

Innate Immune System Activation, Inflammation and Corneal Wound Healing


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction—The Cornea
2. Corneal Wound Healing
2.1. The Healing Process
2.2. Importance of the Limbal Stem Cell Population and Certain Signaling Pathways
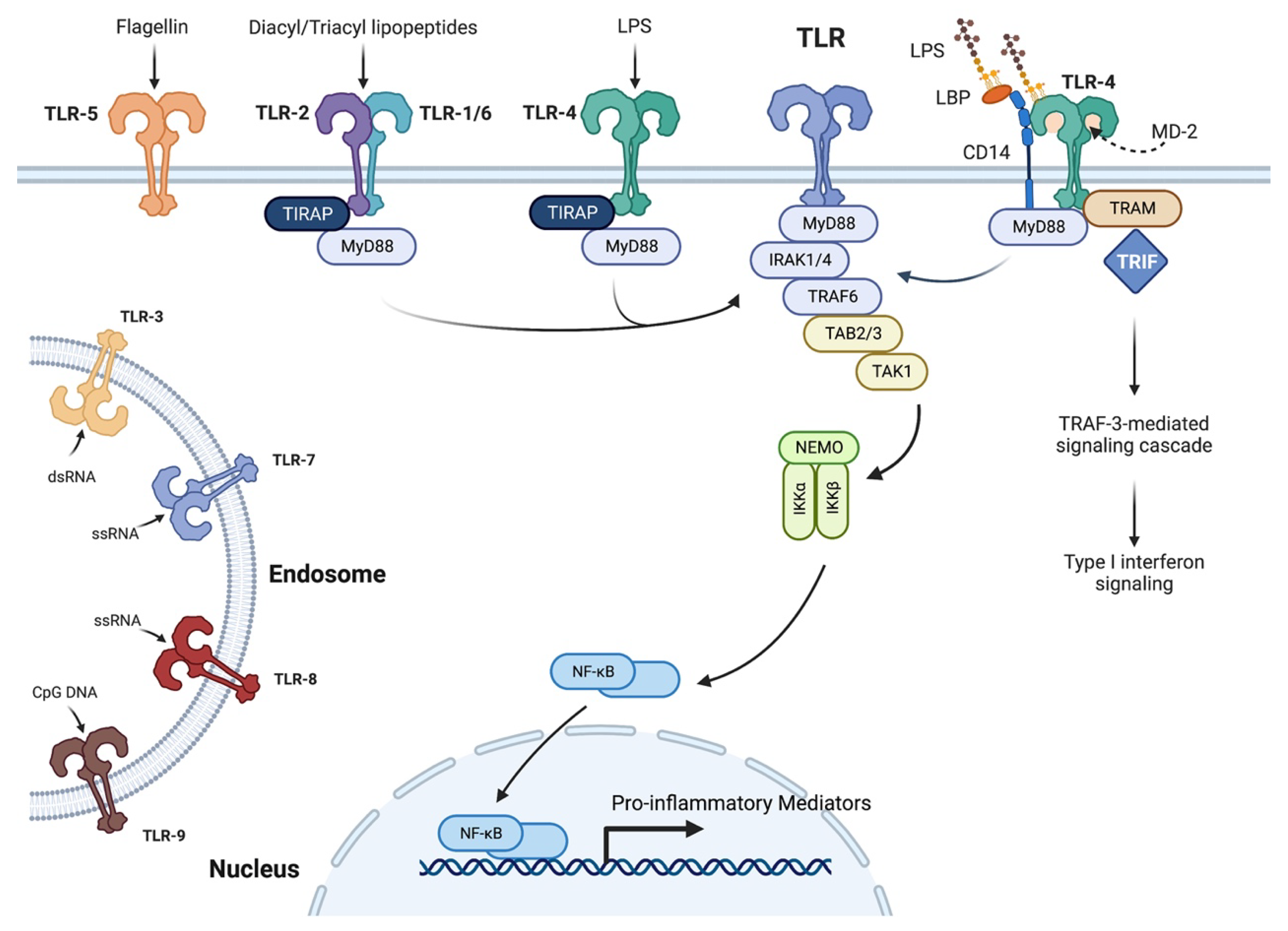
2.3. Importance of the Innate Immune System and Cytokines in Wound Healing and Corneal Inflammation
2.3.1. Role of IL-1 in Wound Healing and Corneal Inflammation
2.3.2. Role of TNF-α and IL-6 in Corneal Inflammation
3. Injury-Induced Corneal Inflammation and Its Role in Wound Healing
3.1. Infectious Corneal Inflammation (Nonsterile)
3.1.1. Bacterial Infection
Pseudomonas aeruginosa
Staphylococcus aureus
3.1.2. Fungal Infection
Aspergillus
Fusarium
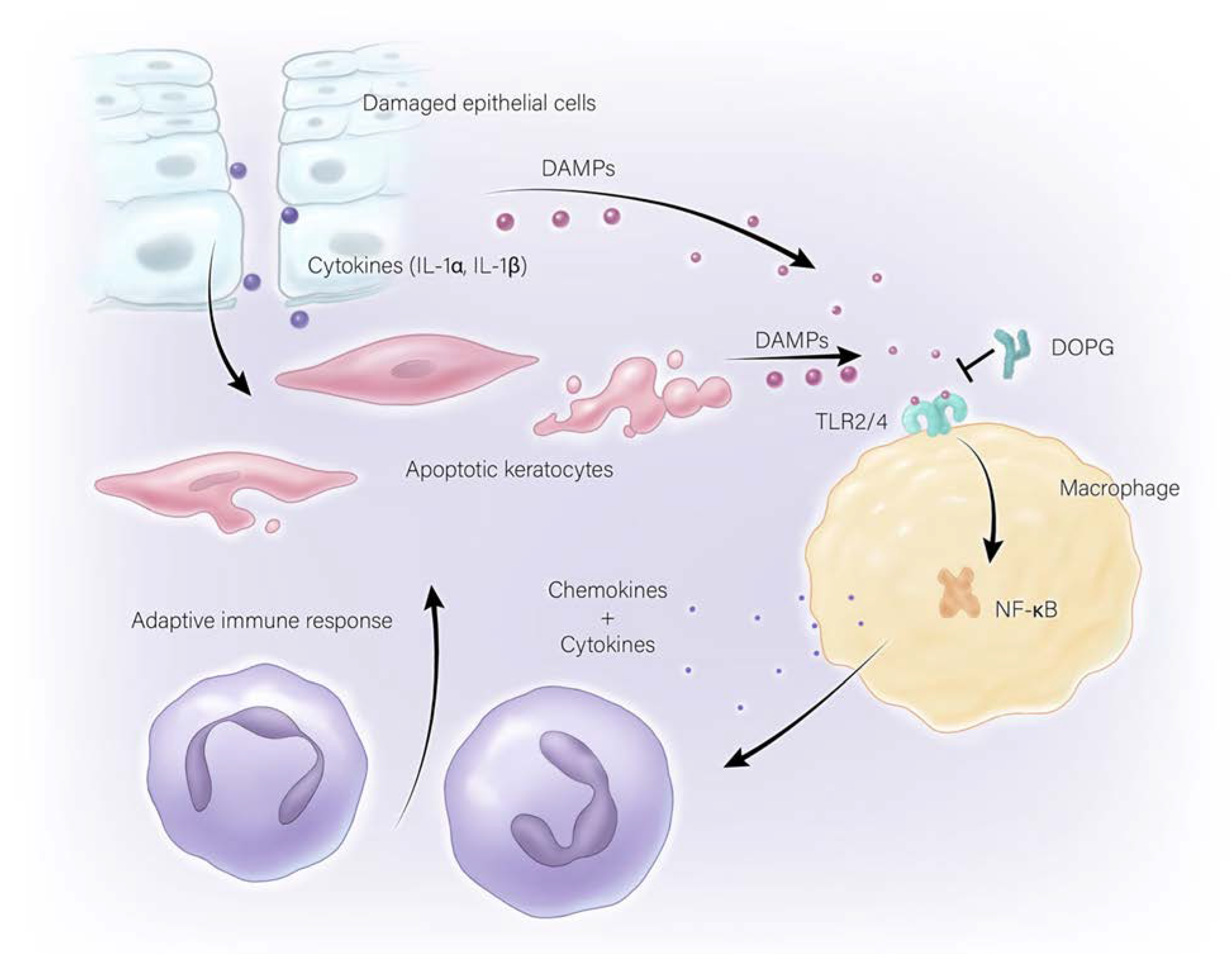
3.2. Sterile Corneal Inflammation (Non-Infectious)
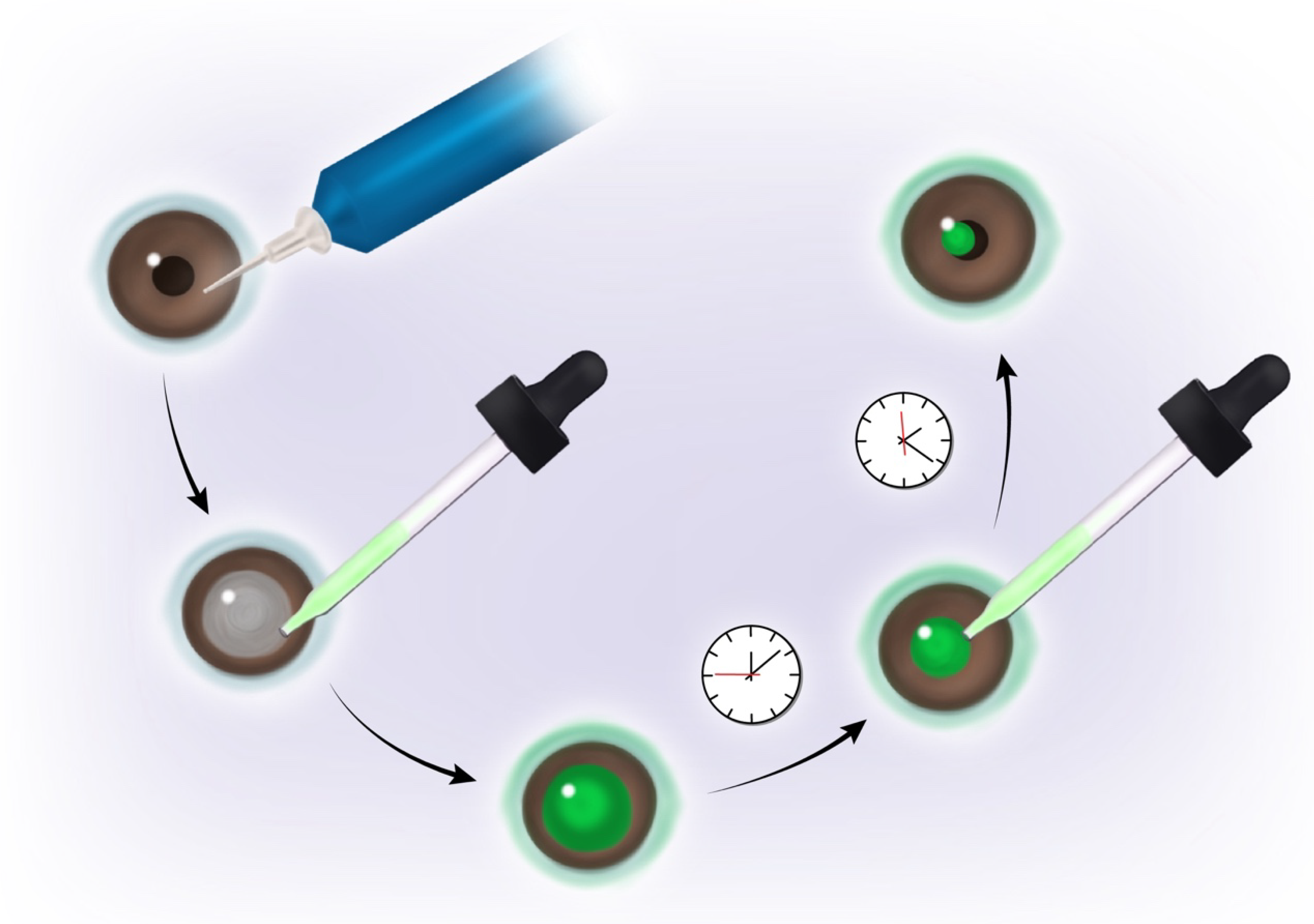
4. Diabetes and Corneal Wound Healing
5. Anti-Inflammatory Actions of Phosphatidylglycerol
6. Phosphatidylglycerol as a Potential Therapeutic Treatment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bollag, W.B.; Olala, L.O.; Xie, D.; Lu, X.; Qin, H.; Choudhary, V.; Patel, R.; Bogorad, D.; Estes, A.; Watsky, M. Dioleoylphosphatidylglycerol Accelerates Corneal Epithelial Wound Healing. Investig. Ophthalmol. Vis. Sci. 2020, 61, 29. [Google Scholar] [CrossRef] [PubMed]
- Dua, H.S.; Gomes, J.A.; Singh, A. Corneal epithelial wound healing. Br. J. Ophthalmol. 1994, 78, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Mescher, A.L. The Eye & Ear: Special Sense Organs. In Junqueira’s Basic Histology: Text and Atlas, 15th ed.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Lee, T.N. The ins and outs of corneal wound healing. Rev. Optom. 2016. Available online: https://www.reviewofoptometry.com/article/the-ins-and-outs-of-corneal-wound-healing (accessed on 23 August 2022).
- Alshamahi, E.Y.A.; Al Nahary, A.A.; Al Shamahy, H.A.; Al Magrami, R.T.F.; Alhowthi, M.A. Epidemiology and aetiological diagnosis of corneal ulceration in Sana’a City, Yemen. World J. Ophthalmol. Vis. Res. 2019, 2. [Google Scholar] [CrossRef]
- McDonald, M.; Patel, D.A.; Keith, M.S.; Snedecor, S.J. Economic and Humanistic Burden of Dry Eye Disease in Europe, North America, and Asia: A Systematic Literature Review. Ocul. Surf. 2016, 14, 144–167. [Google Scholar] [CrossRef] [PubMed]
- Modi, Y.S.; Qurban, Q.; Zlotcavitch, L.; Echeverri, R.J.; Feuer, W.; Florez, H.; Galor, A. Ocular surface symptoms in veterans returning from operation Iraqi freedom and operation enduring freedom. Investig. Ophthalmol. Vis. Sci. 2014, 55, 650–653. [Google Scholar] [CrossRef]
- Galor, A.; Feuer, W.; Lee, D.J.; Florez, H.; Carter, D.; Pouyeh, B.; Prunty, W.J.; Perez, V.L. Prevalence and risk factors of dry eye syndrome in a United States veterans affairs population. Am. J. Ophthalmol. 2011, 152, 377–384.e2. [Google Scholar] [CrossRef] [PubMed]
- Galor, A.; Feuer, W.; Lee, D.J.; Florez, H.; Venincasa, V.D.; Perez, V.L. Ocular surface parameters in older male veterans. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1426–1433. [Google Scholar] [CrossRef]
- Pouyeh, B.; Viteri, E.; Feuer, W.; Lee, D.J.; Florez, H.; Fabian, J.A.; Perez, V.L.; Galor, A. Impact of ocular surface symptoms on quality of life in a United States veterans affairs population. Am. J. Ophthalmol. 2012, 153, 1061–1066.e3. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef]
- Bukowiecki, A.; Hos, D.; Cursiefen, C.; Eming, S.A. Wound-Healing Studies in Cornea and Skin: Parallels, Differences and Opportunities. Int. J. Mol. Sci. 2017, 18, 1257. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, R.C.; Wilson, S.E. Fibrocytes, Wound Healing, and Corneal Fibrosis. Investig. Ophthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef] [PubMed]
- Liu, c.-y.; Kao, W. Corneal Epithelial Wound Healing. Prog. Mol. Biol. Transl. Sci. 2015, 134, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Lassance, L.; Marino, G.K.; Medeiros, C.S.; Thangavadivel, S.; Wilson, S.E. Fibrocyte migration, differentiation and apoptosis during the corneal wound healing response to injury. Exp. Eye Res. 2018, 170, 177–187. [Google Scholar] [CrossRef]
- Chee, K.Y.; Kicic, A.; Wiffen, S.J. Limbal stem cells: The search for a marker. Clin. Exp. Ophthalmol. 2006, 34, 64–73. [Google Scholar] [CrossRef]
- Yoon, J.J.; Ismail, S.; Sherwin, T. Limbal stem cells: Central concepts of corneal epithelial homeostasis. World J. Stem Cells 2014, 6, 391–403. [Google Scholar] [CrossRef]
- Ma, D.H.; Chen, J.K.; Zhang, F.; Lin, K.Y.; Yao, J.Y.; Yu, J.S. Regulation of corneal angiogenesis in limbal stem cell deficiency. Prog. Retin. Eye Res. 2006, 25, 563–590. [Google Scholar] [CrossRef]
- Ksander, B.R.; Kolovou, P.E.; Wilson, B.J.; Saab, K.R.; Guo, Q.; Ma, J.; McGuire, S.P.; Gregory, M.S.; Vincent, W.J.; Perez, V.L.; et al. ABCB5 is a limbal stem cell gene required for corneal development and repair. Nature 2014, 511, 353–357. [Google Scholar] [CrossRef]
- Wilson, S.E.; Esposito, A. Focus on molecules: Interleukin-1: A master regulator of the corneal response to injury. Exp. Eye Res. 2009, 89, 124–125. [Google Scholar] [CrossRef]
- Peterson, J.L.; Phelps, E.D.; Doll, M.A.; Schaal, S.; Ceresa, B.P. The role of endogenous epidermal growth factor receptor ligands in mediating corneal epithelial homeostasis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2870–2880. [Google Scholar] [CrossRef]
- Xu, K.P.; Ding, Y.; Ling, J.; Dong, Z.; Yu, F.S. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 813–820. [Google Scholar] [CrossRef]
- Shu, D.Y.; Hutcheon, A.E.K.; Zieske, J.D.; Guo, X. Epidermal Growth Factor Stimulates Transforming Growth Factor-Beta Receptor Type II Expression In Corneal Epithelial Cells. Sci. Rep. 2019, 9, 8079. [Google Scholar] [CrossRef] [PubMed]
- Block, E.R.; Klarlund, J.K. Wounding sheets of epithelial cells activates the epidermal growth factor receptor through distinct short- and long-range mechanisms. Mol. Biol. Cell 2008, 19, 4909–4917. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.H.; Seong, S.Y. CD14 but not MD2 transmit signals from DAMP. Int. Immunopharmacol. 2010, 10, 98–106. [Google Scholar] [CrossRef] [PubMed]
- van Bergenhenegouwen, J.; Plantinga, T.S.; Joosten, L.A.; Netea, M.G.; Folkerts, G.; Kraneveld, A.D.; Garssen, J.; Vos, A.P. TLR2 & Co: A critical analysis of the complex interactions between TLR2 and coreceptors. J. Leukoc. Biol. 2013, 94, 885–902. [Google Scholar] [CrossRef]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Dana, R. Comparison of topical interleukin-1 vs tumor necrosis factor-alpha blockade with corticosteroid therapy on murine corneal inflammation, neovascularization, and transplant survival (an American Ophthalmological Society thesis). Trans. Am. Ophthalmol. Soc. 2007, 105, 330–343. [Google Scholar]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef]
- Choudhary, V.; Uaratanawong, R.; Patel, R.R.; Patel, H.; Bao, W.; Hartney, B.; Cohen, E.; Chen, X.; Zhong, Q.; Isales, C.M.; et al. Phosphatidylglycerol Inhibits Toll-Like Receptor-Mediated Inflammation by Danger-Associated Molecular Patterns. J. Investig. Dermatol. 2019, 139, 868–877. [Google Scholar] [CrossRef]
- Mohan, R.R.; Liang, Q.; Kim, W.J.; Helena, M.C.; Baerveldt, F.; Wilson, S.E. Apoptosis in the cornea: Further characterization of Fas/Fas ligand system. Exp. Eye Res. 1997, 65, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Mohan, R.R.; Kim, W.J.; Wilson, S.E. Modulation of TNF-alpha-induced apoptosis in corneal fibroblasts by transcription factor NF-kappaB. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1327–1336. [Google Scholar]
- Alexander, G.; Carlsen, H.; Blomhoff, R. Corneal NF-kappaB activity is necessary for the retention of transparency in the cornea of UV-B-exposed transgenic reporter mice. Exp. Eye Res. 2006, 82, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Fujita, S.; Saika, S.; Kao, W.W.; Fujita, K.; Miyamoto, T.; Ikeda, K.; Nakajima, Y.; Ohnishi, Y. Endogenous TNFalpha suppression of neovascularization in corneal stroma in mice. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3051–3055. [Google Scholar] [CrossRef]
- Sugaya, S.; Sakimoto, T.; Shoji, J.; Sawa, M. Regulation of soluble interleukin-6 (IL-6) receptor release from corneal epithelial cells and its role in the ocular surface. Jpn. J. Ophthalmol. 2011, 55, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Austin, A.; Lietman, T.; Rose-Nussbaumer, J. Update on the Management of Infectious Keratitis. Ophthalmology 2017, 124, 1678–1689. [Google Scholar] [CrossRef] [PubMed]
- Rajaiya, J.; Saha, A.; Ismail, A.M.; Zhou, X.; Su, T.; Chodosh, J. Adenovirus and the Cornea: More Than Meets the Eye. Viruses 2021, 13, 293. [Google Scholar] [CrossRef]
- Bartimote, C.; Foster, J.; Watson, S. The spectrum of microbial keratitis: An updated review. Open Ophthalmol. J. 2019, 13, 100–130. [Google Scholar] [CrossRef]
- Ting, D.S.J.; Ho, C.S.; Deshmukh, R.; Said, D.G.; Dua, H.S. Infectious keratitis: An update on epidemiology, causative microorganisms, risk factors, and antimicrobial resistance. Eye 2021, 35, 1084–1101. [Google Scholar] [CrossRef]
- Egrilmez, S.; Yildirim-Theveny, S. Treatment-Resistant Bacterial Keratitis: Challenges and Solutions. Clin. Ophthalmol. 2020, 14, 287–297. [Google Scholar] [CrossRef]
- Ung, L.; Bispo, P.J.M.; Shanbhag, S.S.; Gilmore, M.S.; Chodosh, J. The persistent dilemma of microbial keratitis: Global burden, diagnosis, and antimicrobial resistance. Surv. Ophthalmol. 2019, 64, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Al-Mujaini, A.; Al-Kharusi, N.; Thakral, A.; Wali, U.K. Bacterial keratitis: Perspective on epidemiology, clinico-pathogenesis, diagnosis and treatment. Sultan Qaboos Univ. Med. J. 2009, 9, 184–195. [Google Scholar] [PubMed]
- Lakhundi, S.; Siddiqui, R.; Khan, N.A. Pathogenesis of microbial keratitis. Microb. Pathog. 2017, 104, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Karmakar, M.; Roy, S.; Ramadan, R.T.; Williams, S.R.; Howell, S.; Shive, C.L.; Han, Y.; Stopford, C.M.; Rietsch, A.; et al. TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J. Immunol. 2010, 185, 4272–4283. [Google Scholar] [CrossRef]
- Huang, X.; Du, W.; McClellan, S.A.; Barrett, R.P.; Hazlett, L.D. TLR4 is required for host resistance in Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4910–4916. [Google Scholar] [CrossRef]
- McDermott, A.M. The role of antimicrobial peptides at the ocular surface. Ophthalmic Res. 2009, 41, 60–75. [Google Scholar] [CrossRef]
- Pearlman, E.; Sun, Y.; Roy, S.; Karmakar, M.; Hise, A.G.; Szczotka-Flynn, L.; Ghannoum, M.; Chinnery, H.R.; McMenamin, P.G.; Rietsch, A. Host defense at the ocular surface. Int. Rev. Immunol. 2013, 32, 4–18. [Google Scholar] [CrossRef]
- Kumar, A.; Hazlett, L.D.; Yu, F.S. Flagellin suppresses the inflammatory response and enhances bacterial clearance in a murine model of Pseudomonas aeruginosa keratitis. Infect. Immun. 2008, 76, 89–96. [Google Scholar] [CrossRef]
- Ekanayaka, S.A.; McClellan, S.A.; Peng, X.; Barrett, R.P.; Francis, R.; Hazlett, L.D. HMGB1 Antagonist, Box A, Reduces TLR4, RAGE, and Inflammatory Cytokines in the Cornea of P. aeruginosa-Infected Mice. J. Ocul. Pharmacol. Ther. 2018, 34, 659–669. [Google Scholar] [CrossRef]
- Huang, X.; Barrett, R.P.; McClellan, S.A.; Hazlett, L.D. Silencing Toll-like receptor-9 in Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4209–4216. [Google Scholar] [CrossRef]
- Astley, R.; Miller, F.C.; Mursalin, M.H.; Coburn, P.S.; Callegan, M.C. An Eye on Staphylococcus aureus Toxins: Roles in Ocular Damage and Inflammation. Toxins 2019, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hise, A.G.; Kalsow, C.M.; Pearlman, E. Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect. Immun. 2006, 74, 5325–5332. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Pergolizzi, S.; Lauriano, E.R.; Santoro, G.; Spataro, F.; Cimino, F.; Speciale, A.; Nostro, A.; Bisignano, G. TLR2 activation in corneal stromal cells by Staphylococcus aureus-induced keratitis. Apmis 2015, 123, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Hoshino, K.; Akira, S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol. 2000, 165, 5392–5396. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Qin, Q.; Tu, L.; Zhou, X.; Lin, Y.; Qu, J. Toll-like receptors (TLRs) expression and function in response to inactivate hyphae of Fusarium solani in immortalized human corneal epithelial cells. Mol. Vis. 2007, 13, 1953–1961. [Google Scholar]
- Niu, L.; Liu, X.; Ma, Z.; Yin, Y.; Sun, L.; Yang, L.; Zheng, Y. Fungal keratitis: Pathogenesis, diagnosis and prevention. Microb. Pathog. 2020, 138, 103802. [Google Scholar] [CrossRef]
- Peng, L.; Zhong, J.; Xiao, Y.; Wang, B.; Li, S.; Deng, Y.; He, D.; Yuan, J. Therapeutic effects of an anti-IL-6 antibody in fungal keratitis: Macrophage inhibition and T cell subset regulation. Int. Immunopharmacol. 2020, 85, 106649. [Google Scholar] [CrossRef]
- Sun, Y.; Chandra, J.; Mukherjee, P.; Szczotka-Flynn, L.; Ghannoum, M.A.; Pearlman, E. A murine model of contact lens-associated fusarium keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1511–1516. [Google Scholar] [CrossRef]
- Gao, X.; Zhao, G.; Li, C.; Lin, J.; Jiang, N.; Wang, Q.; Hu, L.; Xu, Q.; Peng, X.; He, K.; et al. LOX-1 and TLR4 affect each other and regulate the generation of ROS in A. fumigatus keratitis. Int. Immunopharmacol. 2016, 40, 392–399. [Google Scholar] [CrossRef]
- Jiang, J.Q.; Li, C.; Cui, C.X.; Ma, Y.N.; Zhao, G.Q.; Peng, X.D.; Xu, Q.; Wang, Q.; Zhu, G.Q.; Li, C.Y. Inhibition of LOX-1 alleviates the proinflammatory effects of high-mobility group box 1 in Aspergillus fumigatus keratitis. Int. J. Ophthalmol. 2019, 12, 898–903. [Google Scholar] [CrossRef]
- Liu, M.; Li, C.; Zhao, G.Q.; Lin, J.; Che, C.Y.; Xu, Q.; Wang, Q.; Xu, R.; Niu, Y.W. Boxb mediate BALB/c mice corneal inflammation through a TLR4/MyD88-dependent signaling pathway in Aspergillus fumigatus keratitis. Int. J. Ophthalmol. 2018, 11, 548–552. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Yue, L.H.; Zhao, G.Q.; Li, C.; Lin, J.; Jiang, N.; Wang, Q.; Xu, Q.; Peng, X.D.; Hu, L.T.; et al. The role of LOX-1 on innate immunity against Aspergillus keratitis in mice. Int. J. Ophthalmol. 2016, 9, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Lin, J.; Zhao, G.Q.; Li, C.; Che, C.Y.; Xu, Q.; Liu, M. Production of interleukin-1beta related to mammalian target of rapamycin/Toll-like receptor 4 signaling pathway during Aspergillus fumigatus infection of the mouse cornea. Int. J. Ophthalmol. 2018, 11, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, J.; Wu, X. Toll-like receptor 2 siRNA suppresses corneal inflammation and attenuates Aspergillus fumigatus keratitis in rats. Immunol. Cell Biol. 2012, 90, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Xin, Z.; Wu, X. The crosstalk between TLR2 and NOD2 in Aspergillus fumigatus keratitis. Mol. Immunol. 2015, 64, 235–243. [Google Scholar] [CrossRef]
- Huang, W.; Ling, S.; Jia, X.; Lin, B.; Huang, X.; Zhong, J.; Li, W.; Lin, X.; Sun, Y.; Yuan, J. Tacrolimus (FK506) suppresses TREM-1 expression at an early but not at a late stage in a murine model of fungal keratitis. PLoS ONE 2014, 9, e114386. [Google Scholar] [CrossRef]
- Wu, M.; Peng, A.; Sun, M.; Deng, Q.; Hazlett, L.D.; Yuan, J.; Liu, X.; Gao, Q.; Feng, L.; He, J.; et al. TREM-1 amplifies corneal inflammation after Pseudomonas aeruginosa infection by modulating Toll-like receptor signaling and Th1/Th2-type immune responses. Infect. Immun. 2011, 79, 2709–2716. [Google Scholar] [CrossRef]
- Zhong, J.; Huang, W.; Deng, Q.; Wu, M.; Jiang, H.; Lin, X.; Sun, Y.; Huang, X.; Yuan, J. Inhibition of TREM-1 and Dectin-1 Alleviates the Severity of Fungal Keratitis by Modulating Innate Immune Responses. PLoS ONE 2016, 11, e0150114. [Google Scholar] [CrossRef]
- Tarabishy, A.B.; Aldabagh, B.; Sun, Y.; Imamura, Y.; Mukherjee, P.K.; Lass, J.H.; Ghannoum, M.A.; Pearlman, E. MyD88 regulation of Fusarium keratitis is dependent on TLR4 and IL-1R1 but not TLR2. J. Immunol. 2008, 181, 593–600. [Google Scholar] [CrossRef]
- Alexandrakis, G.; Jalali, S.; Gloor, P. Diagnosis of Fusarium keratitis in an animal model using the polymerase chain reaction. Br. J. Ophthalmol. 1998, 82, 306–311. [Google Scholar] [CrossRef]
- Guo, H.; Wu, X.; Yu, F.S.; Zhao, J. Toll-like receptor 2 mediates the induction of IL-10 in corneal fibroblasts in response to Fusarium solu. Immunol. Cell Biol. 2008, 86, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ashby, B.D.; Garrett, Q.; Willcox, M.D.P. Corneal injuries and wound healing—Review of processes and therapies. Austin J. Clin. Ophthalmol. 2014, 1, 1017. [Google Scholar]
- Eslani, M.; Movahedan, A.; Afsharkhamseh, N.; Sroussi, H.; Djalilian, A.R. The role of toll-like receptor 4 in corneal epithelial wound healing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6108–6115. [Google Scholar] [CrossRef]
- Blakytny, R.; Jude, E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet. Med. 2006, 23, 594–608. [Google Scholar] [CrossRef]
- Frykberg, R.G.; Banks, J. Challenges in the Treatment of Chronic Wounds. Adv. Wound Care 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Choi, H.; Lee, R.H.; Roddy, G.W.; Ylostalo, J.H.; Wawrousek, E.; Prockop, D.J. Identification of the HSPB4/TLR2/NF-kappaB axis in macrophage as a therapeutic target for sterile inflammation of the cornea. EMBO Mol. Med. 2012, 4, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.Y.; Roddy, G.W.; Choi, H.; Lee, R.H.; Ylostalo, J.H.; Rosa, R.H., Jr.; Prockop, D.J. Anti-inflammatory protein TSG-6 reduces inflammatory damage to the cornea following chemical and mechanical injury. Proc. Natl. Acad. Sci. USA 2010, 107, 16875–16880. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Lan, W.; Lim, R.R.; Chaurasia, S.S. S100A proteins as molecular targets in the ocular surface inflammatory diseases. Ocul. Surf. 2014, 12, 23–31. [Google Scholar] [CrossRef]
- Shimizu, H.; Sakimoto, T.; Yamagami, S. Pro-inflammatory role of NLRP3 inflammasome in experimental sterile corneal inflammation. Sci. Rep. 2019, 9, 9596. [Google Scholar] [CrossRef] [PubMed]
- Bian, F.; Xiao, Y.; Zaheer, M.; Volpe, E.A.; Pflugfelder, S.C.; Li, D.Q.; de Paiva, C.S. Inhibition of NLRP3 Inflammasome Pathway by Butyrate Improves Corneal Wound Healing in Corneal Alkali Burn. Int. J. Mol. Sci. 2017, 18, 562. [Google Scholar] [CrossRef]
- Johnson, A.C.; Heinzel, F.P.; Diaconu, E.; Sun, Y.; Hise, A.G.; Golenbock, D.; Lass, J.H.; Pearlman, E. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.T.; Xiao, Y.; Pflugfelder, S.C.; de Paiva, C.S. Inflammatory Response to Lipopolysaccharide on the Ocular Surface in a Murine Dry Eye Model. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.C.; Lam, K.S.; Tong, L. A systematic review on the impact of diabetes mellitus on the ocular surface. Nutr. Diabetes 2017, 7, e251. [Google Scholar] [CrossRef] [PubMed]
- Bikbova, G.; Oshitari, T.; Baba, T.; Bikbov, M.; Yamamoto, S. Diabetic corneal neuropathy: Clinical perspectives. Clin. Ophthalmol. 2018, 12, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Claramonte, P.J.; Ruiz-Moreno, J.M.; Sanchez-Perez, S.I.; Leon, M.; Grino, C.; Cervino, V.D.; Alio, J.L. Variation of central corneal thickness in diabetic patients as detected by ultrasonic pachymetry. Arch. Soc. Esp. Oftalmol. 2006, 81, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Azar, D.T.; Spurr-Michaud, S.J.; Tisdale, A.S.; Gipson, I.K. Decreased penetration of anchoring fibrils into the diabetic stroma. A morphometric analysis. Arch. Ophthalmol. 1989, 107, 1520–1523. [Google Scholar] [CrossRef]
- Gekka, M.; Miyata, K.; Nagai, Y.; Nemoto, S.; Sameshima, T.; Tanabe, T.; Maruoka, S.; Nakahara, M.; Kato, S.; Amano, S. Corneal epithelial barrier function in diabetic patients. Cornea 2004, 23, 35–37. [Google Scholar] [CrossRef]
- Sady, C.; Khosrof, S.; Nagaraj, R. Advanced Maillard reaction and crosslinking of corneal collagen in diabetes. Biochem. Biophys. Res. Commun. 1995, 214, 793–797. [Google Scholar] [CrossRef]
- Herse, P.R. Corneal hydration control in normal and alloxan-induced diabetic rabbits. Investig. Ophthalmol. Vis. Sci. 1990, 31, 2205–2213. [Google Scholar]
- Whikehart, D.R. The inhibition of sodium, potassium-stimulated ATPase and corneal swelling: The role played by polyols. J. Am. Optom. Assoc. 1995, 66, 331–333. [Google Scholar]
- Tasli, N.G.; Icel, E.; Karakurt, Y.; Ucak, T.; Ugurlu, A.; Yilmaz, H.; Akbas, E.M. The findings of corneal specular microscopy in patients with type-2 diabetes mellitus. BMC Ophthalmol. 2020, 20, 214. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Zhu, M.; Petroll, W.M.; Koppaka, V.; Robertson, D.M. The impact of type 1 diabetes mellitus on corneal epithelial nerve morphology and the corneal epithelium. Am. J. Pathol. 2014, 184, 2662–2670. [Google Scholar] [CrossRef] [PubMed]
- Cameron, N.E.; Eaton, S.; Cotter, M.A.; Tesfaye, S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 2001, 44, 1973–1988. [Google Scholar] [CrossRef] [PubMed]
- Babizhayev, M.A.; Strokov, I.A.; Nosikov, V.V.; Savel’yeva, E.L.; Sitnikov, V.F.; Yegorov, Y.E.; Lankin, V.Z. The role of oxidative stress in diabetic neuropathy: Generation of free radical species in the glycation reaction and gene polymorphisms encoding antioxidant enzymes to genetic susceptibility to diabetic neuropathy in population of type I diabetic patients. Cell Biochem. Biophys. 2015, 71, 1425–1443. [Google Scholar] [CrossRef]
- Kim, J.; Kim, C.-S.; Sohn, E.; Jeong, I.-H.; Kim, H.; Kim, J.S. Involvement of advanced glycation end products, oxidative stress and nuclear factor-kappaB in the development of diabetic keratopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2011, 249, 529–536. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, J.; Zhang, X.; Liu, Z.; Yang, Y.; Gong, Q.; Ren, B. The Role of HMGB1 in the Pathogenesis of Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 2543268. [Google Scholar] [CrossRef]
- Di, G.; Du, X.; Qi, X.; Zhao, X.; Duan, H.; Li, S.; Xie, L.; Zhou, Q. Mesenchymal Stem Cells Promote Diabetic Corneal Epithelial Wound Healing Through TSG-6-Dependent Stem Cell Activation and Macrophage Switch. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4344–4354. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef]
- Ikegami, M.; Weaver, T.E.; Grant, S.N.; Whitsett, J.A. Pulmonary surfactant surface tension influences alveolar capillary shape and oxygenation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 433–439. [Google Scholar] [CrossRef]
- Kuronuma, K.; Mitsuzawa, H.; Takeda, K.; Nishitani, C.; Chan, E.D.; Kuroki, Y.; Nakamura, M.; Voelker, D.R. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J. Biol. Chem. 2009, 284, 25488–25500. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Choudhary, V.; Seremwe, M.; Edwards, J.G.; Wang, A.; Emmons, A.C.; Bollag, K.A.; Johnson, M.H.; Bollag, W.B. Soy Phosphatidylglycerol Reduces Inflammation in a Contact Irritant Ear Edema Mouse Model In Vivo. J. Pharmacol. Exp. Ther. 2018, 366, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.E.; Mauch, S.; Rieckmann, M.; Martinez, D.G.; Hause, G.; Noutsias, M.; Hofmann, U.; Lucas, H.; Meister, A.; Ramos, G.; et al. Phosphatidylserine (PS) and phosphatidylglycerol (PG) nanodispersions as potential anti-inflammatory therapeutics: Comparison of in vitro activity and impact of pegylation. Nanomedicine 2020, 23, 102096. [Google Scholar] [CrossRef]
- Klein, M.E.; Rieckmann, M.; Lucas, H.; Meister, A.; Loppnow, H.; Mader, K. Phosphatidylserine (PS) and phosphatidylglycerol (PG) enriched mixed micelles (MM): A new nano-drug delivery system with anti-inflammatory potential? Eur. J. Pharm. Sci. 2020, 152, 105451. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; Medjane, S.; Chabot, S.; Kubrusly, F.S.; Raw, I.; Chignard, M.; Touqui, L. Surfactant protein-A and phosphatidylglycerol suppress type IIA phospholipase A2 synthesis via nuclear factor-kappaB. Am. J. Respir. Crit. Care Med. 2003, 168, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Havet, N.; Vial, D.; Arbibe, L.; Dumarey, C.; Watson, M.L.; Touqui, L. Dioleylphosphatidylglycerol inhibits the expression of type II phospholipase A2 in macrophages. Am. J. Respir. Crit. Care Med. 1999, 159, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, V.; Griffith, S.; Chen, X.; Bollag, W.B. Pathogen-Associated Molecular Pattern-Induced TLR2 and TLR4 Activation Increases Keratinocyte Production of Inflammatory Mediators and is Inhibited by Phosphatidylglycerol. Mol. Pharmacol. 2020, 97, 324–335. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Kiatsurayanon, C.; Chieosilapatham, P.; Ogawa, H. Friends or Foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp. Dermatol. 2017, 26, 989–998. [Google Scholar] [CrossRef]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef]
- Numata, M.; Chu, H.W.; Dakhama, A.; Voelker, D.R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc. Natl. Acad. Sci. USA 2010, 107, 320–325. [Google Scholar] [CrossRef]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Posey, J.; Hartshorn, K.; Woodland, D.; Voelker, D.R. Phosphatidylglycerol suppresses influenza A virus infection. Am. J. Respir. Cell Mol. Biol. 2012, 46, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Mitchell, J.R.; Tipper, J.L.; Brand, J.D.; Trombley, J.E.; Nagashima, Y.; Kandasamy, P.; Chu, H.W.; Harrod, K.S.; Voelker, D.R. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J. Biol. Chem. 2020, 295, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, P.; Numata, M.; Berry, K.Z.; Fickes, R.; Leslie, C.C.; Murphy, R.C.; Voelker, D.R. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J. Lipid Res. 2016, 57, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.W.; Chao, Y.J.; Chang, W.H.; Chan, J.F.; Hsu, Y.H. Phosphatidylglycerol Incorporates into Cardiolipin to Improve Mitochondrial Activity and Inhibits Inflammation. Sci. Rep. 2018, 8, 4919. [Google Scholar] [CrossRef]
- Shaban, H.; Borras, C.; Vina, J.; Richter, C. Phosphatidylglycerol potently protects human retinal pigment epithelial cells against apoptosis induced by A2E, a compound suspected to cause age-related macula degeneration. Exp. Eye Res. 2002, 75, 99–108. [Google Scholar] [CrossRef]
- Pietromonaco, S.F.; Simons, P.C.; Altman, A.; Elias, L. Protein kinase C-q phosphorylation of moesin in the actin-binding sequence. J. Biol. Chem. 1998, 273, 7594–7603. [Google Scholar] [CrossRef]
- Murray, N.R.; Fields, A.P. Phosphatidylglycerol is a physiologic activator of nuclear protein kinase C. J. Biol. Chem. 1998, 273, 11514–11520. [Google Scholar] [CrossRef]
- Gökmen-Polar, Y.; Fields, A.P. Mapping of a molecular determinant for protein kinase C bII isozyme function. J. Biol. Chem. 1998, 273, 20261–20266. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Committee on Fetus and Newborn. Surfactant replacement therapy for respiratory distress syndrome. Pediatrics 1999, 103, 684–685. [Google Scholar]
- Preuss, S.; Scheiermann, J.; Stadelmann, S.; Omam, F.D.; Winoto-Morbach, S.; Lex, D.; von Bismarck, P.; Adam-Klages, S.; Knerlich-Lukoschus, F.; Wesch, D.; et al. 18:1/18:1-Dioleoyl-phosphatidylglycerol prevents alveolar epithelial apoptosis and profibrotic stimulus in a neonatal piglet model of acute respiratory distress syndrome. Pulm. Pharmacol. Ther. 2014, 28, 25–34. [Google Scholar] [CrossRef]
- Available online: https://systane.myalcon.com/products/systane-complete-preservative-free/?gclid=Cj0KCQjw9ZGYBhCEARIsAEUXITXwmQ3sCyn9DpEojcTQriStANvN6rJ4A6TU8tvHqAgxgI72LW_VLI8aAviSEALw_wcB&gclsrc=aw.ds (accessed on 23 August 2022).
- Rohit, A.; Willcox, M.D.P.; Mitchell, T.; Stapleton, F. Effect of a lipid emulsion drop on tear film characteristics of habitual contact lens wearers. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3156a. [Google Scholar]
- Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021119s022lbl.pdf (accessed on 23 August 2022).
- Sachedina, S.; Greiner, J.V.; Glonek, T. Membrane phospholipids of the ocular tunica fibrosa. Investig. Ophthalmol. Vis. Sci. 1991, 32, 625–632. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortingo, N.; Melnyk, S.; Sutton, S.H.; Watsky, M.A.; Bollag, W.B. Innate Immune System Activation, Inflammation and Corneal Wound Healing. Int. J. Mol. Sci. 2022, 23, 14933. https://doi.org/10.3390/ijms232314933
Fortingo N, Melnyk S, Sutton SH, Watsky MA, Bollag WB. Innate Immune System Activation, Inflammation and Corneal Wound Healing. International Journal of Molecular Sciences. 2022; 23(23):14933. https://doi.org/10.3390/ijms232314933
Chicago/Turabian StyleFortingo, Nyemkuna, Samuel Melnyk, Sarah H. Sutton, Mitchell A. Watsky, and Wendy B. Bollag. 2022. "Innate Immune System Activation, Inflammation and Corneal Wound Healing" International Journal of Molecular Sciences 23, no. 23: 14933. https://doi.org/10.3390/ijms232314933
APA StyleFortingo, N., Melnyk, S., Sutton, S. H., Watsky, M. A., & Bollag, W. B. (2022). Innate Immune System Activation, Inflammation and Corneal Wound Healing. International Journal of Molecular Sciences, 23(23), 14933. https://doi.org/10.3390/ijms232314933