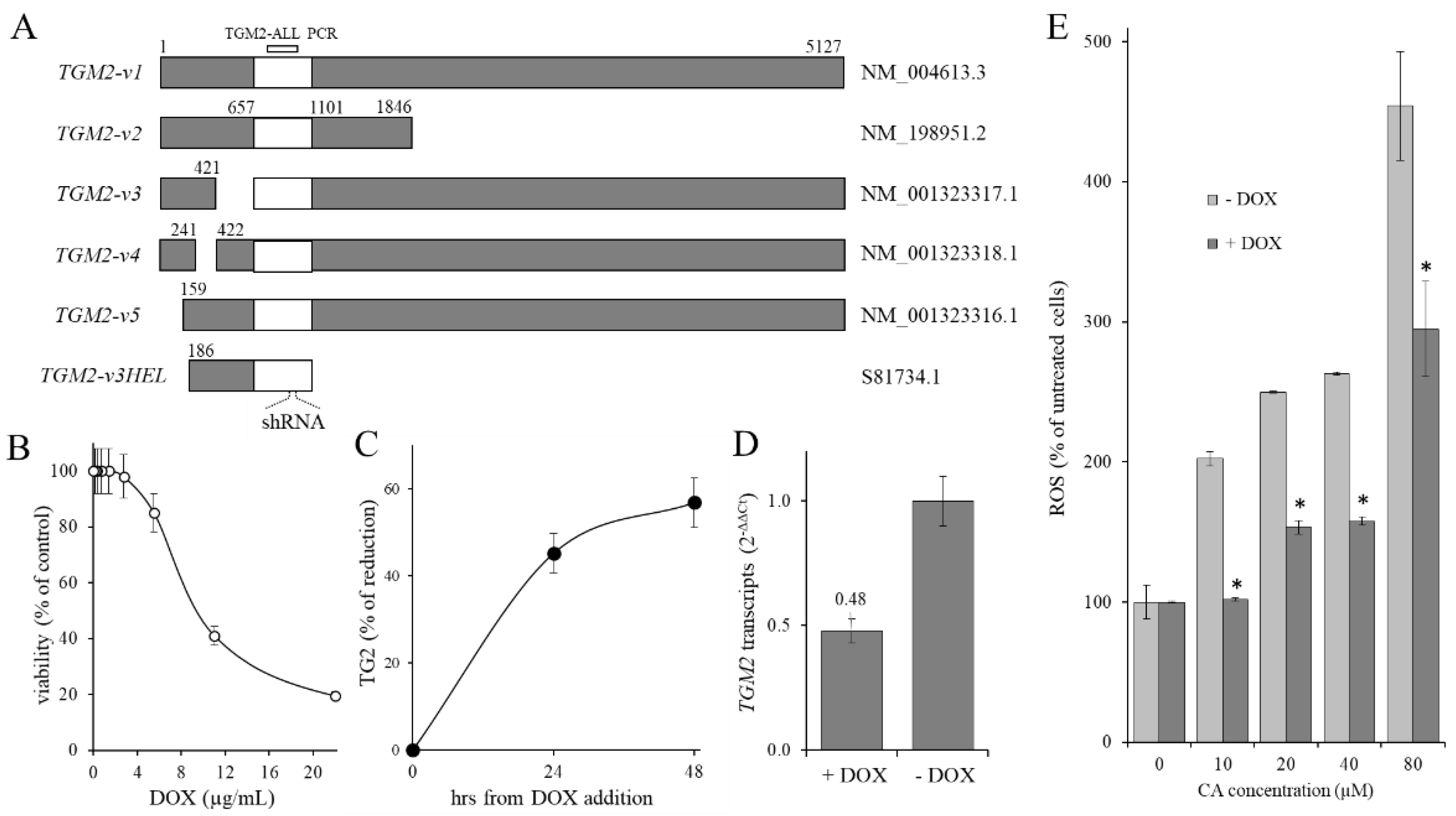
2.4. Contribution of TG2 on CA-Induced ROS Level Increase
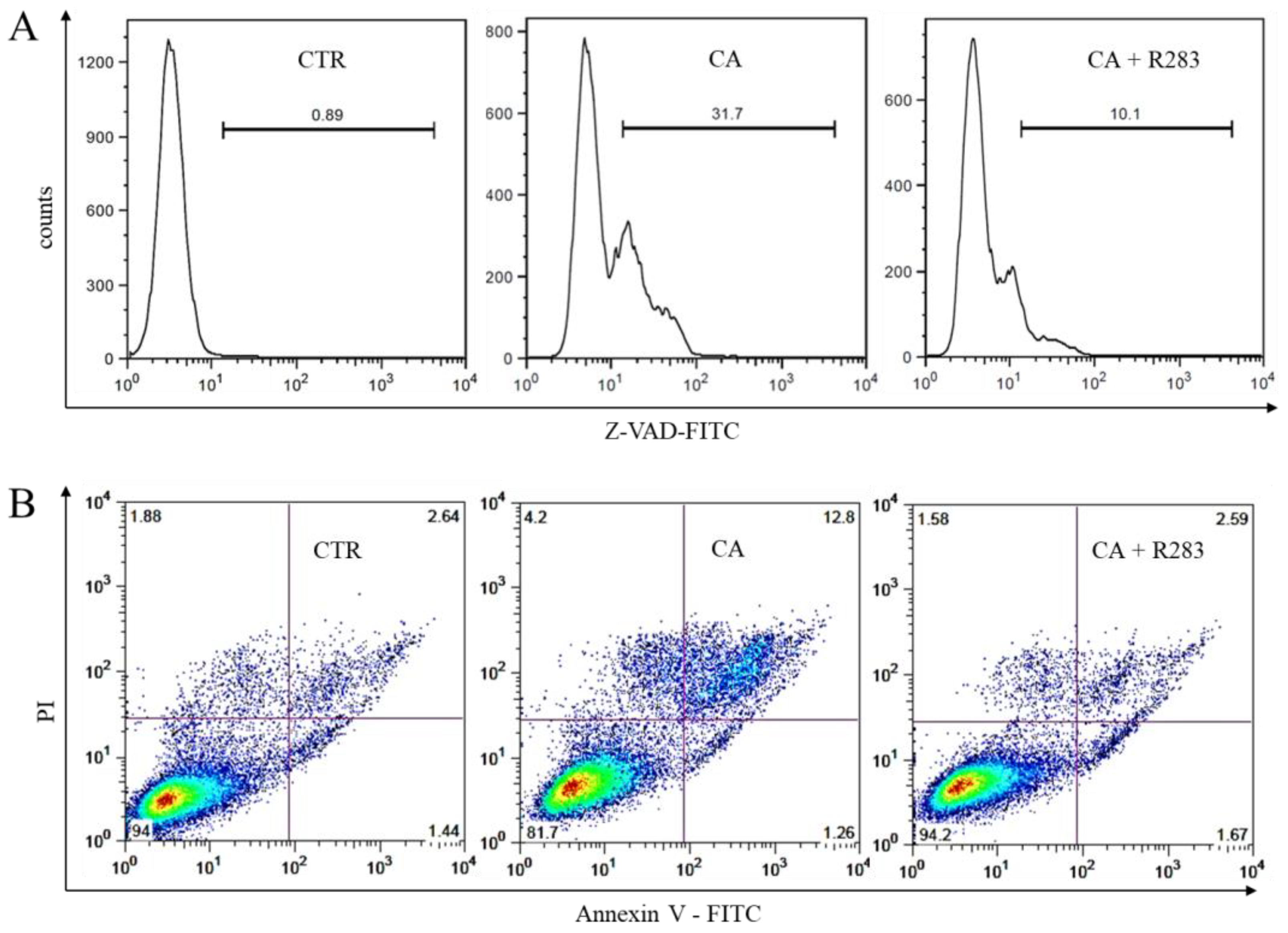
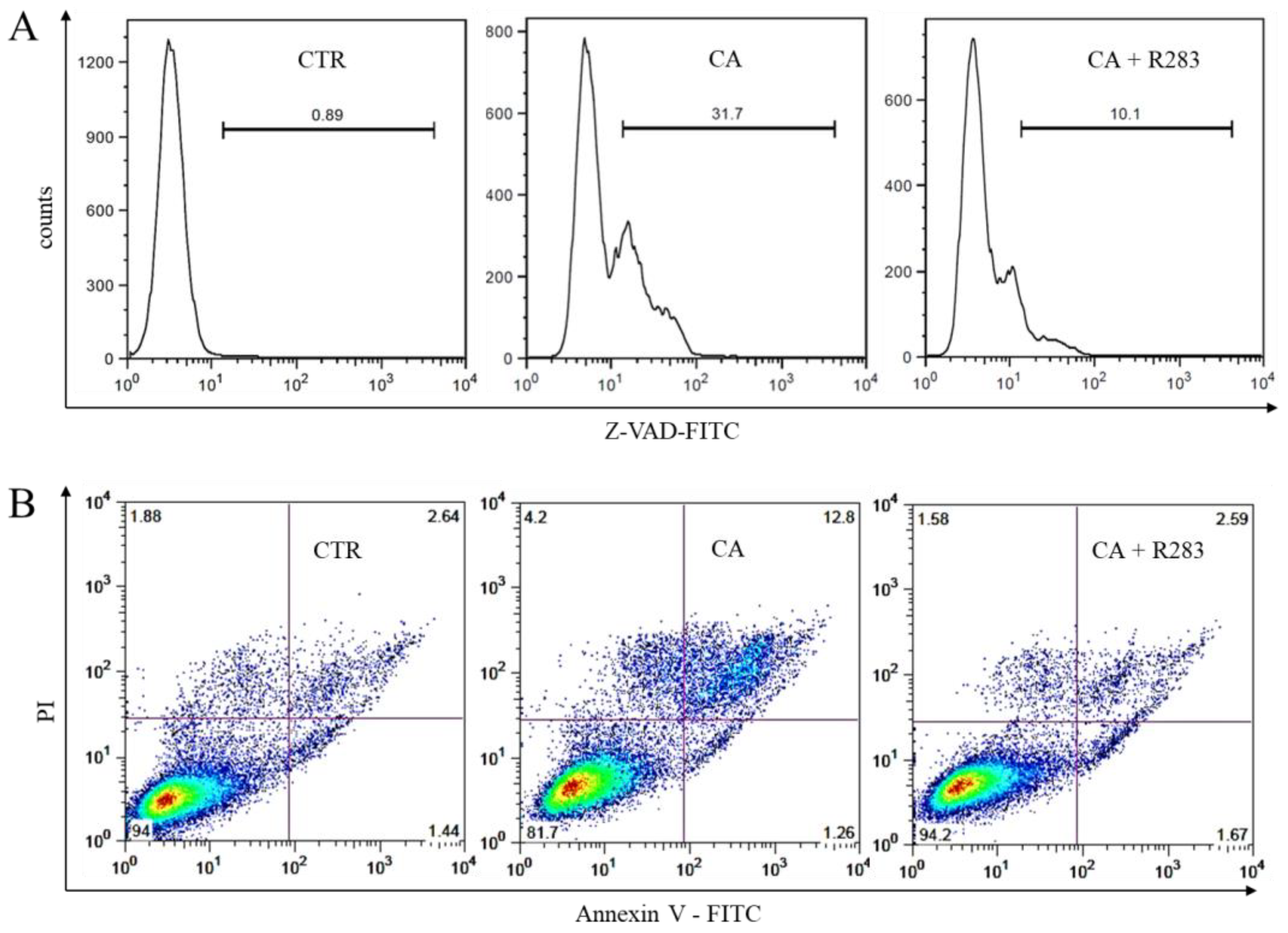
2.5. TG2 Activity Sustained the CA-Triggered Apoptotic Mechanism
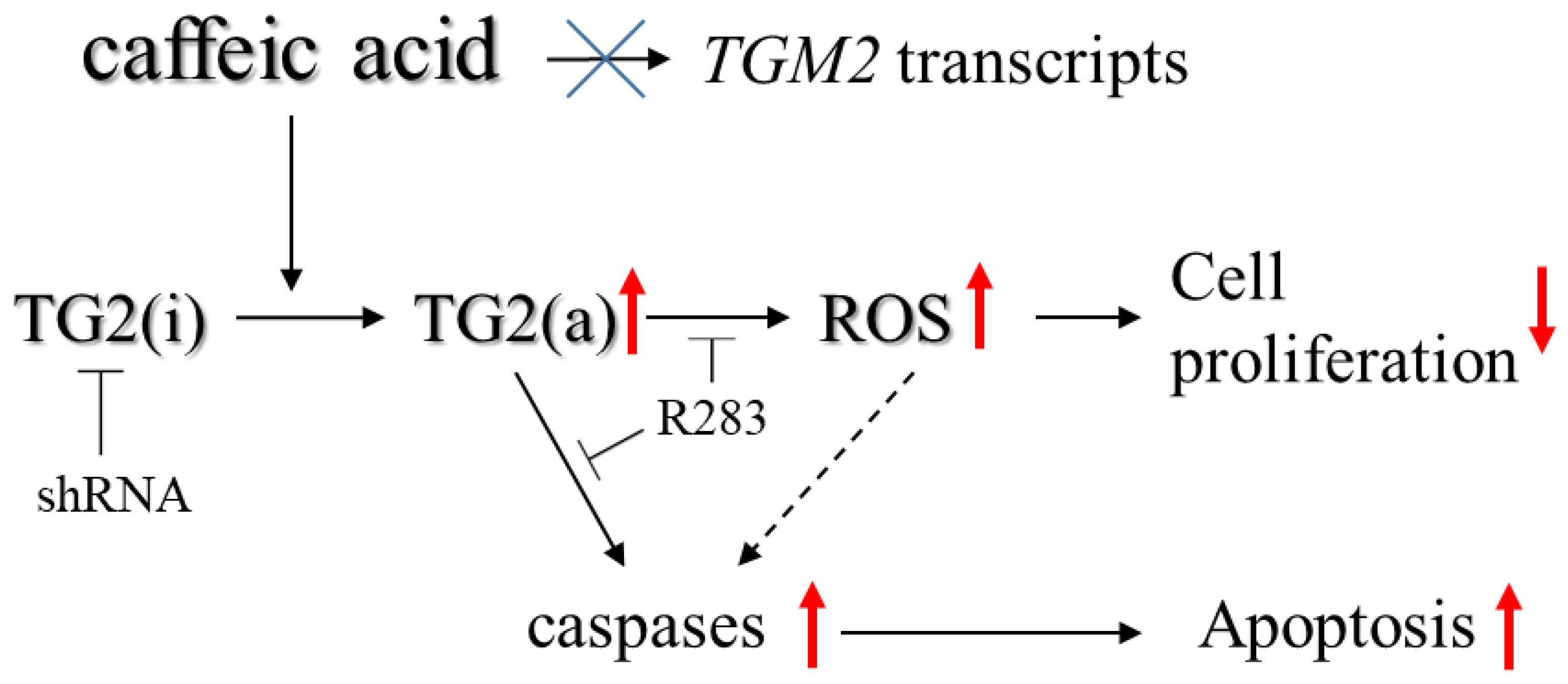
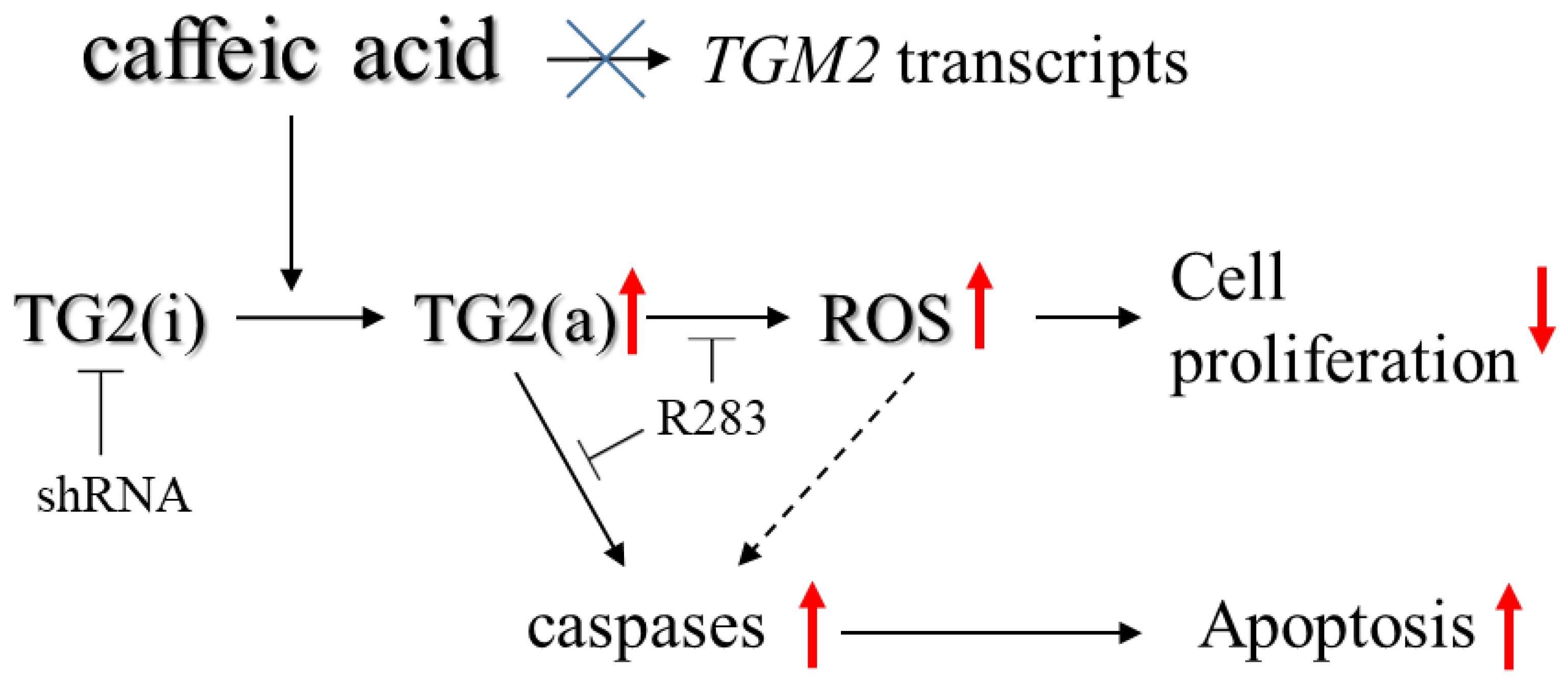
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Viability Test
4.3. RNA Isolation and RT-PCR
4.4. TG2 Activity Assay
4.5. KD-TGM2 Cells Preparation
4.6. Apoptosis Measurement
4.7. Caspase Activity Assay
4.8. Assessment of Intracellular Oxygen-Derived Free Radicals
4.9. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yung, Y.; Lee, E.; Chu, H.T.; Yip, P.K.; Gill, H. Targeting Abnormal Hematopoietic Stem Cells in Chronic Myeloid Leukemia and Philadelphia Chromosome-Negative Classical Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 659. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, C.; De Martino, A.; Mischiati, C.; Feriotto, G.; Beninati, S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med. Sci. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentini, A.; Abbruzzese, A.; Provenzano, B.; Tabolacci, C.; Beninati, S. Transglutaminases: Key regulators of cancer metastasis. Amino Acids. 2013, 44, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, K.; Kojima, S.; Fesus, L. Transglutaminases, Multiple Functional Modifiers and Targets for New Drug Discovery, 1st ed.; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Tatsukawa, H.; Sano, T.; Fukaya, Y.; Ishibashi, N.; Watanabe, M.; Okuno, M.; Moriwaki, H.; Kojima, S. Dual induction of caspase 3- and transglutaminase-dependent apoptosis by acyclic retinoid in hepatocellular carcinoma cells. Mol. Cancer 2011, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katt, W.P.; Antonyak, M.A.; Cerione, R.A. The diamond anniversary of tissue transglutaminase: A protein of many talents. Drug Discov. Today 2018, 23, 575–591. [Google Scholar] [CrossRef] [PubMed]
- Birckbichler, P.J.; Orr, G.R.; Conway, E.; Patterson, M.K., Jr. Transglutaminase activity in normal and transformed cells. Cancer Res. 1977, 37, 1340–1344. [Google Scholar]
- Barnes, R.N.; Bungay, P.J.; Elliott, B.M.; Walton, P.L.; Griffin, M. Alterations in the distribution and activity of transglutam-inase during tumour growth and metastasis. Carcinogenesis 1985, 6, 459–463. [Google Scholar] [CrossRef]
- Mangala, L.S.; Mehta, K. Tissue transglutaminase (TG2) in cancer biology. Prog. Exp. Tumor Res. 2005, 38, 125–138. [Google Scholar]
- Delaine-Smith, R.; Wright, N.; Hanley, C.; Hanwell, R.; Bhome, R.; Bullock, M.; Drifka, C.; Eliceiri, K.; Thomas, G.; Knight, M.; et al. Transglutaminase-2 Mediates the Biomechanical Properties of the Colorectal Cancer Tissue Microenvironment that Contribute to Disease Progression. Cancers 2019, 11, 701. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Moon, Y.; Choi, J.; Yoon, S.; Jung, K.H.; Cheon, J.; Kim, W.; Kim, D.; Lee, C.H.; Kim, S.W.; et al. The GTP binding activity of transglutaminase 2 promotes bone metastasis of breast cancer cells by downregulating mi-croRNA-205. Am. J. Cancer Res. 2019, 9, 597–607. [Google Scholar]
- Roncoroni, L.; Elli, L.; Braidotti, P.; Tosi, D.; Vaira, V.; Tacchini, L.; Lombardo, V.; Branchi, F.; Scricciolo, A.; Doneda, L. Tran-sglutaminase 2 Mediates the Cytotoxicity of Resveratrol in a Human Cholangiocarcinoma and Gallbladder Cancer Cell Lines. Nutr. Cancer 2018, 70, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Suedhoff, T.; Birckbichler, P.J.; Lee, K.N.; Conway, E.; Patterson, M.K., Jr. Differential expression of transglutaminase in human erythroleukemia cells in response to retinoic acid. Cancer Res. 1990, 50, 7830–7834. [Google Scholar] [PubMed]
- Berntorp, E.; Seiving, B.; Stenberg, P. Characterization of transglutaminases in normal and malignant human leukocytes. Scand. J. Haematol. 1985, 34, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.O.; Lim, Y.C.; Kim, Y.M.; Ha, K.S. Transglutaminase 2 promotes both caspase-dependent and caspase-independent apoptotic cell death via the calpain/Bax protein signaling pathway. J. Biol. Chem. 2012, 287, 14377–14388. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, D.; Currò, M.; Ferlazzo, N.; Condello, S.; Ientile, R. Monitoring of transglutaminase 2 under different oxidative stress conditions. Amino Acids 2012, 42, 1037–1043. [Google Scholar] [CrossRef]
- Fraij, B.M.; Birckbichler, P.J.; Patterson, M.K.; Lee, K.N.; Gonzales, R.G. A retinoic acid-inducible mRNA from human erythroleukemia cells encodes a novel tissue transglutaminase homologue. J. Biol. Chem. 1992, 267, 22616–22673. [Google Scholar] [CrossRef]
- Fraij, B.M.; Gonzales, R.A. A third human tissue transglutaminase homologue as a result of alternative gene transcripts. Biochim. Biophys. Acta 1996, 1306, 63–74. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, Z.; Liu, H.; Guo, J.; Long, S. Hybrid molecules based on caffeic acid as potential therapeutics: A focused review. Eur. J. Med. Chem. 2022, 243, 114745. [Google Scholar] [CrossRef]
- Alam, M.; Ahmed, S.; Elasbali, A.M.; Adnan, M.; Alam, S.; Hassan, M.I.; Pasupuleti, V.R. Therapeutic Implications of Caffeic Acid in Cancer and Neurological Diseases. Front. Oncol. 2022, 12, 860508. [Google Scholar] [CrossRef]
- Feriotto, G.; Tagliati, F.; Giriolo, R.; Casciano, F.; Tabolacci, C.; Beninati, S.; Khan, M.T.H.; Mischiati, C. Caffeic Acid Enhances the Anti-Leukemic Effect of Imatinib on Chronic Myeloid Leukemia Cells and Triggers Apoptosis in Cells Sensitive and Resistant to Imatinib. Int. J. Mol. Sci. 2021, 22, 1644. [Google Scholar] [CrossRef]
- Meshram, D.D.; Pike, C.V.S.; Coussons, P.J. Inhibition of Transglutaminase 2 activity increases cisplatin cytotoxicity in a model of human hepatocarcinoma chemotherapy. Eur. J. Pharmacol. 2017, 815, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraij, B.M. The 55 kDa tissue transglutaminase cross-linking active isoform TG induces cell death. Mol. Carcinog. 2015, 54, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.M.; Kim, I. Regulation of Transglutaminase 2 by Oxidative Stress. In Transglutaminases; Springer: Berlin/Heidelberg, Germany, 2015; pp. 315–331. [Google Scholar]
- Phatak, V.M.; Croft, S.M.; Setty, S.G.R.; Scarpellini, A.; Hughes, D.C.; Rees, R.; McArdle, S.; Verderio, E.A. Expres-sion of transglutaminase-2 isoforms in normal human tissues and cancer cell lines: Dysregulation of alternative splicing in cancer. Amino Acids 2013, 44, 33–44. [Google Scholar] [CrossRef]
- Forni, C.; Rossi, M.; Borromeo, I.; Feriotto, G.; Platamone, G.; Tabolacci, C.; Mischiati, C.; Beninati, S. Flavonoids: A Myth or a Reality for Cancer Therapy? Molecules 2021, 26, 3583. [Google Scholar] [CrossRef]
- Min, J.; Shen, H.; Xi, W.; Wang, Q.; Yin, L.; Zhang, Y.; Yu, Y.; Yang, Q.; Wang, Z.N. Synergistic anticancer activity of combined use of caffeic acid with paclitaxel enhances apoptosis of non-small-cell lung cancer H1299 cells in vivo and in vitro. Cell. Physiol. Biochem. 2018, 48, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Camarasa, J.; Escubedo, E.; Adzet, T. Pharmacokinetics of caffeic acid in rats by a high-performance liquid chromatography method. J. Pharm. Biomed. Anal. 1988, 6, 503–510. [Google Scholar] [CrossRef]
- Gonthier, M.P.; Verny, M.A.; Besson, C.; Rémésy, C.; Scalbert, A. Chlorogenic acid bioavailability largely depends on its metabolism by the gut microflora in rats. J. Nutr. 2003, 133, 1853–1859. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Goel, N. Phenolic acids: Natural versatile molecules with promising therapeutic applications. Biotechnol. Rep. 2019, 24, e00370. [Google Scholar] [CrossRef]
- Espíndola, K.M.M.; Ferreira, R.G.; Narvaez, L.E.M.; Rosario, A.C.R.S.; da Silva, A.H.M.; Silva, A.G.B.; Vieira, A.P.O.; Monteiro, M.C. Chemical and Pharmacological Aspects of Caffeic Acid and Its Activity in Hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [Green Version]
- Pelinson, L.P.; Assmann, C.E.; Palma, T.V.; da Cruz, I.; Pillat, M.M.; Mânica, A.; Stefanello, N.; Weis, G.C.C.; de Oliveira Alves, A.; De Andrade, C.M.; et al. Antiproliferative and apoptotic effects of caffeic acid on SK-Mel-28 human melanoma cancer cells. Mol. Biol. Rep. 2019, 46, 2085–2092. [Google Scholar] [CrossRef]
- Khan, F.A.; Maalik, A.; Murtaza, G. Inhibitory mechanism against oxidative stress of caffeic acid. J. Food Drug Anal. 2016, 24, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Chen, I.L.; Chou, C.T.; Liang, W.Z.; Kuo, D.H.; Shieh, P.; Jan, C.R. Effect of caffeic acid on Ca(2+) homeostasis and apoptosis in SCM1 human gastric cancer cells. Arch. Toxicol. 2013, 87, 2141–2150. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Shin, D.H.; Zheng, H.; Kang, J.S.; Kim, W.K.; Kim, S.J. Inhibition of store-operated Ca2+ entry channels and K+ channels by caffeic acid phenethylester in T lymphocytes. Eur. J. Pharmacol. 2009, 612, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bose, J.S.; Gangan, V.; Jain, S.K.; Manna, S.K. Novel caffeic acid ester derivative induces apoptosis by expressing FasL and downregulating NF-KappaB: Potentiation of cell death mediated by chemotherapeutic agents. J. Cell. Physiol. 2009, 218, 653–662. [Google Scholar] [CrossRef]
- Slaughter, T.F.; Achyuthan, K.E.; Lai, T.S.; Greenberg, C.S. A microtiter plate transglutaminase assay utilizing 5-(biotinamido)pentylamine as substrate. Anal. Biochem. 1992, 205, 166–171. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feriotto, G.; Tagliati, F.; Brunello, A.; Beninati, S.; Tabolacci, C.; Mischiati, C. A Central Contribution of TG2 Activity to the Antiproliferative and Pro-Apoptotic Effects of Caffeic Acid in K562 Cells of Human Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 15004. https://doi.org/10.3390/ijms232315004
Feriotto G, Tagliati F, Brunello A, Beninati S, Tabolacci C, Mischiati C. A Central Contribution of TG2 Activity to the Antiproliferative and Pro-Apoptotic Effects of Caffeic Acid in K562 Cells of Human Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2022; 23(23):15004. https://doi.org/10.3390/ijms232315004
Chicago/Turabian StyleFeriotto, Giordana, Federico Tagliati, Arianna Brunello, Simone Beninati, Claudio Tabolacci, and Carlo Mischiati. 2022. "A Central Contribution of TG2 Activity to the Antiproliferative and Pro-Apoptotic Effects of Caffeic Acid in K562 Cells of Human Chronic Myeloid Leukemia" International Journal of Molecular Sciences 23, no. 23: 15004. https://doi.org/10.3390/ijms232315004