
Structural Anomaly in Glasses: Molecular Dynamics Study of Organic Radical in Dibutylphthalate at Different Temperatures


Abstract
:1. Introduction
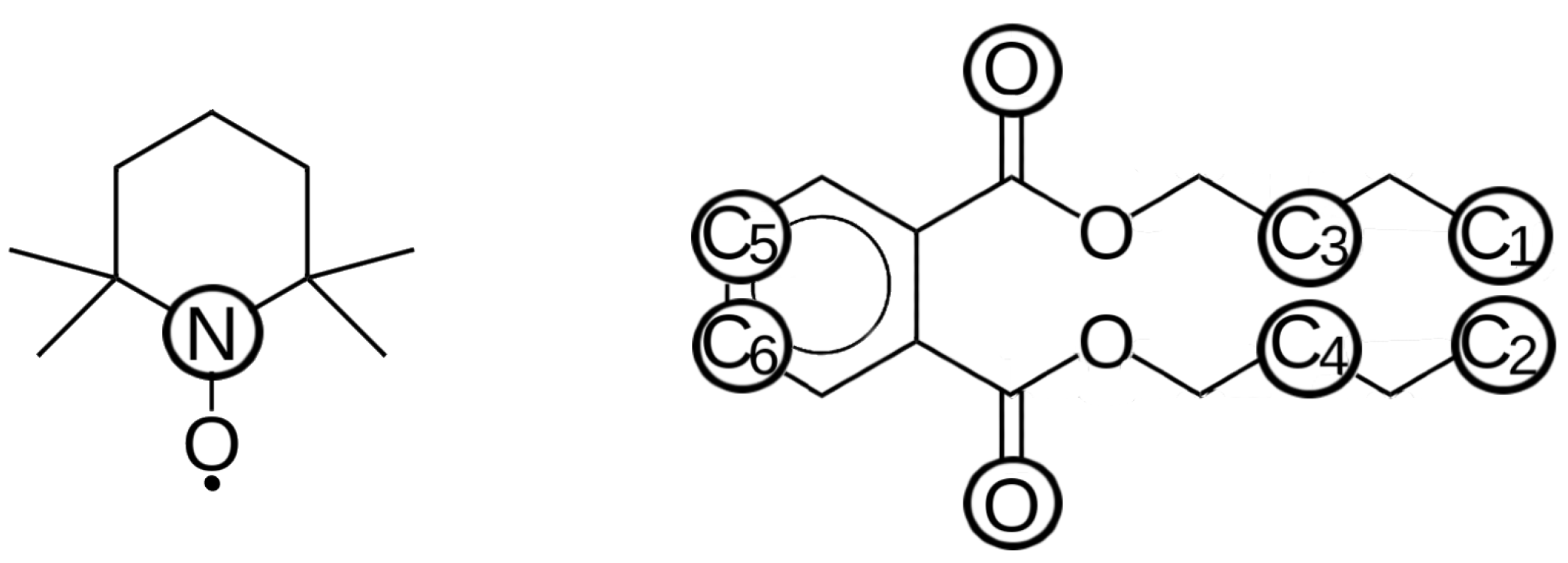
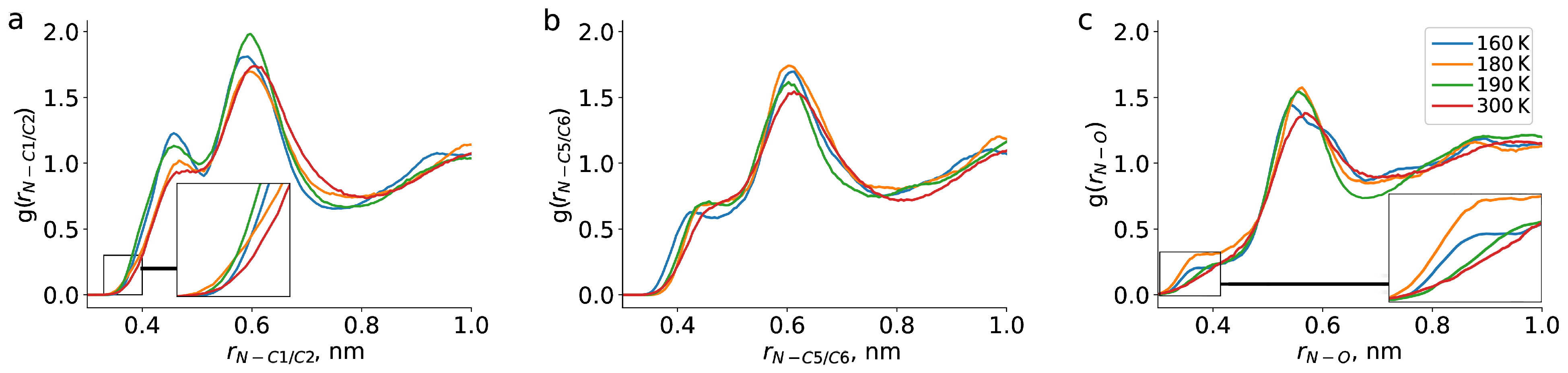
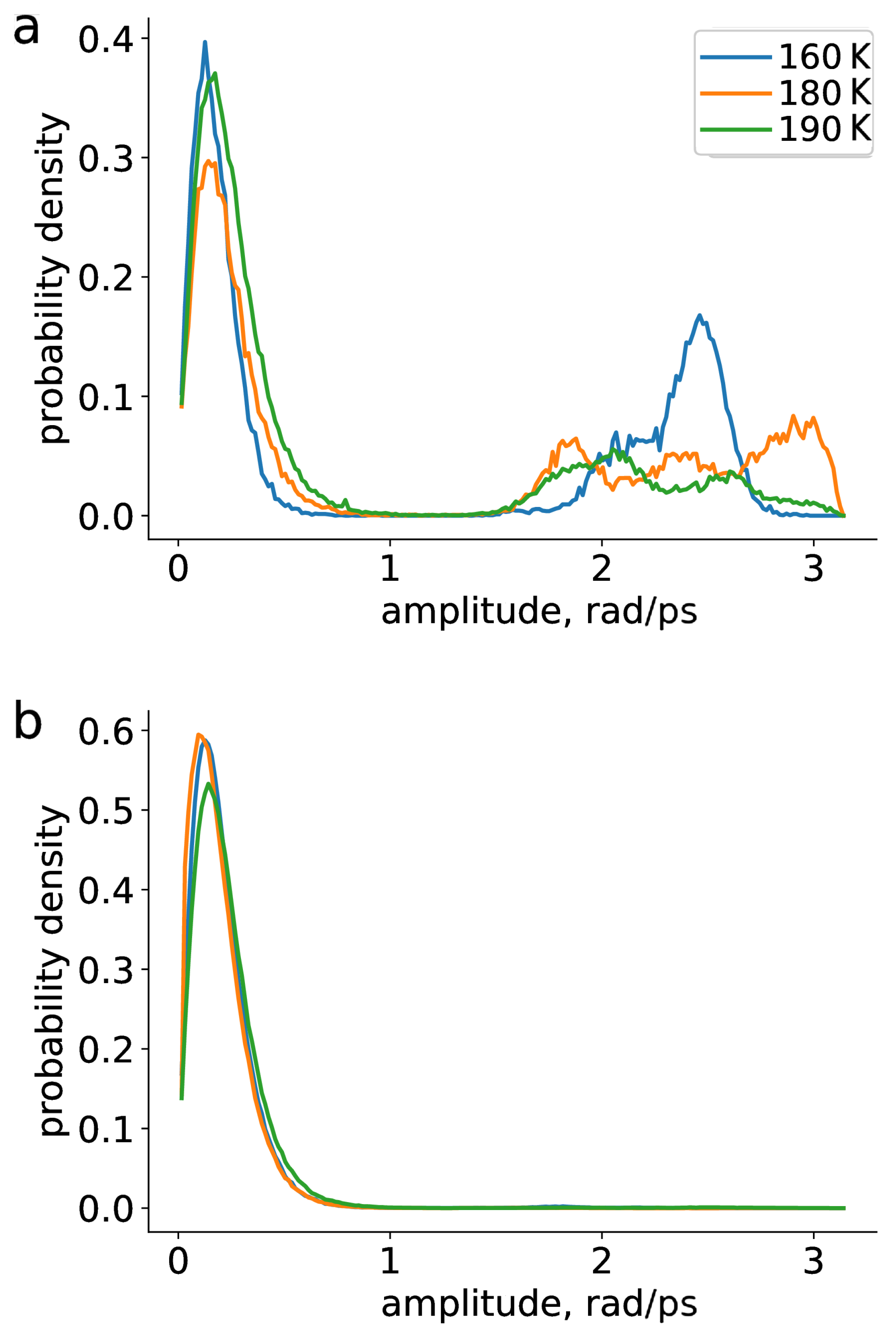
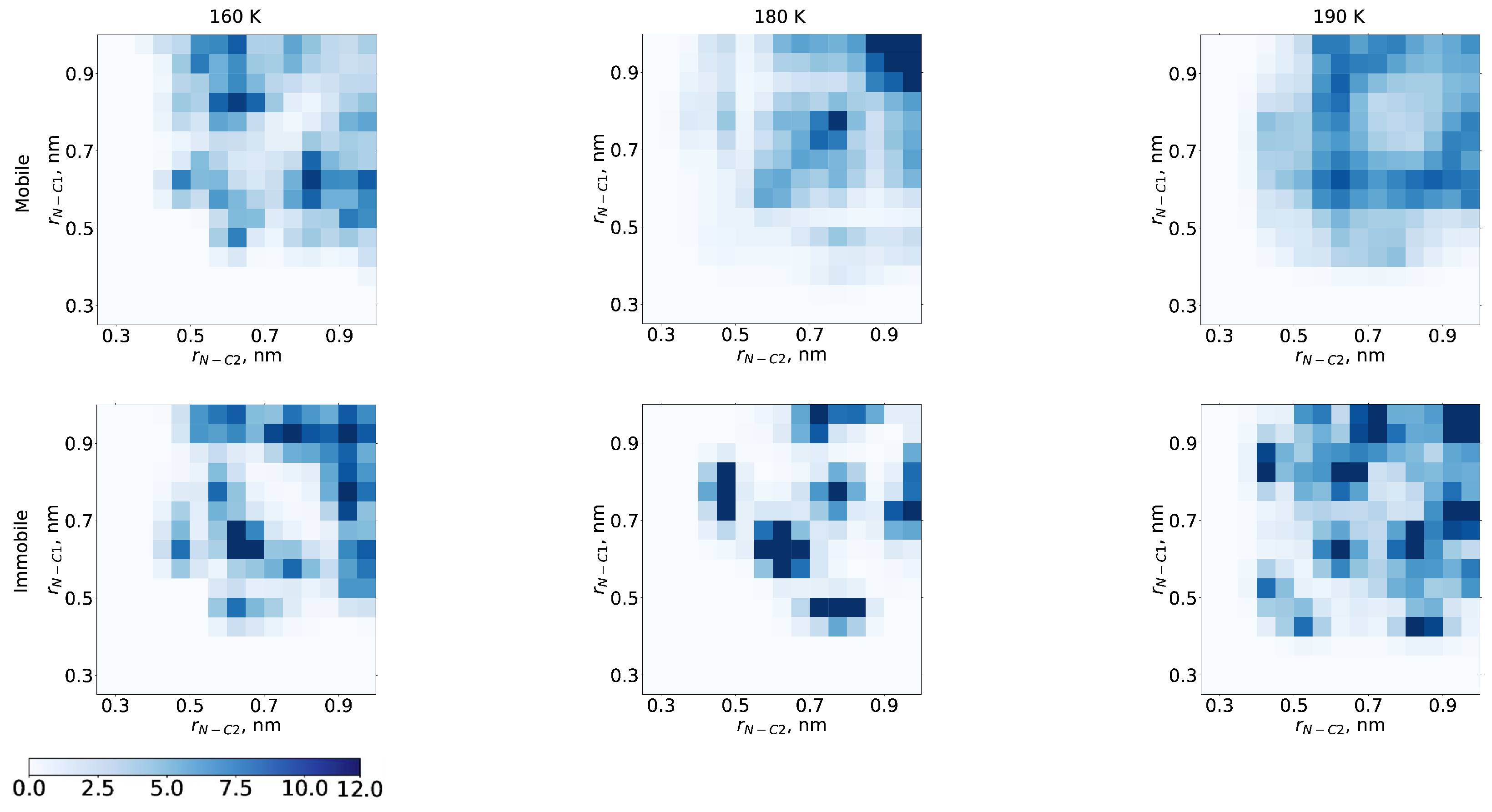
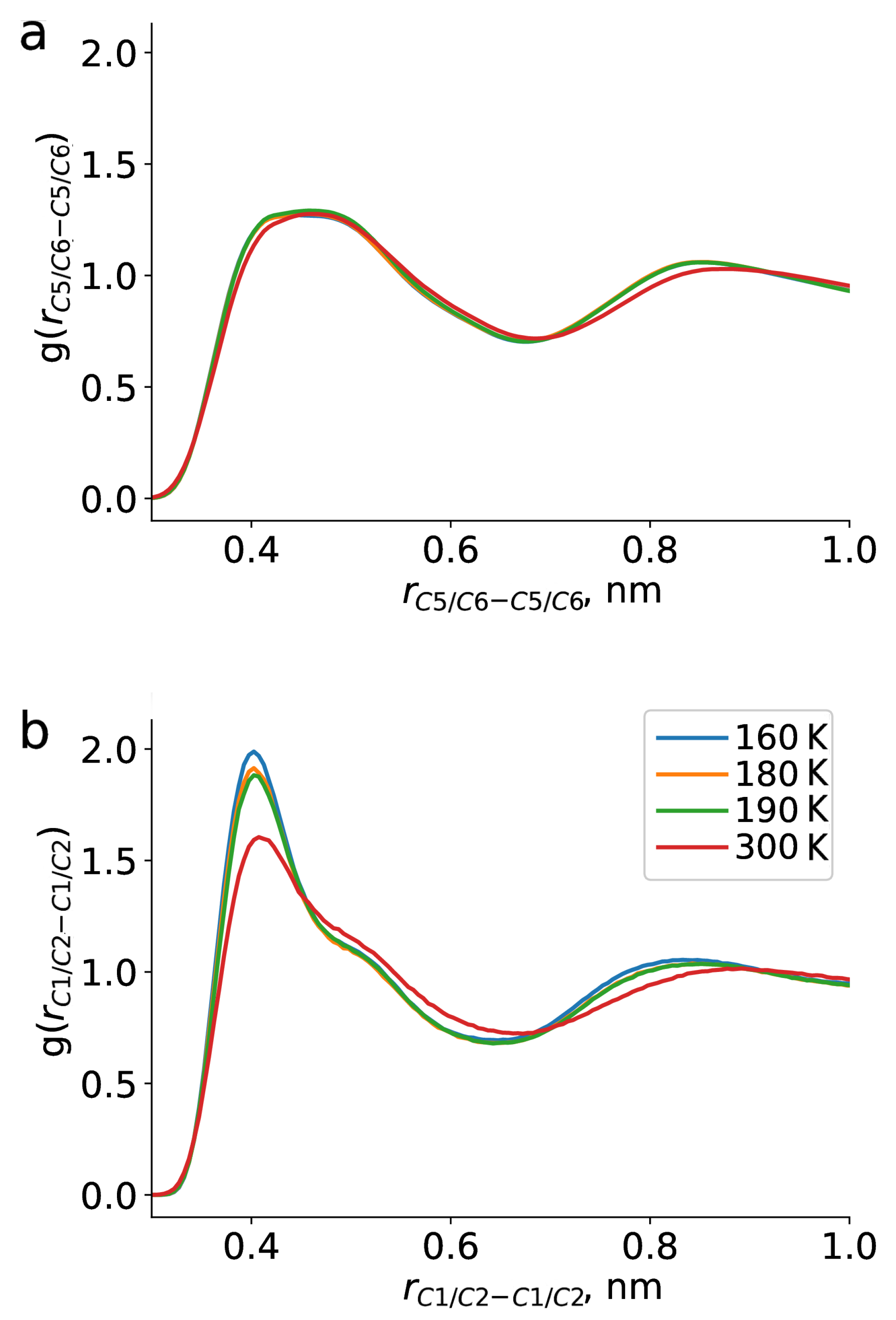
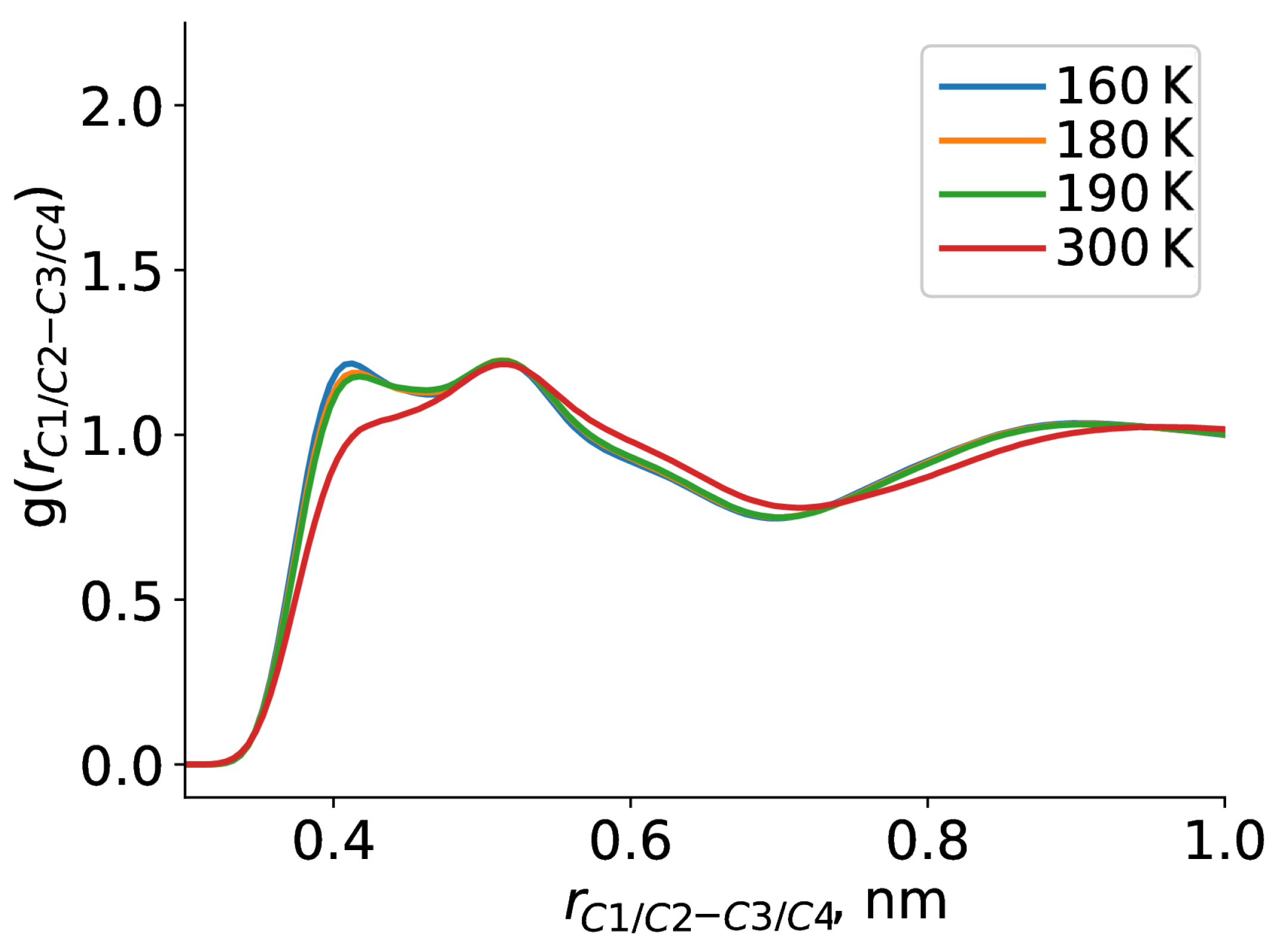
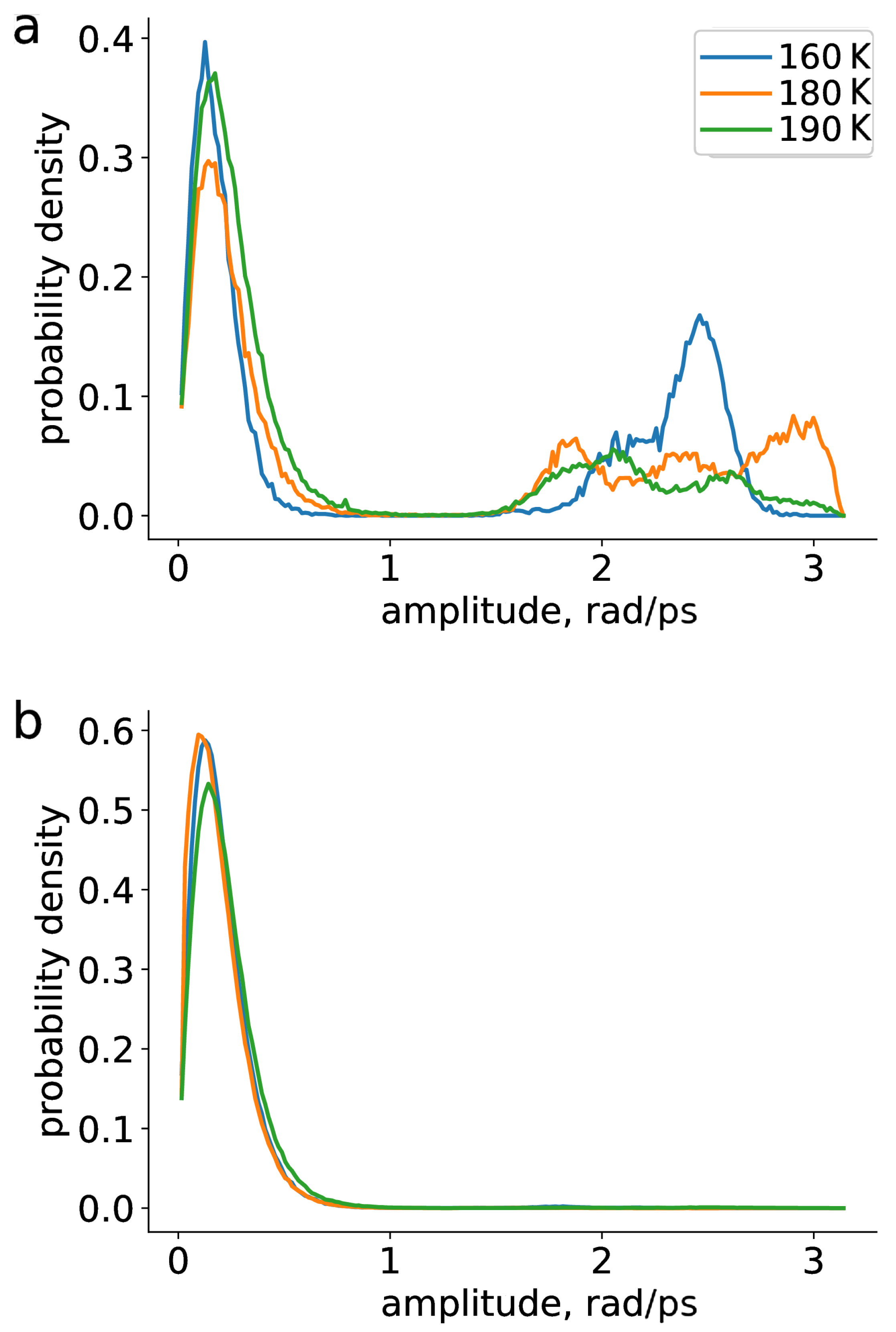
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sillescu, H. Heterogeneity at the glass transition: A review. J. Non-Cryst. Solids 1999, 243, 81–108. [Google Scholar] [CrossRef]
- Wisitsorasak, A.; Wolynes, P.G. Dynamical Heterogeneity of the Glassy State. J. Phys. Chem. B 2014, 118, 7835–7847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ediger, M.D. Spatially Heterogeneous Dynamics in Supercooled Liquids. Annu. Rev. Phys. Chem. 2000, 51, 99–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biroli, G.; Garrahan, J.P. Perspective: The glass transition. J. Chem. Phys. 2013, 138, 12A301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brace, D.D.; Gottke, S.D.; Cang, H.; Fayer, M.D. Orientational dynamics of the glass forming liquid, dibutylphthalate: Time domain experiments and comparison to mode coupling theory. J. Chem. Phys. 2002, 116, 1598–1606. [Google Scholar] [CrossRef]
- Wang, Y.L.; Li, B.; Sarman, S.; Mocci, F.; Lu, Z.Y.; Yuan, J.; Laaksonen, A.; Fayer, M.D. Microstructural and Dynamical Heterogeneities in Ionic Liquids. Chem. Rev. 2020, 120, 5798–5877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russina, O.; Triolo, A.; Gontrani, L.; Caminiti, R. Mesoscopic Structural Heterogeneities in Room-Temperature Ionic Liquids. J. Phys. Chem. Lett. 2012, 3, 27–33. [Google Scholar] [CrossRef]
- Ji, Y.; Shi, R.; Wang, Y.; Saielli, G. Effect of the Chain Length on the Structure of Ionic Liquids: From Spatial Heterogeneity to Ionic Liquid Crystals. J. Phys. Chem. B 2013, 117, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Khudozhitkov, A.E.; Stange, P.; Bonsa, A.M.; Overbeck, V.; Appelhagen, A.; Stepanov, A.G.; Kolokolov, D.I.; Paschek, D.; Ludwig, R. Dynamical heterogeneities in ionic liquids as revealed from deuteron NMR. Chem. Commun. 2018, 54, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.Y.; Prikhod’ko, S.A.; Adonin, N.Y.; Kirilyuk, I.A.; Adichtchev, S.V.; Surovtsev, N.V.; Dzuba, S.A.; Fedin, M.V. Structural Anomalies in Ionic Liquids near the Glass Transition Revealed by Pulse EPR. J. Phys. Chem. Lett. 2018, 9, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Bakulina, O.D.; Ivanov, M.Y.; Prikhod’ko, S.A.; Pylaeva, S.; Zaytseva, I.V.; Surovtsev, N.V.; Adonin, N.Y.; Fedin, M.V. Nanocage formation and structural anomalies in imidazolium ionic liquid glasses governed by alkyl chains of cations. Nanoscale 2020, 12, 19982–19991. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.Y.; Prikhod’ko, S.A.; Bakulina, O.D.; Kiryutin, A.S.; Adonin, N.Y.; Fedin, M.V. Validation of Structural Grounds for Anomalous Molecular Mobility in Ionic Liquid Glasses. Molecules 2021, 26, 5828. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.Y.; Poryvaev, A.S.; Polyukhov, D.M.; Prikhod’ko, S.A.; Adonin, N.Y.; Fedin, M.V. Nanoconfinement effects on structural anomalies in imidazolium ionic liquids. Nanoscale 2020, 12, 23480–23487. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.Y.; Bakulina, O.D.; Alimov, D.V.; Prikhod’ko, S.A.; Veber, S.L.; Pylaeva, S.; Adonin, N.Y.; Fedin, M.V. Inherent heterogeneities and nanostructural anomalies in organic glasses revealed by EPR. Nanoscale Adv. 2021, 3, 4973–4978. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.A.; Pádua, A.A.H. Nanostructural Organization in Ionic Liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [Google Scholar] [CrossRef]
- Wang, Y.; Voth, G.A. Unique Spatial Heterogeneity in Ionic Liquids. J. Am. Chem. Soc. 2005, 127, 12192–12193. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Weber, H.; Thomas, M.; Hollóczki, O.; Kirchner, B. Domain Analysis in Nanostructured Liquids: A Post-Molecular Dynamics Study at the Example of Ionic Liquids. ChemPhysChem 2015, 16, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, S.; von Domaros, M.; Clark, R.; Hollóczki, O.; Brehm, M.; Welton, T.; Luzar, A.; Kirchner, B. Structure and lifetimes in ionic liquids and their mixtures. Faraday Discuss. 2018, 206, 219–245. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, V.S. EPR spectroscopy and molecular dynamics modelling: A combined approach to study liquid crystals. Liq. Cryst. 2018, 45, 2139–2157. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Margulis, C.J. Heterogeneity in a room-temperature ionic liquid: Persistent local environments and the red-edge effect. Proc. Natl. Acad. Sci. USA 2006, 103, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Margulis, C.J.; Li, Y.; Berne, B.J. Why Is the Partial Molar Volume of CO2 So Small When Dissolved in a Room Temperature Ionic Liquid? Structure and Dynamics of CO2 Dissolved in [Bmim+] [PF6-]. J. Am. Chem. Soc. 2005, 127, 17842–17851. [Google Scholar] [CrossRef] [PubMed]
- Margulis, C.J.; Stern, H.A.; Berne, B.J. Computer Simulation of a “Green Chemistry” Room-Temperature Ionic Solvent. J. Phys. Chem. B 2002, 106, 12017–12021. [Google Scholar] [CrossRef] [Green Version]
- Araque, J.C.; Hettige, J.J.; Margulis, C.J. Modern Room Temperature Ionic Liquids, a Simple Guide to Understanding Their Structure and How It May Relate to Dynamics. J. Phys. Chem. B 2015, 119, 12727–12740. [Google Scholar] [CrossRef]
- Amith, W.D.; Araque, J.C.; Margulis, C.J. A Pictorial View of Viscosity in Ionic Liquids and the Link to Nanostructural Heterogeneity. J. Phys. Chem. Lett. 2020, 11, 2062–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumble, C.A.; Kaintz, A.; Yadav, S.K.; Conway, B.; Araque, J.C.; Baker, G.A.; Margulis, C.; Maroncelli, M. Rotational Dynamics in Ionic Liquids from NMR Relaxation Experiments and Simulations: Benzene and 1-Ethyl-3-Methylimidazolium. J. Phys. Chem. B 2016, 120, 9450–9467. [Google Scholar] [CrossRef]
- Amith, W.D.; Araque, J.C.; Margulis, C.J. Relationship between the Relaxation of Ionic Liquid Structural Motifs and That of the Shear Viscosity. J. Phys. Chem. B 2021, 125, 6264–6271. [Google Scholar] [CrossRef]
- Li, Q.; Deng, X.; Liu, Y.; Cheng, Q.; Liu, C. Gelation of waxy crude oil system with ethylene-vinyl acetate on solid surface: A molecular dynamics study. J. Mol. Liq. 2021, 331, 115816. [Google Scholar] [CrossRef]
- Cook, R.L.; King, H.E.; Herbst, C.A.; Herschbach, D.R. Pressure and temperature dependent viscosity of two glass forming liquids: Glycerol and dibutyl phthalate. J. Chem. Phys. 1994, 100, 5178–5189. [Google Scholar] [CrossRef]
- Dufour, J.; Jorat, L.; Bondeau, A.; Siblini, A.; Noyel, G. Shear viscosity and dielectric relaxanon time of dibutyl phthalate down to glass transition temperature. J. Mol. Liq. 1994, 62, 75–82. [Google Scholar] [CrossRef]
- Lindahl, A.; van der Spoel, H. GROMACS 2020 Source Code; Zenodo: Geneve, Switzerland, 2020. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Dodda, L.S.; Cabeza de Vaca, I.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Sezer, D.; Freed, J.H.; Roux, B. Parametrization, Molecular Dynamics Simulation, and Calculation of Electron Spin Resonance Spectra of a Nitroxide Spin Label on a Polyalanine α-Helix. J. Phys. Chem. B 2008, 112, 5755–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T, | M, | L, | M, | L, |
|---|---|---|---|---|
| K | a.u. | a.u. | a.u. | rad/ps |
| 160 | 0 | 2.3 | 0.06 | 7.5 |
| 180 | 0.2 | 2.1 | 0.17 | 6.3 |
| 190 | 0.4 | 3.3 | 0.33 | 14.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alimov, D.V.; Ivanov, M.Y.; Pylaeva, S.; Fedin, M.V. Structural Anomaly in Glasses: Molecular Dynamics Study of Organic Radical in Dibutylphthalate at Different Temperatures. Int. J. Mol. Sci. 2022, 23, 14859. https://doi.org/10.3390/ijms232314859
Alimov DV, Ivanov MY, Pylaeva S, Fedin MV. Structural Anomaly in Glasses: Molecular Dynamics Study of Organic Radical in Dibutylphthalate at Different Temperatures. International Journal of Molecular Sciences. 2022; 23(23):14859. https://doi.org/10.3390/ijms232314859
Chicago/Turabian StyleAlimov, Dmitry V., Mikhail Yu. Ivanov, Svetlana Pylaeva, and Matvey V. Fedin. 2022. "Structural Anomaly in Glasses: Molecular Dynamics Study of Organic Radical in Dibutylphthalate at Different Temperatures" International Journal of Molecular Sciences 23, no. 23: 14859. https://doi.org/10.3390/ijms232314859
APA StyleAlimov, D. V., Ivanov, M. Y., Pylaeva, S., & Fedin, M. V. (2022). Structural Anomaly in Glasses: Molecular Dynamics Study of Organic Radical in Dibutylphthalate at Different Temperatures. International Journal of Molecular Sciences, 23(23), 14859. https://doi.org/10.3390/ijms232314859