


Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease
Abstract
1. Introduction
2. Case Presentation
2.1. Methods
2.2. Results
2.2.1. Study Cohort
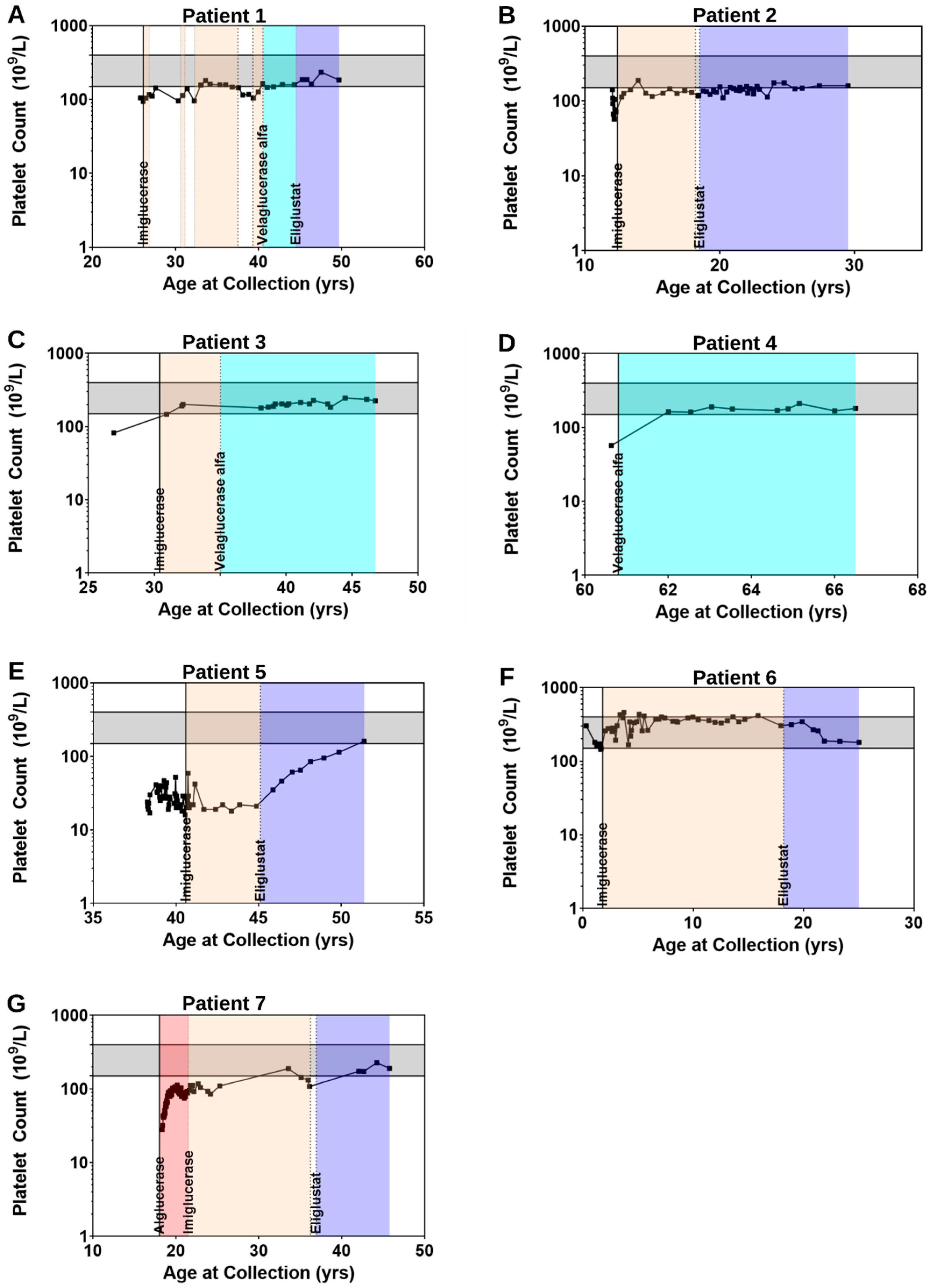
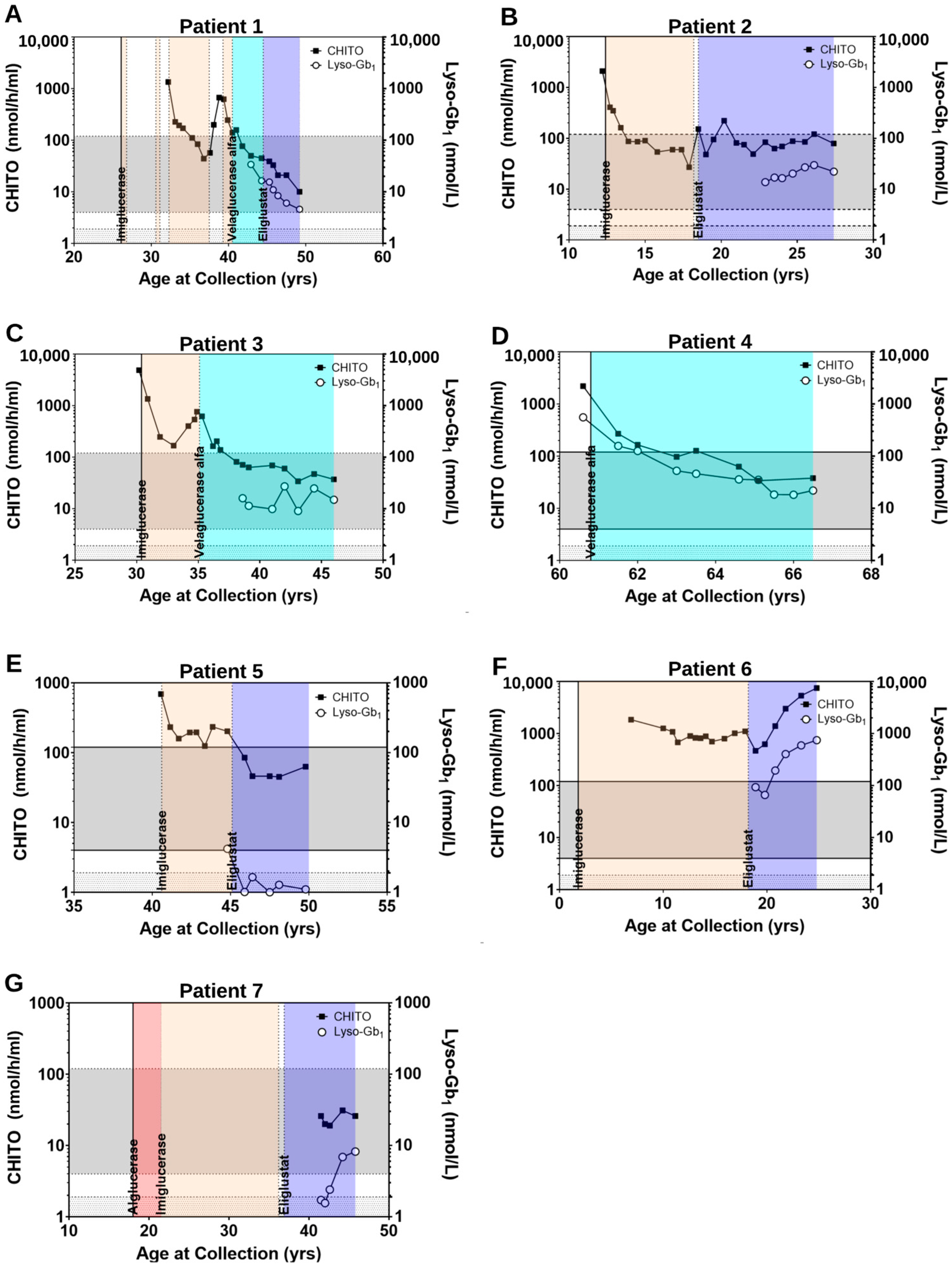
2.2.2. Clinical Parameters Improve and CHITO Activity Normalizes with Treatment
2.2.3. Clinical Parameters Improve and Biomarker Levels Normalize upon Switch from ERT to SRT
2.2.4. Clinical Parameters and Biomarker Levels Worsening upon Transition to SRT: Situations of Non-Compliance
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Gaucher disease. In Advances in Human Genetics 21; Springer: Berlin/Heidelberg, Germany, 1993; pp. 377–441. [Google Scholar]
- Hollak, C.E.; van Weely, S.; van Oers, M.H.; Aerts, J.M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Investig. 1994, 93, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Boot, R.G.; Renkema, G.H.; Van Weely, S.; Jones, S.; Hollak, C.E.; van Oers, M. Molecular and biochemical abnormalities of Gaucher disease: Chitotriosidase, a newly identified biochemical marker. In Seminars in Hematology; Elsevier: Amsterdam, The Netherlands, 1995; pp. 10–13. [Google Scholar]
- Guo, Y.; He, W.; Boer, A.M.; Wevers, R.A.; de Bruijn, A.M.; Groener, J.E.; Hollak, C.E.; Aerts, J.M.; Galjaard, H.; van Diggelen, O.P. Elevated plasma chitotriosidase activity in various lysosomal storage disorders. J. Inherit. Metab. Dis. 1995, 18, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.; Cameli, P.; Lanzarone, N.; Carobene, L.; Bianchi, N.; Fui, A.; Rizzi, L.; Bergantini, L.; Cillis, G.; d’Alessandro, M.; et al. Chitotriosidase: A biomarker of activity and severity in patients with sarcoidosis. Respir. Res. 2020, 21, 6. [Google Scholar] [CrossRef]
- Boot, R.G.; Renkema, G.H.; Verhoek, M.; Strijland, A.; Bliek, J.; de Meulemeester, T.M.; Mannens, M.M.; Aerts, J.M. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J. Biol. Chem. 1998, 273, 25680–25685. [Google Scholar] [CrossRef]
- Deegan, P.B.; Moran, M.T.; McFarlane, I.; Schofield, J.P.; Boot, R.G.; Aerts, J.M.; Cox, T.M. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol. Dis. 2005, 35, 259–267. [Google Scholar] [CrossRef]
- van Dussen, L.; Hendriks, E.J.; Groener, J.E.; Boot, R.G.; Hollak, C.E.; Aerts, J.M. Value of plasma chitotriosidase to assess non-neuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J. Inherit. Metab. Dis. 2014, 37, 991–1001. [Google Scholar] [CrossRef]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Aerts, J.; Kuo, C.L.; Lelieveld, L.T.; Boer, D.E.C.; van der Lienden, M.J.C.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Mellgard, B.; Dinh, Q.; Lan, L.; Qiu, Y.; Cozma, C.; Eichler, S.; Böttcher, T.; Zimran, A. Reductions in glucosylsphingosine (lyso-Gb1) in treatment-naïve and previously treated patients receiving velaglucerase alfa for type 1 Gaucher disease: Data from phase 3 clinical trials. Mol. Genet. Metab. 2017, 122, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [PubMed]
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long Term Follow-Up of 103 Untreated Adult Patients with Type 1 Gaucher Disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef] [PubMed]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment Efficiency in Gaucher Patients Can Reliably Be Monitored by Quantification of Lyso-Gb1 Concentrations in Dried Blood Spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef] [PubMed]
- Dinur, T.; Grittner, U.; Revel-Vilk, S.; Becker-Cohen, M.; Istaiti, M.; Cozma, C.; Rolfs, A.; Zimran, A. Impact of Long-Term Enzyme Replacement Therapy on Glucosylsphingosine (Lyso-Gb1) Values in Patients with Type 1 Gaucher Disease: Statistical Models for Comparing Three Enzymatic Formulations. Int. J. Mol. Sci. 2021, 22, 7699. [Google Scholar] [CrossRef]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef]
- Kartha, R.V.; Terluk, M.R.; Brown, R.; Travis, A.; Mishra, U.R.; Rudser, K.; Lau, H.; Jarnes, J.R.; Cloyd, J.C.; Weinreb, N.J. Patients with Gaucher disease display systemic oxidative stress dependent on therapy status. Mol. Genet. Metab. Rep. 2020, 25, 100667. [Google Scholar] [CrossRef]
- Smid, B.E.; Ferraz, M.J.; Verhoek, M.; Mirzaian, M.; Wisse, P.; Overkleeft, H.S.; Hollak, C.E.; Aerts, J.M. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J. Rare Dis. 2016, 11, 28. [Google Scholar] [CrossRef]
- Mistry, P.K.; Balwani, M.; Charrow, J.; Kishnani, P.; Niederau, C.; Underhill, L.H.; McClain, M.R. Real-world effectiveness of eliglustat in treatment-naïve and switch patients enrolled in the International Collaborative Gaucher Group Gaucher Registry. Am. J. Hematol. 2020, 95, 1038–1046. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Aggio, M.C.; Andersson, H.C.; Andria, G.; Charrow, J.; Clarke, J.T.; Erikson, A.; Giraldo, P.; Goldblatt, J.; Hollak, C.; et al. Gaucher disease type 1: Revised recommendations on evaluations and monitoring for adult patients. Semin. Hematol. 2004, 41 (Suppl. S5), 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, P.; Baris, H.; De Meirleir, L.; Di Rocco, M.; El-Beshlawy, A.; Huemer, M.; Martins, A.M.; Nascu, I.; Rohrbach, M.; Steinbach, L.; et al. Revised recommendations for the management of Gaucher disease in children. Eur. J. Pediatr. 2013, 172, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Prakash-Cheng, A.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: Consensus recommendations. J. Pediatr. 2004, 144, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Baldellou, A.; Andria, G.; Campbell, P.E.; Charrow, J.; Cohen, I.J.; Grabowski, G.A.; Harris, C.M.; Kaplan, P.; McHugh, K.; Mengel, E.; et al. Paediatric non-neuronopathic Gaucher disease: Recommendations for treatment and monitoring. Eur. J. Pediatr. 2004, 163, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Beasley, J.; McCaw, P.; Zhang, H.; Young, S.P.; Stiles, A.R. Combined analysis of plasma or serum glucosylsphingosine and globotriaosylsphingosine by UPLC-MS/MS. Clin. Chim. Acta 2020, 511, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Thielke, R.; Payne, J.; Gonzalez, N.; Conde, J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009, 42, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Taylor, R.; Minor, B.L.; Elliott, V.; Fernandez, M.; O’Neal, L.; McLeod, L.; Delacqua, G.; Delacqua, F.; Kirby, J.; et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019, 95, 103208. [Google Scholar] [CrossRef]
- Hollak, C.E.; vom Dahl, S.; Aerts, J.M.; Belmatoug, N.; Bembi, B.; Cohen, Y.; Collin-Histed, T.; Deegan, P.; van Dussen, L.; Giraldo, P.; et al. Force majeure: Therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol. Dis. 2010, 44, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.I.; DeArmey, S.; Cope, H.; Rairikar, M.; Kishnani, P.S. Treatment of profound thrombocytopenia in a patient with Gaucher disease type 1: Is there a role for substrate reduction therapy. Mol. Genet. Metab. Rep. 2017, 12, 82–84. [Google Scholar] [CrossRef]
- Weinreb, N.J.; Goker-Alpan, O.; Kishnani, P.S.; Longo, N.; Burrow, T.A.; Bernat, J.A.; Gupta, P.; Henderson, N.; Pedro, H.; Prada, C.E.; et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol. Genet. Metab. 2022, 136, 4–21. [Google Scholar] [CrossRef]
- Pavlova, E.V.; Archer, J.; Wang, S.; Dekker, N.; Aerts, J.M.; Karlsson, S.; Cox, T.M. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J. Pathol. 2015, 235, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Kuter, D.J.; Salek, S.S.; Belmatoug, N.; Bembi, B.; Bright, J.; Vom Dahl, S.; Deodato, F.; Di Rocco, M.; Göker-Alpan, O.; et al. Presenting signs and patient co-variables in Gaucher disease: Outcome of the Gaucher Earlier Diagnosis Consensus (GED-C) Delphi initiative. Intern. Med. J. 2019, 49, 578–591. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Eliglustat: A Review in Gaucher Disease Type 1. Drugs 2015, 75, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, J.; Underhill, L.H.; Taylor, J.S.; Angell, J.; Peterschmitt, M.J. Clinical response to eliglustat in treatment-naïve patients with Gaucher disease type 1: Post-hoc comparison to imiglucerase-treated patients enrolled in the International Collaborative Gaucher Group Gaucher Registry. Mol. Genet. Metab. Rep. 2016, 8, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Rhead, W.; Charrow, J.; Shankar, S.P.; Bavdekar, A.; Longo, N.; Mardach, R.; Harmatz, P.; Hangartner, T.; Lee, H.M.; et al. Long-term velaglucerase alfa treatment in children with Gaucher disease type 1 naïve to enzyme replacement therapy or previously treated with imiglucerase. Mol. Genet. Metab. 2016, 117, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.E.; Balwani, M.; Nazarenko, I.; Prakash-Cheng, A.; Desnick, R.J. Type 1 Gaucher disease: Null and hypomorphic novel chitotriosidase mutations-implications for diagnosis and therapeutic monitoring. Hum. Mutat. 2007, 28, 866–873. [Google Scholar] [CrossRef]
- Schoonhoven, A.; Rudensky, B.; Elstein, D.; Zimran, A.; Hollak, C.E.M.; Groener, J.E.; Aerts, J.M.F.G. Monitoring of Gaucher patients with a novel chitotriosidase assay. Clin. Chim. Acta 2007, 381, 136–139. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Fuller, M.; Zimran, A. Value of Glucosylsphingosine (Lyso-Gb1) as a Biomarker in Gaucher Disease: A Systematic Literature Review. Int. J. Mol. Sci. 2020, 21, 7159. [Google Scholar] [CrossRef]
- Pleat, R.; Cox, T.M.; Burrow, T.A.; Giraldo, P.; Goker-Alpan, O.; Rosenbloom, B.E.; Croal, L.R.; Underhill, L.H.; Gaemers, S.J.; Peterschmitt, M.J. Stability is maintained in adults with Gaucher disease type 1 switched from velaglucerase alfa to eliglustat or imiglucerase: A sub-analysis of the eliglustat ENCORE trial. Mol. Genet. Metab. Rep. 2016, 9, 25–28. [Google Scholar] [CrossRef]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristic | Patients (n = 7) |
|---|---|
| Age at diagnosis in years, mean (range) | 23.3 (1.5–60) |
| Sex, n (%) | |
| Male | 5 (71) |
| Female | 2 (29) |
| Race, n (%) | |
| Caucasian | 7 (100) |
| Subtype, n (%) | |
| Type 1 | 6 (86) |
| Type 3 | 1 (14) |
| Genotype, n (%) | |
| p.Asn409Ser/p.Asn409Ser | 5 (29) |
| p.Leu483Pro/p.Leu483Pro | 1 (14) |
| p.Asn409Ser/p.Arg159Trp | 1 (14) |
| Splenectomy, n (%) | 0 |
| Naïve & treatment status, n (%) | |
| Naïve | 3 (43) |
| Treated non-naïve | 4 (57) |
| Treatment type, n (%) | |
| ERT | 1 (14) |
| ERT to ERT switch | 1 (14) |
| ERT to ERT to SRT switch | 1 (14) |
| ERT to SRT switch | 4 (57) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gayed, M.M.; Jung, S.-H.; Huggins, E.; Rodriguez-Rassi, E.; DeArmey, S.; Kishnani, P.S.; Stiles, A.R. Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease. Int. J. Mol. Sci. 2022, 23, 14938. https://doi.org/10.3390/ijms232314938
Gayed MM, Jung S-H, Huggins E, Rodriguez-Rassi E, DeArmey S, Kishnani PS, Stiles AR. Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease. International Journal of Molecular Sciences. 2022; 23(23):14938. https://doi.org/10.3390/ijms232314938
Chicago/Turabian StyleGayed, Matthew M., Seung-Hye Jung, Erin Huggins, Eleanor Rodriguez-Rassi, Stephanie DeArmey, Priya Sunil Kishnani, and Ashlee R. Stiles. 2022. "Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease" International Journal of Molecular Sciences 23, no. 23: 14938. https://doi.org/10.3390/ijms232314938
APA StyleGayed, M. M., Jung, S.-H., Huggins, E., Rodriguez-Rassi, E., DeArmey, S., Kishnani, P. S., & Stiles, A. R. (2022). Glucosylsphingosine (Lyso-Gb1): An Informative Biomarker in the Clinical Monitoring of Patients with Gaucher Disease. International Journal of Molecular Sciences, 23(23), 14938. https://doi.org/10.3390/ijms232314938