
ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster

Abstract
:1. Introduction
2. Results
2.1. Nanopore Sequencing
- Data set I, represented by a concatenated FASTQ file that contains all the trimmed reads;
- Data set II, where the concatenated FASTQ file contains only trimmed and filtered reads.
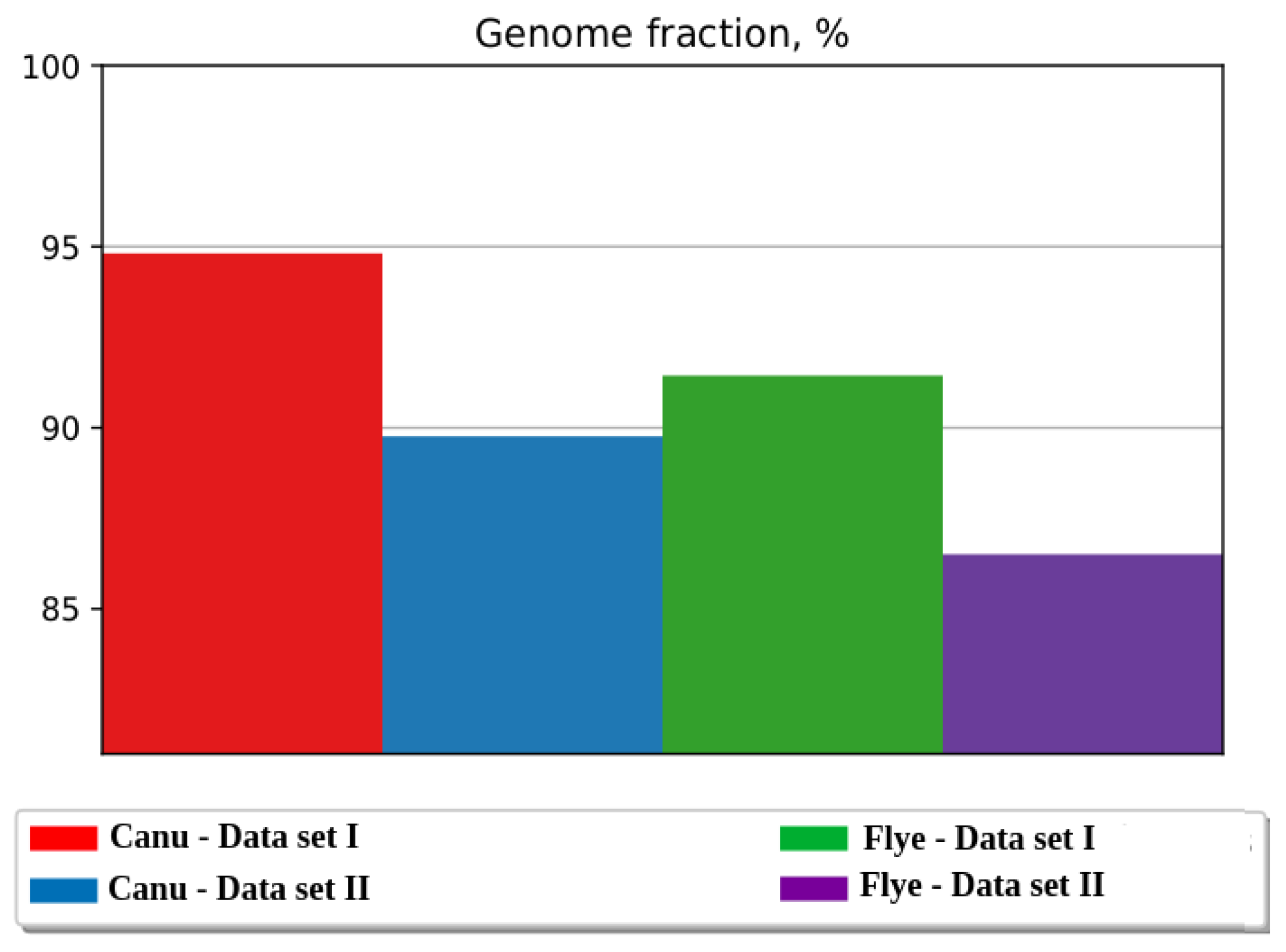
2.2. De Novo Assembly
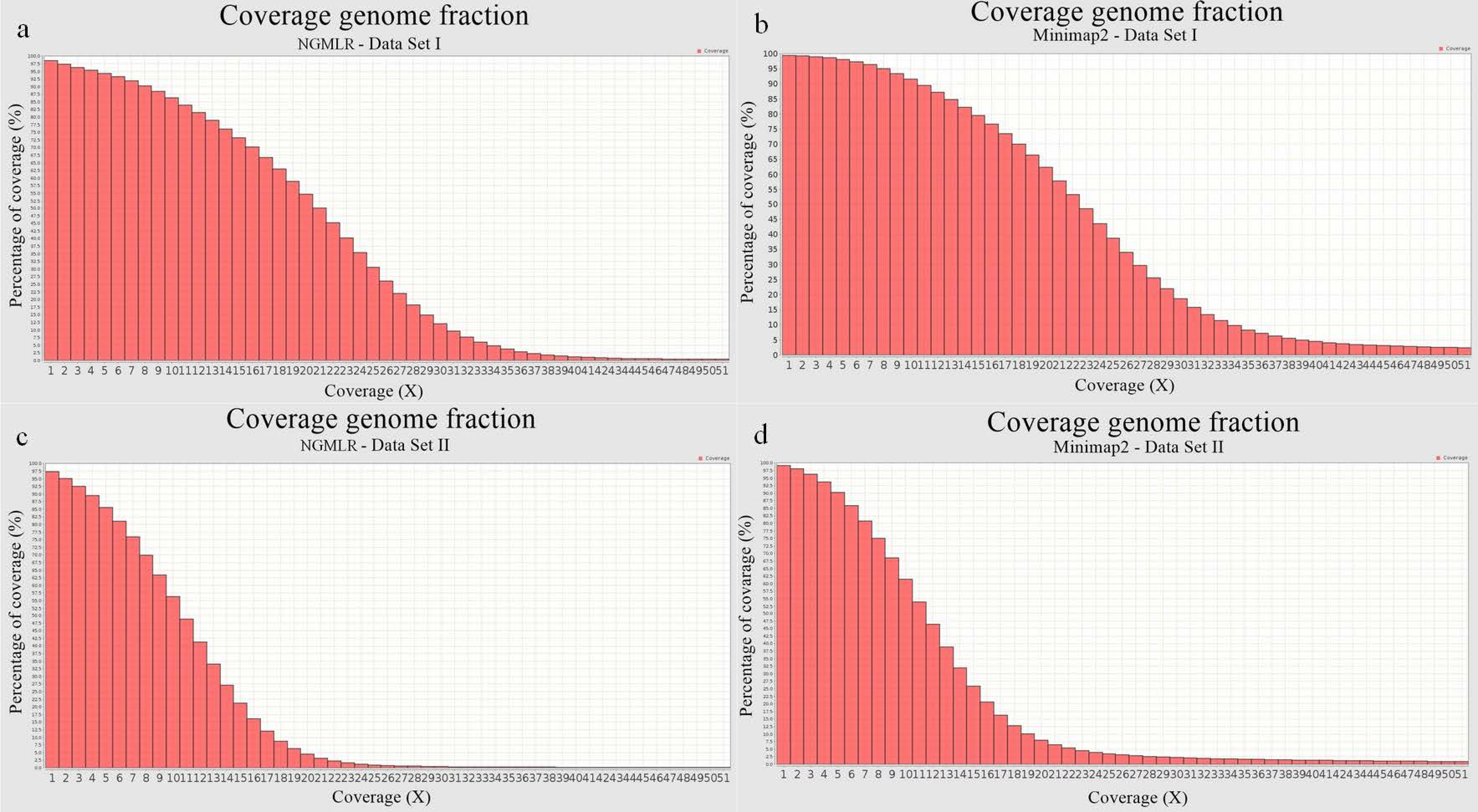
2.3. Guided Assembly versus the Reference Genome of D. melanogaster
3. Discussion
4. Materials and Methods
4.1. Fly Stock
4.2. DNA Isolation and Quantification
4.3. Nanopore Library Preparation, Sequencing, and Basecalling
4.4. Computational Environment
4.5. Data Processing and Quality Control
4.6. De Novo Assembly
4.7. Assembly versus the Reference Genome of D. melanogaster
- Minimap2 version 2.20 (accessed on 20 June 2021)—a bioinformatics application designed to align long ONT and PacBio reads to a reference sequence. The program quickly aligns the nucleotide sequences with each other to identify overlapped regions and aligns the reads to the reference genome [7].
- NGMLR version 0.2.8 (accessed on 21 June 2021)—a bioinformatics tool able to map ONT reads to a large reference genome. NGMLR application provides quick and accurate nucleotide sequences alignments, taking into account both possible sequencing errors and genomic variations [8].
- SAMtools version 1.7 (accessed on 20 June 2021)—a suite of programs dedicated to process high-throughput sequencing data [41].
4.8. Assessing the Quality of Generated Assemblies
- QUAST-LG (accessed on 4 August 2021) is one of the best-known tools for evaluating the quality of de novo genome assemblies. The application can also be used with a reference genome and supports multiple assemblies at the same time, which makes it suitable for comparative analyses [11];
- BUSCO version 5.2.2 (accessed on 3 December 2021) searches in de novo assemblies for highly conserved USCOs. We used the metazoa_odb10 database, which contains 954 USCOs likely to be present in many metazoan genomes [12];
- Qualimap version 2.2.1 (accessed on 19 July 2021) is a Java application that allows qualitative evaluation of the assemblies resulting following reads alignment to a reference genome. Guided assembly data (BAM files) are used to obtain a qualitative report that includes graphs and statistical parameters of the assembly [24];
- BAMstats version 1.25 (accessed on 20 July 2021) is a graphical interface program used to calculate mapping statistics of reads from a BAM file. This application provides an overview of the query/reference genome alignment quality [25].
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nie, F.; Xie, S.Q.; Zheng, Y.F.; Dai, Q.; Bray, T.; Wang, Y.X.; Xing, J.F.; Huang, Z.J.; Wang, D.P.; et al. Efficient assembly of nanopore reads via highly accurate and intact error correction. Nat. Commun. 2021, 12, 60. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Shafin, K.; Pesout, T.; Lorig-Roach, R.; Haukness, M.; Olsen, H.E.; Bosworth, C.; Armstrong, J.; Tigyi, K.; Maurer, N.; Koren, S.; et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. 2020, 38, 1044–1053. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. Genom. 2017, 3, e000132. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chifiriuc, M.C.; Bologa, A.M.; Ratiu, A.C.; Ionascu, A.; Ecovoiu, A.A. Mutations of gammaCOP Gene Disturb Drosophila melanogaster Innate Immune Response to Pseudomonas aeruginosa. Int. J. Mol. Sci. 2022, 23, 6499. [Google Scholar] [CrossRef]
- Smith, R.D.; Puzey, J.R.; Conradi Smith, G.D. Population genetics of transposable element load: A mechanistic account of observed overdispersion. PLoS ONE 2022, 17, e0270839. [Google Scholar] [CrossRef]
- Lerat, E.; Goubert, C.; Guirao-Rico, S.; Merenciano, M.; Dufour, A.B.; Vieira, C.; Gonzalez, J. Population-specific dynamics and selection patterns of transposable element insertions in European natural populations. Mol. Ecol. 2019, 28, 1506–1522. [Google Scholar] [CrossRef] [Green Version]
- Rech, G.E.; Radio, S.; Guirao-Rico, S.; Aguilera, L.; Horvath, V.; Green, L.; Lindstadt, H.; Jamilloux, V.; Quesneville, H.; Gonzalez, J. Population-scale long-read sequencing uncovers transposable elements associated with gene expression variation and adaptive signatures in Drosophila. Nat. Commun. 2022, 13, 1948. [Google Scholar] [CrossRef]
- Solares, E.A.; Chakraborty, M.; Miller, D.E.; Kalsow, S.; Hall, K.; Perera, A.G.; Emerson, J.J.; Hawley, R.S. Rapid Low-Cost Assembly of the Drosophila melanogaster Reference Genome Using Low-Coverage, Long-Read Sequencing. G3 2018, 8, 3143–3154. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: https://www.repeatmasker.org (accessed on 27 October 2022).
- Ecovoiu, A.A.; Bologa, A.M.; Chifiriuc, D.I.M.; Ciuca, A.M.; Constantin, N.D.; Ghionoiu, I.C.; Ghita, I.C.; Ratiu, A.C. Genome ARTIST_v2-An Autonomous Bioinformatics Tool for Annotation of Natural Transposons in Sequenced Genomes. Int. J. Mol. Sci. 2022, 23, 12686. [Google Scholar] [CrossRef]
- Merel, V.; Boulesteix, M.; Fablet, M.; Vieira, C. Transposable elements in Drosophila. Mob. DNA 2020, 11, 23. [Google Scholar] [CrossRef]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3, RESEARCH0084. [Google Scholar] [CrossRef] [Green Version]
- McCullers, T.J.; Steiniger, M. Transposable elements in Drosophila. Mob. Genet. Elements 2017, 7, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.M.; Gotz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.F.; Conesa, A. Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28, 2678–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: http://bamstats.sourceforge.net (accessed on 20 July 2021).
- Courtine, D.; Provaznik, J.; Reboul, J.; Blanc, G.; Benes, V.; Ewbank, J. Long-read only assembly of Drechmeria coniospora genomes reveals widespread chromosome plasticity and illustrates the limitations of current nanopore methods. Gigascience 2020, 9, giaa099. [Google Scholar] [CrossRef]
- Alkan, C.; Sajjadian, S.; Eichler, E.E. Limitations of next-generation genome sequence assembly. Nat. Methods 2011, 8, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszkiewicz, K.; Studholme, D.J. De novo assembly of short sequence reads. Brief. Bioinform. 2010, 11, 457–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.E.; Peluso, P.; Babayan, P.; Yeadon, P.J.; Yu, C.; Fisher, W.W.; Chin, C.S.; Rapicavoli, N.A.; Rank, D.R.; Li, J.; et al. Long-read, whole-genome shotgun sequence data for five model organisms. Sci. Data 2014, 1, 140045. [Google Scholar] [CrossRef] [Green Version]
- Chaisson, M.J.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef]
- Debladis, E.; Llauro, C.; Carpentier, M.C.; Mirouze, M.; Panaud, O. Detection of active transposable elements in Arabidopsis thaliana using Oxford Nanopore Sequencing technology. BMC Genom. 2017, 18, 537. [Google Scholar] [CrossRef] [Green Version]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef]
- Shirasawa, K.; Sasaki, K.; Hirakawa, H.; Isobe, S. Genomic region associated with pod color variation in pea (Pisum sativum). G3 2021, 11, jkab081. [Google Scholar] [CrossRef] [PubMed]
- Chernyavskaya, Y.; Zhang, X.; Liu, J.; Blackburn, J. Long-read sequencing of the zebrafish genome reorganizes genomic architecture. BMC Genom. 2022, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, J.; Ichikawa, K.; Shoura, M.J.; Artiles, K.L.; Gabdank, I.; Wahba, L.; Smith, C.L.; Edgley, M.L.; Rougvie, A.E.; Fire, A.Z.; et al. Recompleting the Caenorhabditis elegans genome. Genome Res. 2019, 29, 1009–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyson, J.R.; O’Neil, N.J.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.E.; Staber, C.; Zeitlinger, J.; Hawley, R.S. Highly Contiguous Genome Assemblies of 15 Drosophila Species Generated Using Nanopore Sequencing. G3 2018, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.Y.; Wang, J.R.; Miller, D.E.; Barmina, O.; Delaney, E.; Thompson, A.; Comeault, A.A.; Peede, D.; D’Agostino, E.R.R.; Pelaez, J.; et al. Highly contiguous assemblies of 101 drosophilid genomes. eLife 2021, 10, e66405. [Google Scholar] [CrossRef] [PubMed]
- Ellison, C.E.; Cao, W. Nanopore sequencing and Hi-C scaffolding provide insight into the evolutionary dynamics of transposable elements and piRNA production in wild strains of Drosophila melanogaster. Nucleic Acids Res. 2020, 48, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; Dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Statistics | Run_1 | Run_2 |
|---|---|---|
| Total number of FAST5 files | 173 | 158 |
| Total read number | 688,560 | 629,570 |
| Size (Gb) | 5 | 4.1 |
| The longest read length (nt) | 121,786 | 112,857 |
| Mean read length | 3548 | 3171 |
| Mean read quality | 10.8 | 8.1 |
| Statistics | Data Set I | Data Set II |
|---|---|---|
| Size (Gb) | 8.9 | 4.1 |
| Total number of reads | 1,317,857 | 590,406 |
| Mean q | 9.3 | 11.8 |
| Mean read length | 3298 | 3356 |
| Longest read | 121,786 | 98,982 |
| Total number of bases | 4,346,556,125 | 1,981,948,635 |
| Assembly Statistics | Canu Data Set I | Canu Data Set II | Flye Data Set I | Flye Data set II |
|---|---|---|---|---|
| No. of contigs | 3202 | 3586 | 1348 | 1531 |
| Largest contig | 4,036,320 | 1,027,435 | 10,359,939 | 3,223,716 |
| N50 | 256,290 | 121,999 | 3,373,574 | 492,599 |
| NG50 | 479,257 | 160,502 | 3,475,578 | 502,738 |
| Total length | 192,838,120 | 164,407,780 | 148,574,057 | 138,855,691 |
| Reference length | 137,567,484 | |||
| Genome fraction (%) | 94.8 | 89.7 | 91.4 | 86.5 |
| No. of misassembled contigs | 1392 | 1408 | 268 | 382 |
| No. of fully unaligned contigs | 493 | 247 | 124 | 99 |
| No. of possible NTs | 434 | 298 | 132 | 90 |
| Canu Data Set I | Canu Data Set II | Flye Data Set I | Flye Data Set II | Minimap/Miniasm ISO1 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Bases Masked | 51769568 bp | 41861639 bp | 20608100 bp | 18111261 bp | 21716093 bp | |||||
| (26.83%) | (25.45%) | (13.83%) | (12.99%) | (16.47%) | ||||||
| No. of Elements | Percentage of Seq (%) | No. of Elements | Percentage of Seq (%) | No. of Elements | Percentage of Seq (%) | No. of Elements | Percentage of Seq (%) | No. of Elements | Percentage of Seq (%) | |
| Retro elements | 22,385 | 18.93 | 19,128 | 18.46 | 11,300 | 8.65 | 10,272 | 8.17 | 9689 | 12.49 |
| LINEs: | 8295 | 6.39 | 7017 | 6.36 | 4001 | 2.95 | 3636 | 2.84 | 3579 | 4.24 |
| L2/CR1/Rex | 1144 | 0.61 | 1001 | 0.63 | 774 | 0.55 | 734 | 0.55 | 648 | 0.53 |
| R1/LOA/Jockey | 1727 | 1.86 | 1434 | 1.84 | 677 | 0.63 | 609 | 0.60 | 819 | 1.26 |
| R2/R4/NeSL | 69 | 0.08 | 57 | 0.07 | 17 | 0.01 | 16 | 0.01 | 18 | 0.03 |
| LTR elements | 14,090 | 12.54 | 12,111 | 12.10 | 7299 | 5.70 | 6636 | 5.33 | 6110 | 8.25 |
| BEL/Pao | 2882 | 2.26 | 2528 | 2.19 | 1836 | 0.96 | 1649 | 0.90 | 1398 | 1.92 |
| Ty1/Copia | 772 | 0.74 | 671 | 0.75 | 283 | 0.26 | 265 | 0.24 | 252 | 0.40 |
| Gypsy | 10,436 | 9.54 | 8912 | 9.17 | 5180 | 4.48 | 4722 | 4.19 | 4460 | 5.93 |
| DNA transposons | 5301 | 1.39 | 4429 | 1.39 | 3178 | 1.01 | 2943 | 1.01 | 2951 | 1.12 |
| hobo-Activator | 286 | 0.07 | 239 | 0.07 | 158 | 0.04 | 147 | 0.05 | 204 | 0.07 |
| Tc1-IS630-Pogo | 1408 | 0.38 | 1098 | 0.33 | 929 | 0.31 | 850 | 0.29 | 930 | 0.40 |
| PiggyBac | 31 | 0.01 | 22 | 0.01 | 21 | 0.01 | 24 | 0.01 | 12 | 0.01 |
| Other (Mirage, P-element, Transib) | 2825 | 0.70 | 2439 | 0.75 | 1559 | 0.48 | 1433 | 0.47 | 1402 | 0.49 |
| Rolling-circles | 5689 | 0.63 | 5071 | 0.65 | 4456 | 0.64 | 4385 | 0.67 | 3213 | 0.53 |
| Unclassified | 473 | 0.04 | 374 | 0.03 | 372 | 0.04 | 301 | 0.04 | 320 | 0.04 |
| Small RNA | 1061 | 0.41 | 761 | 0.36 | 289 | 0.06 | 169 | 0.04 | ||
| Total interspersed repeats | 20.35 | 19.89 | 9.70 | 9.22 | 13.65 | |||||
| Satellites | 1602 | 1.40 | 1280 | 0.95 | 735 | 0.52 | 598 | 0.38 | 739 | 0.34 |
| Simple repeats | 85,262 | 3.79 | 76,658 | 3.34 | 81,534 | 2.60 | 75,898 | 2.37 | 50,764 | 1.64 |
| Low complexity | 9737 | 0.25 | 8827 | 0.25 | 9777 | 0.31 | 9155 | 0.31 | 6613 | 0.25 |
| Type of mdg1 Insertion | Canu Data Set I | Canu Data Set II | Flye Data set I | Flye Data Set II | Minimap/Miniasm ISO1 |
|---|---|---|---|---|---|
| Conserved | 10 | 10 | 7 | 6 | 17 |
| Horezu specific | 29 | 28 | 3 | 4 | - |
| Ambiguous | 5 | - | 1 | 1 | - |
| Unresolved | 7 | 6 | 3 | 1 | 1 |
| Total mapped insertions | 44 | 38 | 11 | 11 | 17 |
| Statistics | Minimap2 Data Set I | Minimap2 Data Set II | NGMLR Data set I | NGMLR Data Set II |
|---|---|---|---|---|
| Mapped reads (%) | 90.27 | 96.45 | 73.93 | 84.56 |
| Mean Coverage | 26.52 | 13.51 | 21.45 | 11.15 |
| Mean Mapping Quality | 50.4 | 51.08 | 47.1 | 48.82 |
| General error rate (%) | 15.82 | 11.76 | 12.85 | 8.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bologa, A.M.; Stoica, I.; Ratiu, A.C.; Constantin, N.D.; Ecovoiu, A.A. ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster. Int. J. Mol. Sci. 2022, 23, 14892. https://doi.org/10.3390/ijms232314892
Bologa AM, Stoica I, Ratiu AC, Constantin ND, Ecovoiu AA. ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster. International Journal of Molecular Sciences. 2022; 23(23):14892. https://doi.org/10.3390/ijms232314892
Chicago/Turabian StyleBologa, Alexandru Marian, Ileana Stoica, Attila Cristian Ratiu, Nicoleta Denisa Constantin, and Alexandru Al. Ecovoiu. 2022. "ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster" International Journal of Molecular Sciences 23, no. 23: 14892. https://doi.org/10.3390/ijms232314892
APA StyleBologa, A. M., Stoica, I., Ratiu, A. C., Constantin, N. D., & Ecovoiu, A. A. (2022). ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster. International Journal of Molecular Sciences, 23(23), 14892. https://doi.org/10.3390/ijms232314892