
Transcriptome Analysis Reveals Potential Mechanism in Storage Protein Trafficking within Developing Grains of Common Wheat

 and
and
Abstract
:1. Introduction
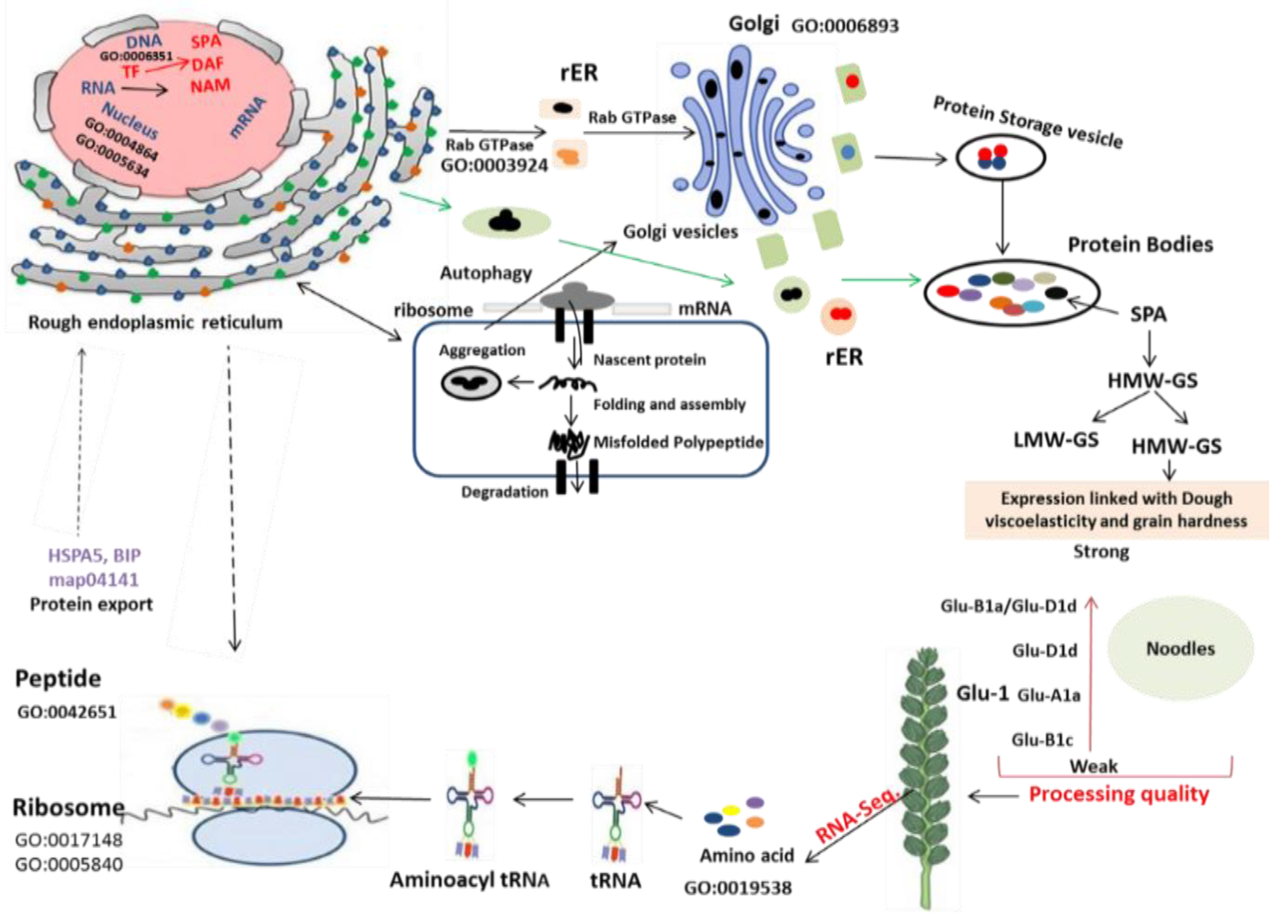
2. Results
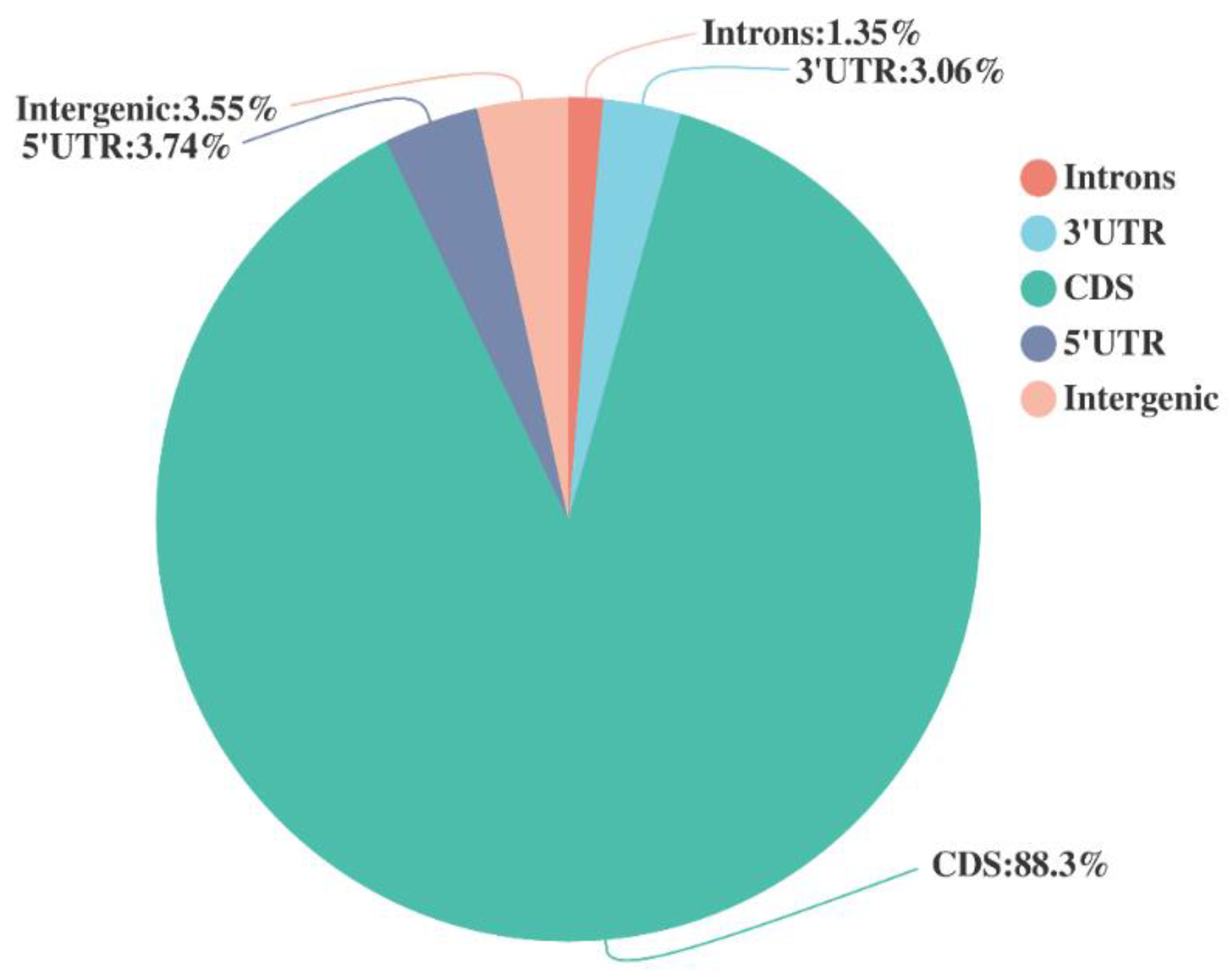
2.1. Sequence Assembly Based on cDNA Libraries
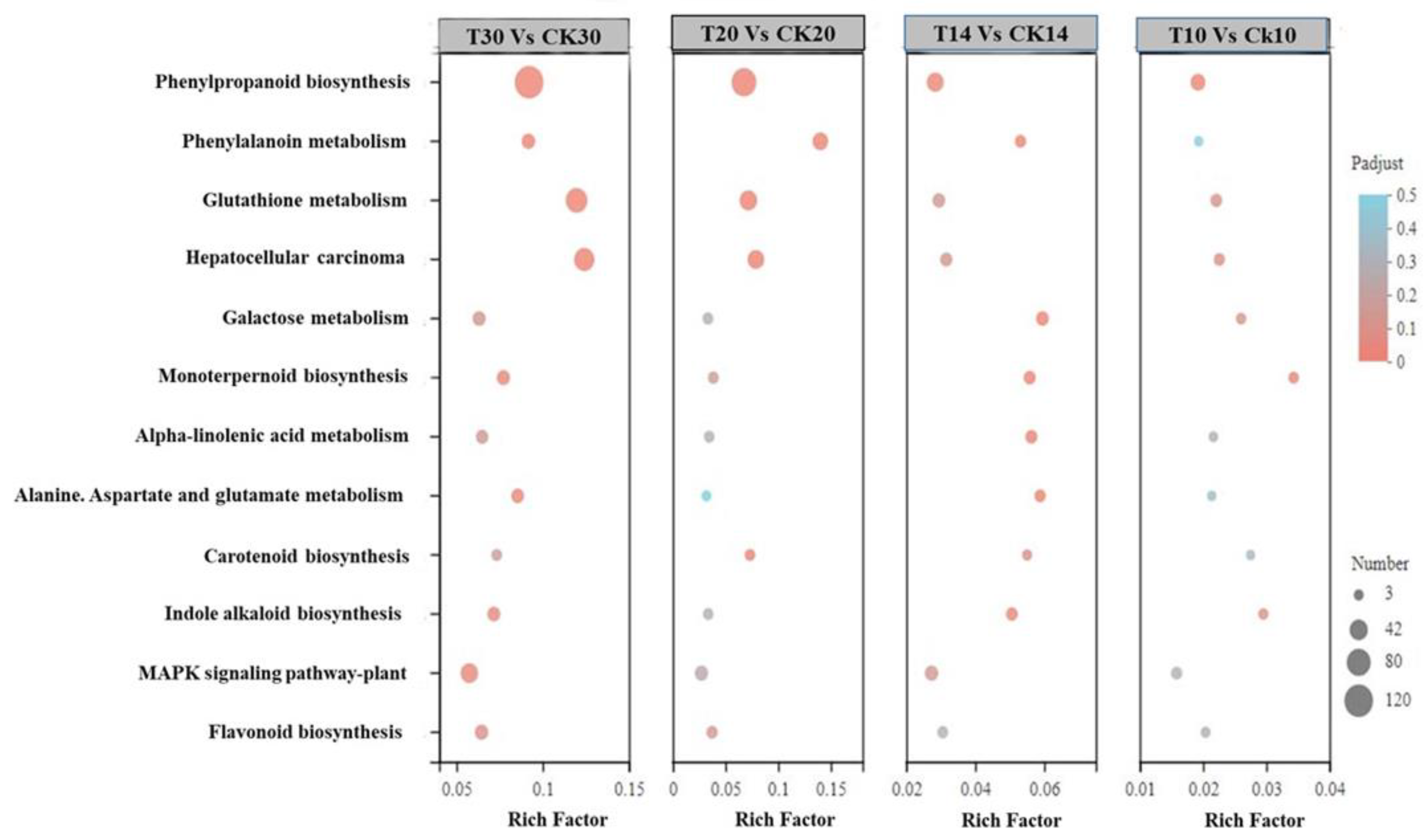
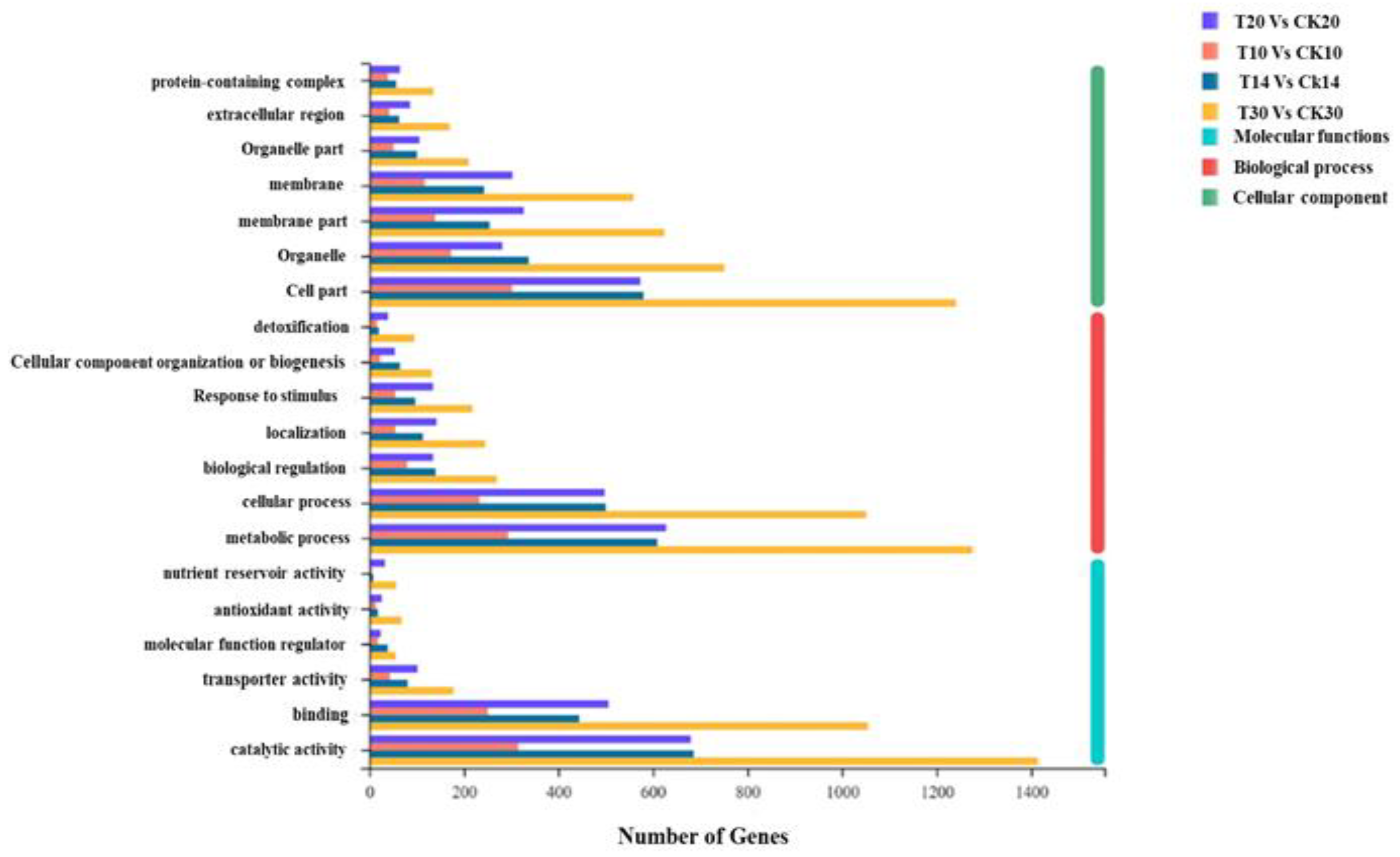
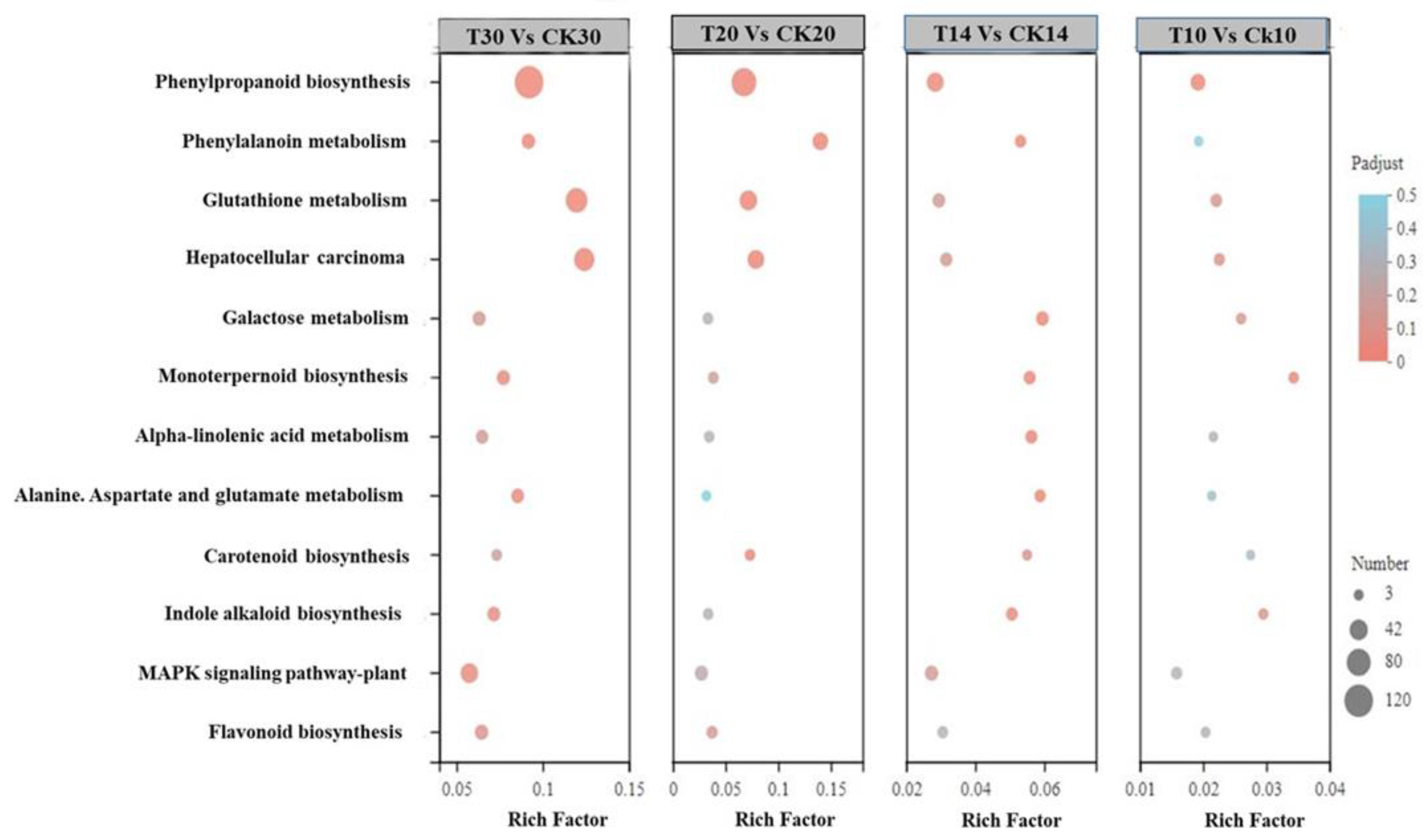
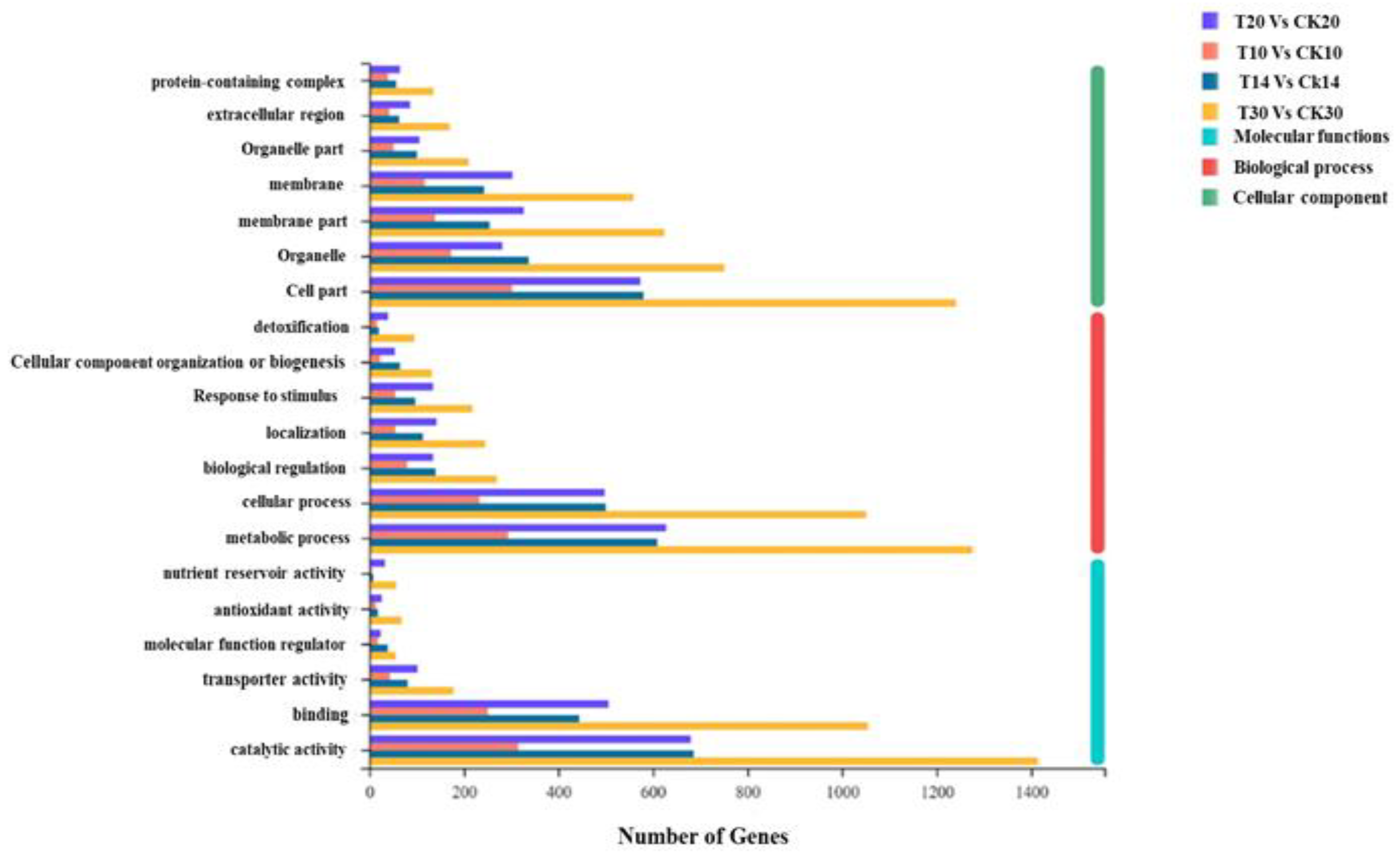
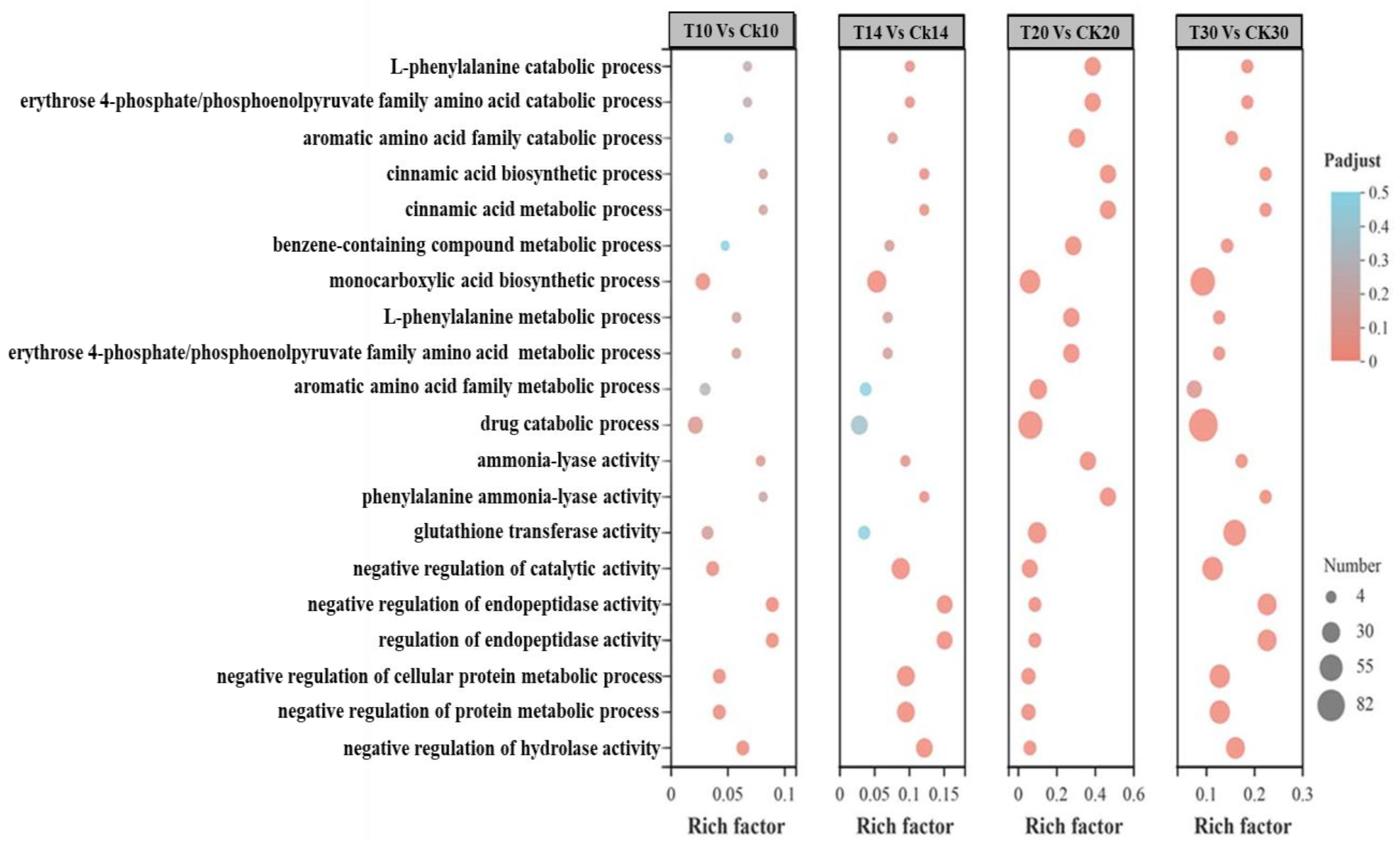
2.2. Functional Annotation between Different Grain Development Stages
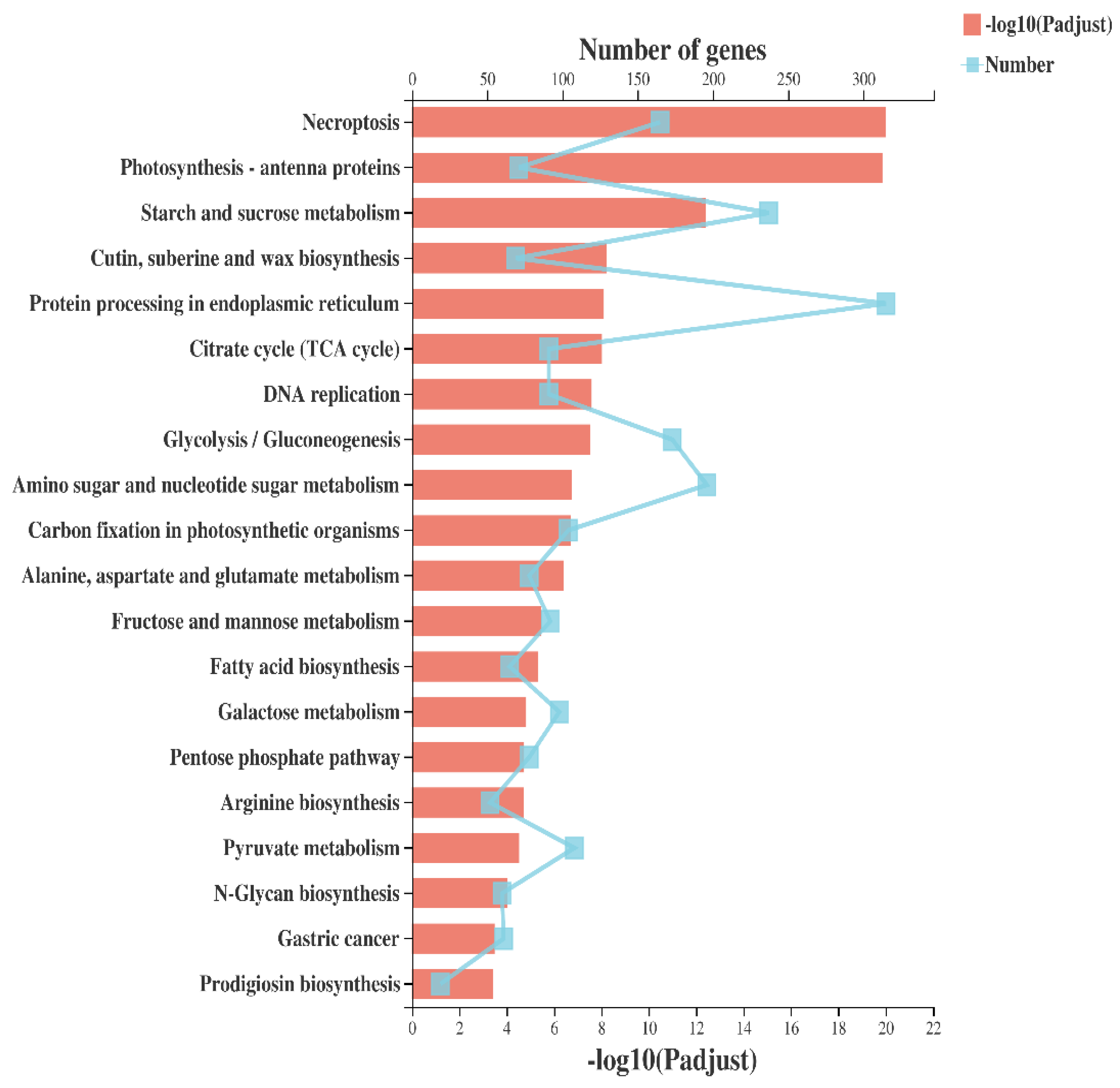
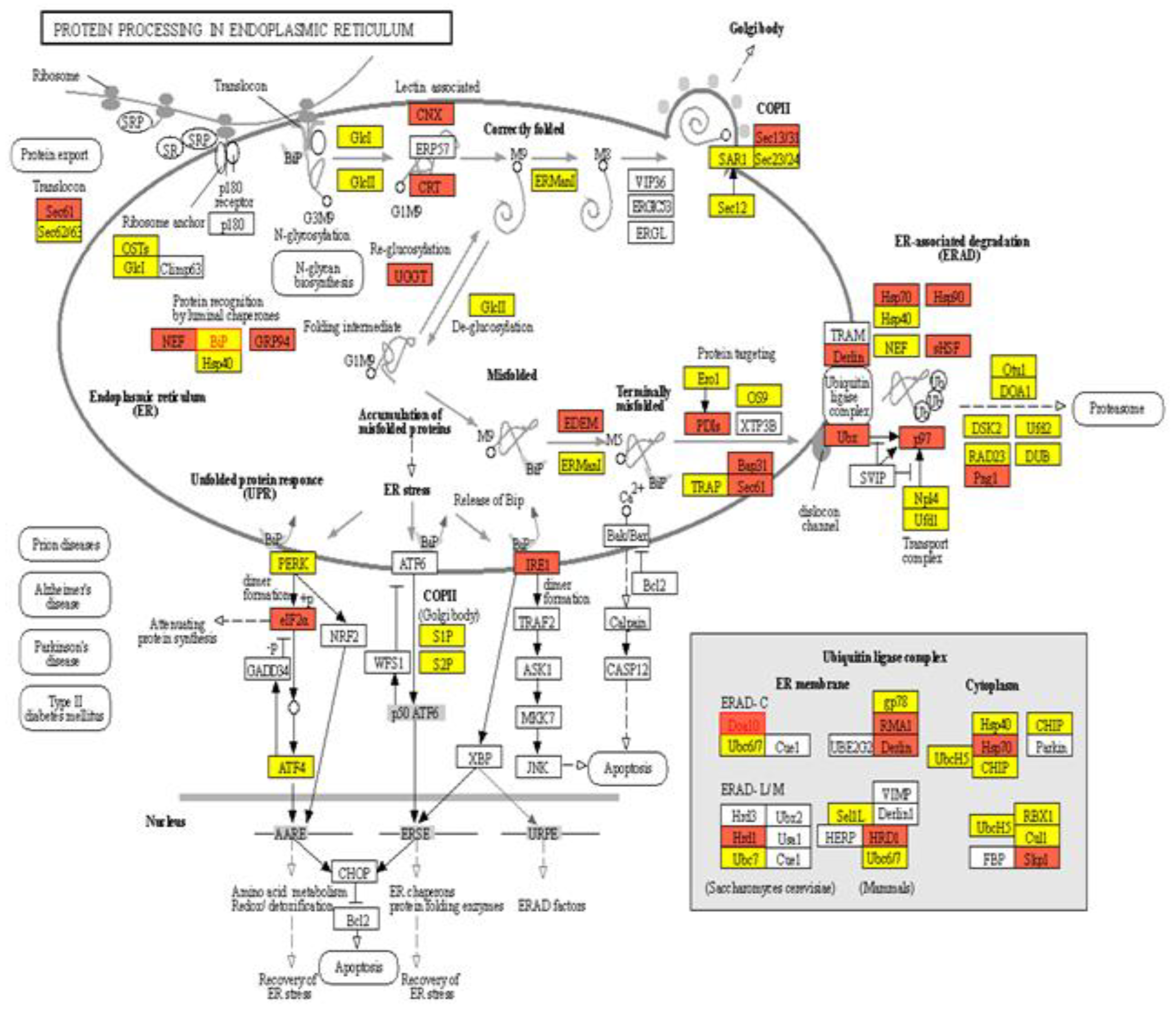
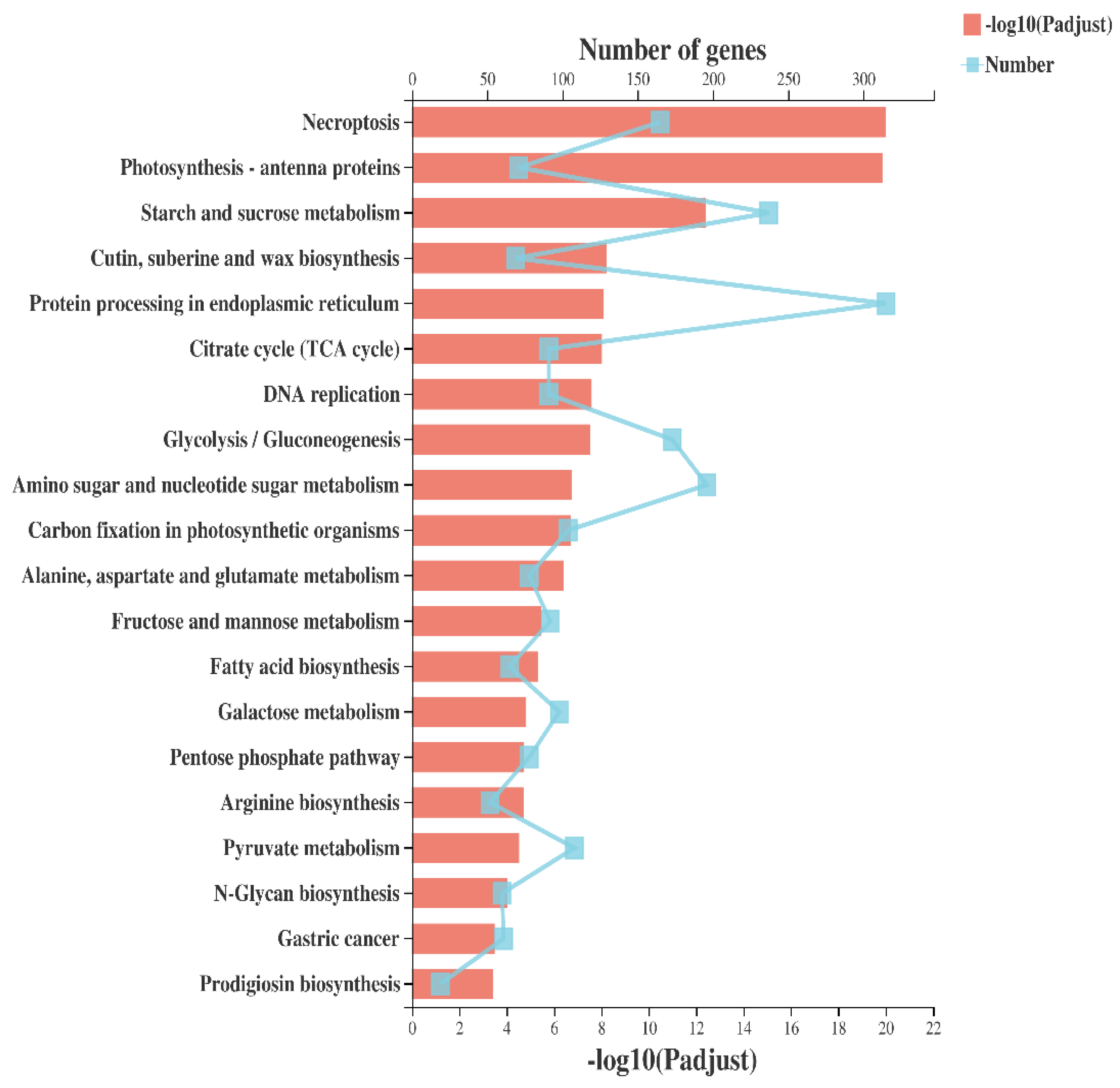
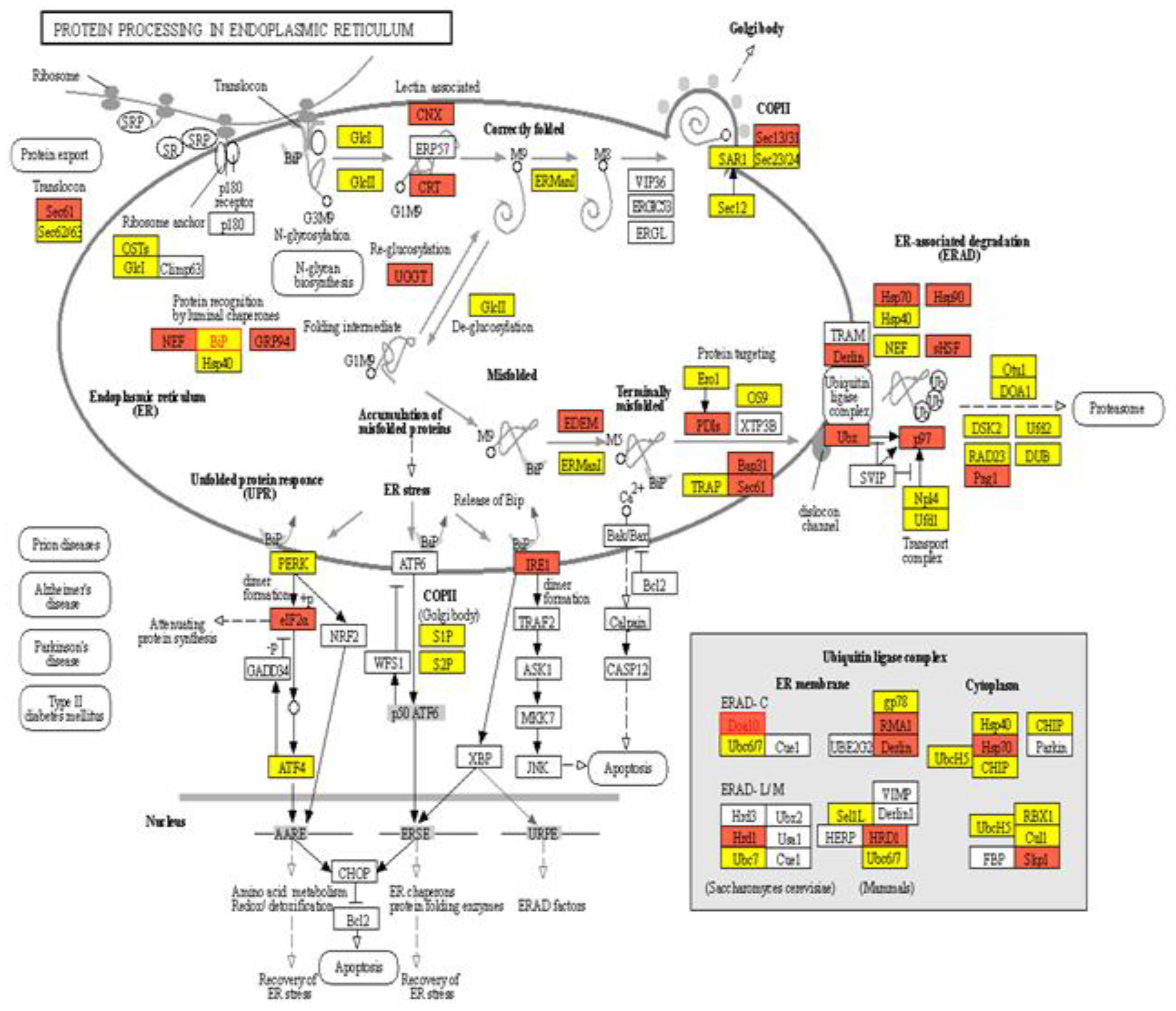
2.3. Pathway Analysis of Protein Processing in Endoplasmic Reticulum (ER)
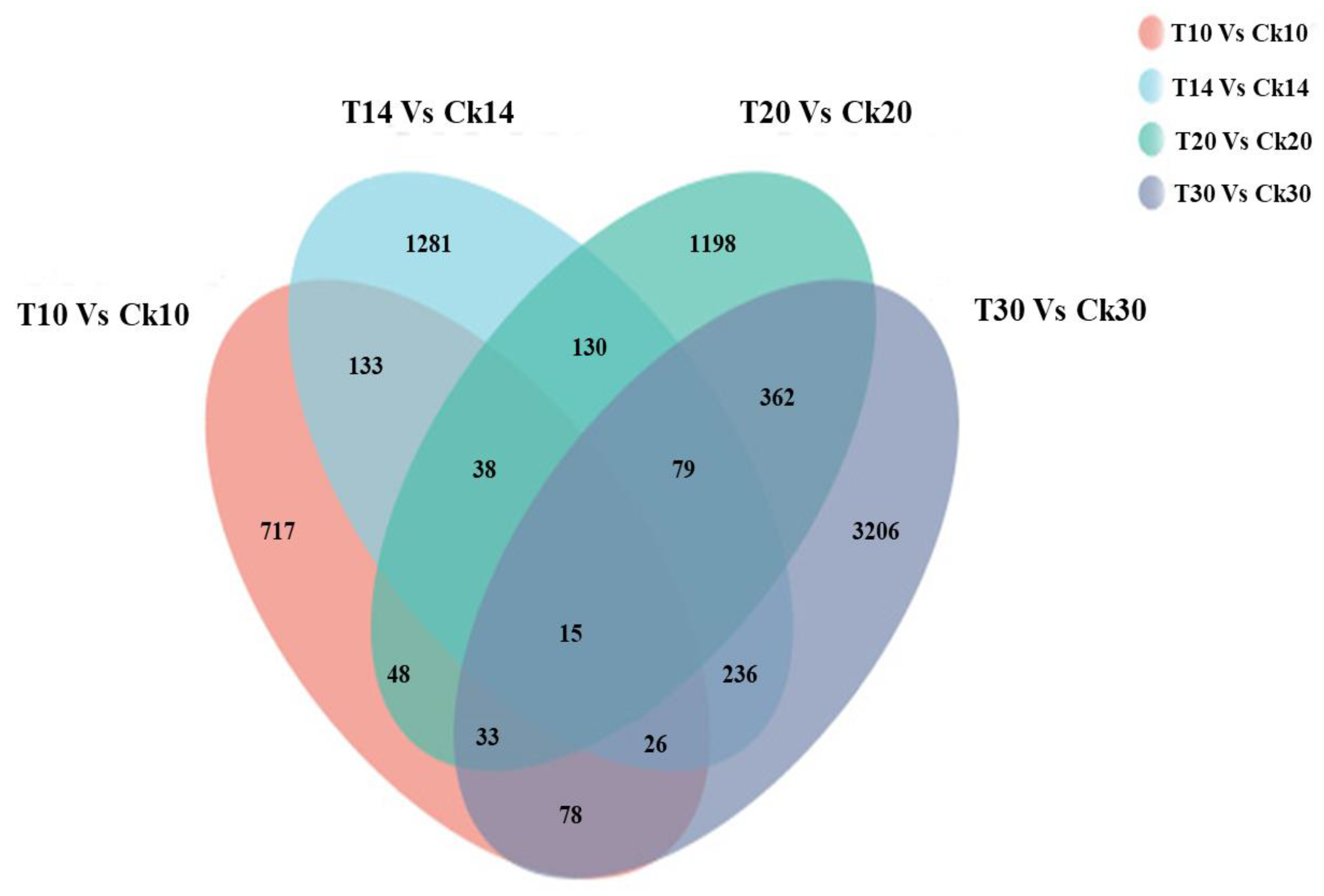
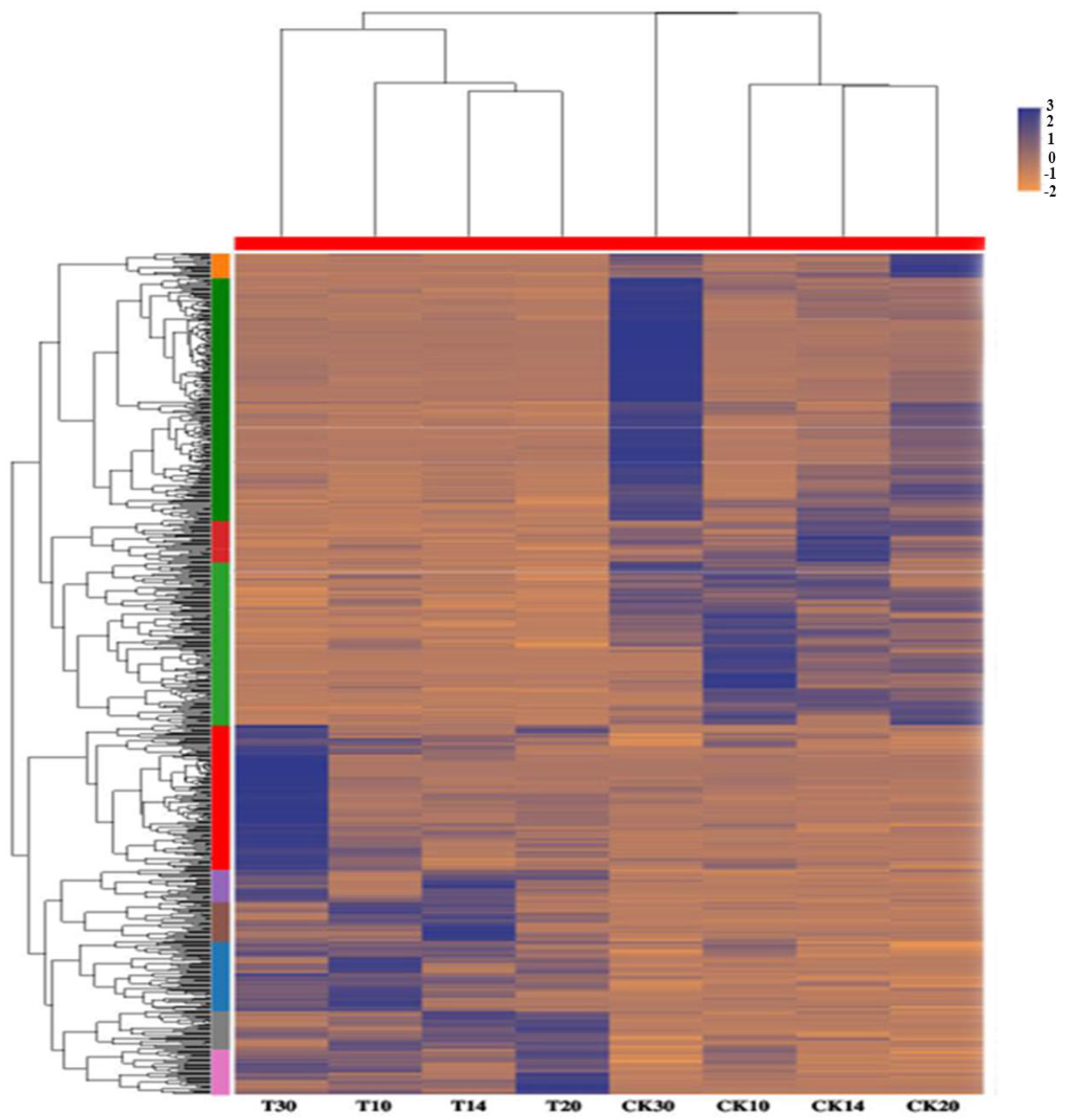
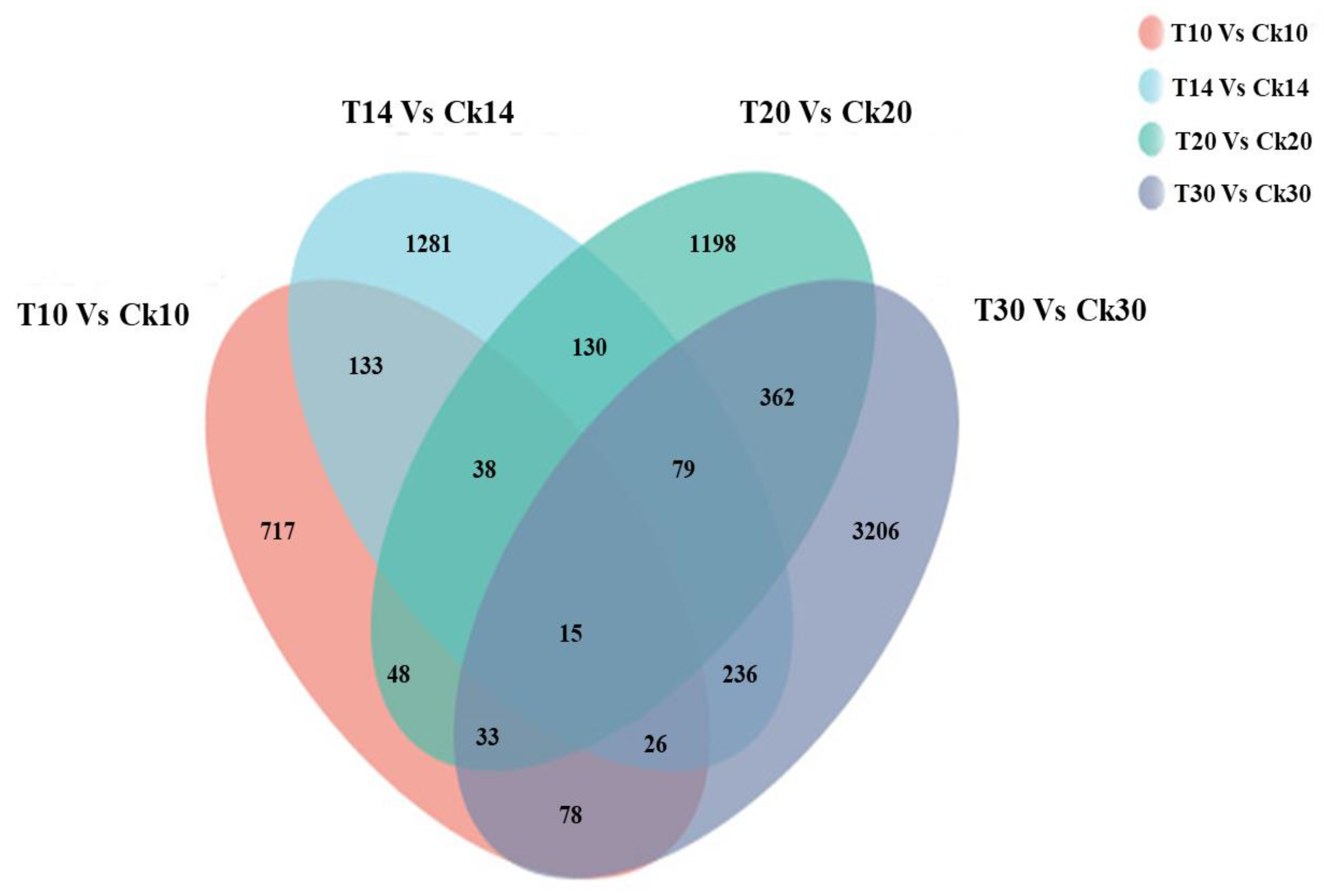
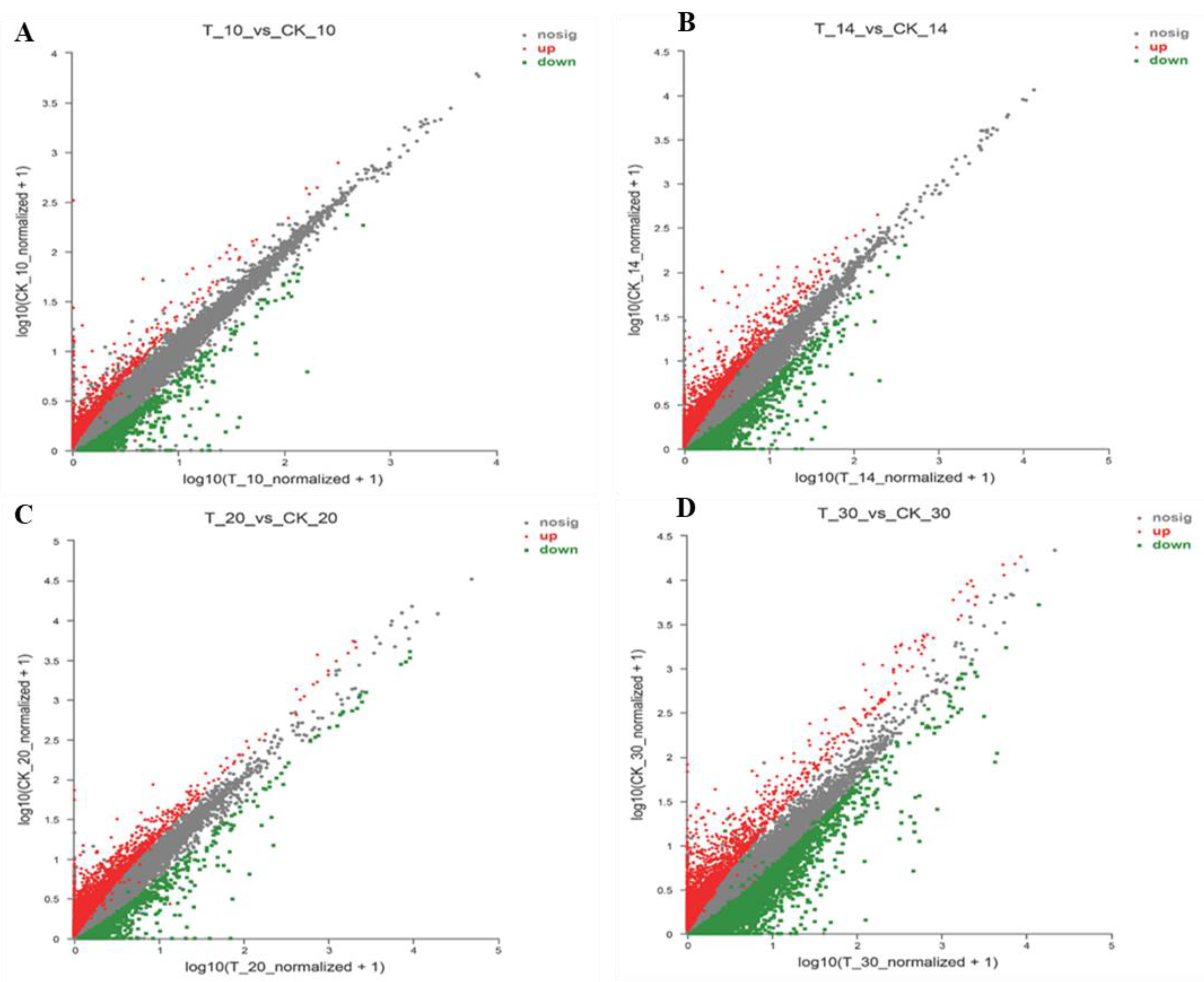
2.4. Analysis of Different Groups of Differently Expressed Genes
2.5. Putative Genes Related to Protein Quality Control
3. Discussion
4. Materials and Method
4.1. Plant Materials
4.2. RNA Extraction
4.3. Library Preparation and Illumina Hiseq4000 Sequencing
4.4. Read Mapping
4.5. Differential Expression Analysis and Functional Enrichment
4.6. New Isoforms Prediction
4.7. Alternative Splice Events Identification
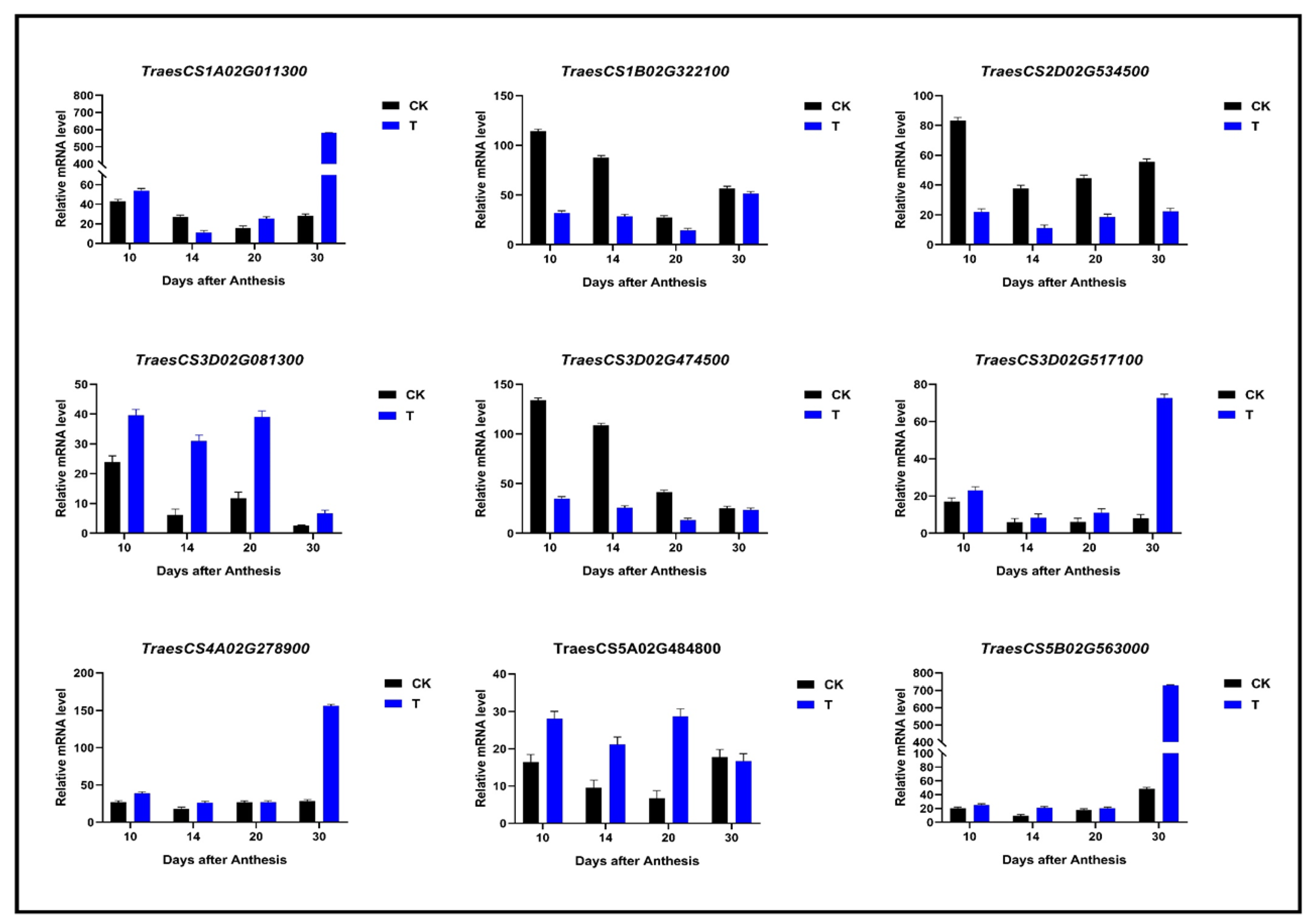
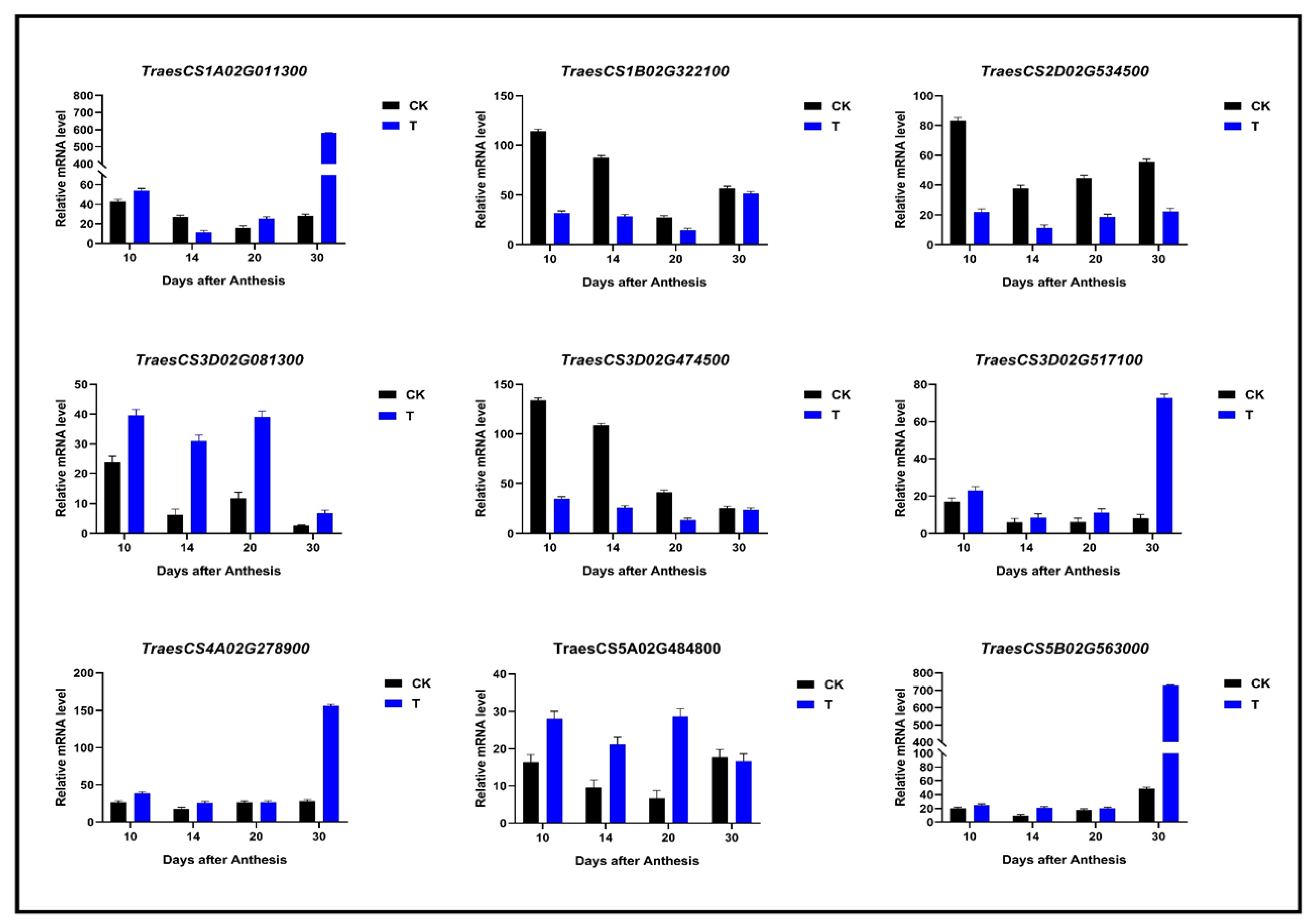
4.8. Gene Expression Level Validation by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Beecher, B.; Bettge, A.; Smidansky, E.; Giroux, M. Expression of wild-type pinB sequence in transgenic wheat complements a hard phenotype. Theor. Appl. Genet. 2002, 105, 870–877. [Google Scholar] [CrossRef]
- Day, L.; Augustin, M.A.; Batey, I.L.; Wrigley, C.W. Wheat-gluten uses and industry needs. Trends Food Sci. Technol. 2006, 17, 82–90. [Google Scholar] [CrossRef]
- Shewry, P.R.; Hey, S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015, 4, 178–202. [Google Scholar] [CrossRef] [PubMed]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Su, L.; Chen, X.; Qi, X. Involvement of auxin in growth and stress response of cucumber. Veg. Res. 2022. [Google Scholar] [CrossRef]
- Curtis, T.; Halford, N.G. Food security: The challenge of increasing wheat yield and the importance of not compromising food safety. Ann. Appl. Biol. 2014, 164, 354–372. [Google Scholar] [CrossRef] [Green Version]
- FAOSTAT. (FAOSTAT Analytical Brief 41. (ISSN2709-006x [Print]. ISSN2709-0078 [Online]). 2018. Available online: http://www.fao.org/faostat/ (accessed on 12 September 2022).
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef]
- Xiang, D.; Quilichini, T.D.; Liu, Z.; Gao, P.; Pan, Y.; Li, Q.; Nilsen, K.T.; Venglat, P.; Esteban, E.; Pasha, A.; et al. The Transcriptional landscape of polypoid wheats and their diploid ancestors during embryogenesis and grain development. Plant Cell 2019, 31, 2888–2911. [Google Scholar] [CrossRef] [Green Version]
- Michel, S.; Gallee, M.; Löschenberger, F.; Buerstmayr, H.; Kummer, C. Improving the baking quality of bread wheat using rapid tests and genomics: The prediction of dough rheological parameters by gluten peak indices and genomic selection models. J. Cereal Sci. 2017, 77, 24–34. [Google Scholar] [CrossRef]
- Wang, D.; Li, F.; Cao, S.; Zhang, K. Genomic and functional genomics analyses of gluten proteins and prospect for simultaneous improvement of end-use and health-related traits in wheat. Theor. Appl. Genet. 2020, 133, 1521–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Liu, C.; Qin, M.; Li, W.; Hou, J.; Shi, X.; Dai, Z.; Yao, W.; Tian, B.; Lei, Z.; et al. Promoter DNA hypermethylation of TaGli-γ-2.1 positively regulates gluten strength in bread wheat. J. Adv. Res. 2022, 36, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Si, X.; Ji, X.; Fan, R.; Liu, J.; Chen., K.; Wang, D.; Gao, C. Genome editing of upstream open reading frames enables translational control in plants. Nat. Biotechnol. 2018, 36, 894–898. [Google Scholar] [CrossRef]
- Li, D.; Jin, H.; Zhang, K.; Wang, Z.; Wang, F.; Zhao, Y.; Huo, N.; Liu, X.; Gu, Y.Q.; Wang, D.; et al. Analysis of the Gli-D2 locus identifies a genetic target for simultaneously improving the bread-making and health-related traits of common wheat. Plant J. 2018, 95, 414–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, Y.; Yang, Y.; Liu, X.; Qin, H.; Dong, Z.; Zheng, S.; Zhang, K.; Wang, D. New insight into the function of wheat glutenin proteins as investigated with two series of genetic mutants. Sci. Rep. 2017, 7, 3428. [Google Scholar] [CrossRef]
- Ma, W.; Yu, Z.; She, M.; Zhao, Y.; Islam, S. Wheat gluten protein and its impacts on wheat processing quality. Front. Agric. Sci. Eng. 2019, 6, 279. [Google Scholar] [CrossRef] [Green Version]
- Anjum, F.M.; Khan, M.R.; Din, A.; Saeed, M.; Pasha, I.; Arshad, M.U. Wheat gluten: High molecular weight glutenin subunits—structure, genetics, and relation to dough elasticity. J. Food Sci. 2007, 72, R56–R63. [Google Scholar] [CrossRef]
- Gupta, R.B.; Shepherd, K.W. Two-step one-dimensional SDS-PAGE analysis of LMW subunits of glutelin. Theor. Appl. Genet. 1990, 80, 183–187. [Google Scholar] [CrossRef]
- Rehman, S.U.; Paterson, A.; Piggott, J.R. Optimization of flours for chapatti preparation using a mixture design. J. Sci. Food Agric. 2007, 87, 425–430. [Google Scholar] [CrossRef]
- Shewry, P.R.; Popineau, Y.; Lafiandra, D.; Belton, P. Wheat glutenin subunits and dough elasticity: Findings of the EUROWHEAT project. Trends Food Sci. Technol. 2000, 11, 433–441. [Google Scholar] [CrossRef]
- Osborne, T.B. The Proteins of the Wheat Kerne; Carnegie Institution of Washington, Publication No.84; Judd & Detweiler, Inc.: Washington, DC, USA, 1907. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R. What is gluten—Why is it special? Front Nutr. 2019, 6, 101. [Google Scholar] [CrossRef] [PubMed]
- Shewry, P.R.; Halford, N.G.; Lafiandra, D. Genetics of wheat gluten proteins. Adv. Genet. 2003, 49, 111–184. [Google Scholar] [PubMed]
- Evers, T.; Millar, S. Cereal Grain Structure and Development: Some Implications for Quality. J. Cereal Sci. 2002, 36, 261–284. [Google Scholar] [CrossRef]
- Laudencia-Chingcuanco, D.L.; Stamova, B.S.; Lazo, G.R.; Cui, X.; Anderson, O.D. Analysis of the wheat endosperm transcriptome. J. Appl. Genet. 2006, 47, 287–302. [Google Scholar] [CrossRef]
- Cao, X.; Dong, Z.; Tian, D.; Dong, L.; Qian, W.; Liu, J.; Liu, X.; Qin, H.; Zhai, W.; Gao, C.; et al. Development and characterization of marker-free and transgene insertion site-defined transgenic wheat with improved grain storability and fatty acid content. Plant Biotechnol. J. 2020, 18, 129–140. [Google Scholar] [CrossRef]
- Tyler, A.M.; Bhandari, D.G.; Poole, M.; Napier, J.A.; Jones, H.D.; Lu, C.; Lycett, G.W. Gluten quality of bread wheat is associated with activity of RabD GTPases. Plant Biotechnol. J. 2015, 13, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batoko, H.; Zheng, H.Q.; Hawes, C.; Moore, I. A Rab1 GTPase is required for transport between the endoplasmic reticulum and Golgi apparatus and for normal Golgi movement in plants. Plant Cell 2000, 12, 2201–2217. [Google Scholar] [CrossRef] [Green Version]
- Adam Micheal Tyler. Manipulating Rab GTPase Activity in Wheat to Alter Gluten Quality for Bread-making. 2012. Available online: http://eprints.nottingham.ac.uk/12914/1/Thesis_Hardbound_AMT_6.pdf (accessed on 12 September 2022).
- Tosi, P.; Parker, M.; Gritsch, C.S.; Carzaniga, R.; Martin, B.; Shewry, P.R. Trafficking of storage proteins in developing grain of wheat. J. Exp. Bot. 2009, 60, 979–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.L. Protein Body Inclusions in Developing Wheat Endosperm. Ann. Bot. 1980, 46, 29–36. [Google Scholar] [CrossRef]
- Zheng, H.; Camacho, L.; Wee, E.; Batoko, H.; Legen, J.; Leaver, C.J.; Malhó, R.; Hussey, P.; Moore, I. A Rab-E GTPase Mutant Acts Downstream of the Rab-D Subclass in Biosynthetic Membrane Traffic to the Plasma Membrane in Tobacco Leaf Epidermis. Plant Cell 2005, 17, 2020–2036. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, F.; Plivelic, T.S.; Kuktaite, R.; Hedenqvist, M.S.; Johansson, E. Unraveling the Structural Puzzle of the Giant Glutenin Polymer—An Interplay between Protein Polymerization, Nano morphology, and Functional Properties in Bioplastic Films. ACS Omega 2018, 3, 5584–5592. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Nilsson, H.; Mazhar, H.; Skerritt, J.; MacRitchie, F.; Svensson, G. Seasonal effects on storage proteins and gluten strength in four Swedish wheat cultivars. J. Sci. Food Agric. 2002, 82, 1305–1311. [Google Scholar] [CrossRef]
- Rasheed, F.; Kuktaite, R.; Hedenqvist, M.S.; Gallstedt, M.; Plivelic, T.S.; Johansson, E. The use of plants as a green factory to produce high strength gluten-based materials. Green Chem. 2016, 18, 2782–2792. [Google Scholar] [CrossRef]
- Vitale, A.; Ceriotti, A.; Denecke, J. The Role of the Endoplasmic Reticulum in Protein Synthesis, Modification and Intracellular Transport. J. Exp. Bot. 1993, 44, 1417–1444. [Google Scholar] [CrossRef]
- Parker, M.L. Protein accumulation in the developing endosperm of a high protein line of Triticum dicoccoides. Plant Cell Environ. 1982, 5, 37–43. [Google Scholar]
- Kim, W.T.; Franceschi, V.R.; Krishnan, H.B.; Okita, T.W.; Okita, T. Formation of wheat protein bodies: Involvement of the Golgi apparatus in gliadin transport. Planta 1988, 176, 173–182. [Google Scholar] [CrossRef]
- Loussert, C.; Popineau, Y.; Mangavel, C. Protein bodies ontogeny and localization of prolamin components in the developing endosperm of wheat caryopses. J. Cereal Sci. 2008, 47, 445–456. [Google Scholar] [CrossRef]
- Grimberg, Å.; Wilkinson, M.; Snell, P.; De Vos, R.P.; González-Thuillier, I.; Tawfike, A.; Ward, J.L.; Carlsson, A.S.; Shewry, P.; Hofvander, P. Transitions in wheat endosperm metabolism upon transcriptional induction of oil accumulation by oat endosperm WRINKLED1. BMC Plant Biol. 2020, 20, 235. [Google Scholar] [CrossRef]
- Cullum, R.; Alder, O.; Hoodless, P.A. The next generation: Using new sequencing technologies to analyses gene regulation. Respirology 2011, 16, 210–222. [Google Scholar] [CrossRef]
- Yu, K.; Liu, D.; Chen, Y.; Wang, D.; Yang, W.; Yang, W.; Yin, L.; Zhang, C.; Zhao, S.; Sun, J.; et al. Unraveling the genetic architecture of grain size in einkorn wheat through linkage and homology mapping and transcriptomic profiling. J. Exp. Bot. 2019, 70, 4671–4688. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Margarido, G.R.A.; Heckerman, D. ConPADE: Genome Assembly Ploidy Estimation from Next-Generation Sequencing Data. PLOS Comput. Biol. 2015, 11, e1004229. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, L.-J.; Bansept-Basler, P.; Olohan, L.; Joynson, R.; Brenchley, R.; Hall, N.; O’Sullivan, D.M.; Hall, A. Mapping-by-sequencing in complex polyploid genomes using genic sequence capture: A case study to map yellow rust resistance in hexaploid wheat. Plant J. 2016, 87, 403–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Poole, R.L.; Huttly, A.K.; Toscano-Underwood, C.; Feeney, K.; Welham, S.; Gooding, M.J.; Mills, C.; Edwards, K.J.; Shewry, P.R.; et al. Transcriptome analysis of grain development in hexaploid wheat. BMC Genom. 2008, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Liu, H.; Wang, K.; Liu, L.; Wang, S.; Zhao, Y.; Yin, J.; Li, Y. Development-associated microRNAs in grains of wheat (Triticum aestivum L.). BMC Plant Biol. 2013, 13, 140. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.; Buell, C.R. Crop domestication: Past, present and future. Plant Cell Physiol. 2022, 63, 1527–1528. [Google Scholar] [CrossRef]
- Brinton, J.; Uauy, C. A reductionist approach to dissecting grain weight and yield in wheat. J. Integr. Plant Biol. 2018, 61, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Adamski, N.M.; Borrill, P.; Brinton, J.; Harrington, S.A.; Marchal, C.; Bentley, A.R.; Bovill, W.D.; Cattivelli, L.; Cockram, J.; Contreras-Moreira, B.; et al. A roadmap for gene functional characterization in crops with large genomes: Lessons from polyploid wheat. eLife 2020, 9, 55646. [Google Scholar] [CrossRef]
- Barrero, J.M.; Cavanagh, C.; Verbyla, K.L.; Tibbits, J.F.; Verbyla, A.P.; Huang, B.E.; Rosewarne, G.M.; Stephen, S.; Wang, P.; Whan, A.; et al. Transcriptomic analysis of wheat near-isogenic lines identifies PM19-A1 and A2 as candidates for a major dormancy QTL. Genome Biol. 2015, 16, 93. [Google Scholar] [CrossRef] [Green Version]
- Zhai, H.; Jiang, C.; Zhao, Y.; Yang, S.; Li, Y.; Yan, K.; Wu, S.; Luo, B.; Du, Y.; Jin, H.; et al. Wheat heat tolerance is impaired by heightened deletions in the distal end of 4AL chromosomal arm. Plant Biotechnol. J. 2021, 19, 1038–1051. [Google Scholar] [CrossRef]
- Arenas-M, A.; Castillo, F.M.; Godoy, D.; Canales, J.; Calderini, D.F. Transcriptomic and Physiological Response of Durum Wheat Grain to Short-Term Heat Stress during Early Grain Filling. Plants 2022, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.O.; Quatrano, R.S. Morphological Staging of Wheat Caryopsis Development. Am. J. Bot. 1983, 70, 308–311. [Google Scholar] [CrossRef]
- Survase, A.R. Genomics of Consumer Traits in Chapatti Quality. Queensland Alliance for Agriculture and Food Innovation. 2017. Available online: https://espace.library.uq.edu.au/data/UQ_701980/s4306877_thesis.pdf (accessed on 12 September 2022).
- Ravel, C.; Martre, P.; Romeuf, I.; Dardevet, M.; El-Malki, R.; Bordes, J.; Charmet, G. Nucleotide Polymorphism in the Wheat Transcriptional Activator Spa Influences Its Pattern of Expression and Has Pleiotropic Effects on Grain Protein Composition, Dough Viscoelasticity, and Grain Hardness. Plant Physiol. 2009, 151, 2133–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oszvald, M.; Tamas, L.; Shewry, P.R.; Tosi, P. The trafficking pathway of a wheat storage protein in transgenic rice endosperm. Ann. Bot. 2014, 113, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, G.; Sengupta-Gopalan, C.; Ceriotti, A. The endoplasmic reticulum of plant cells and its role in protein maturation and biogenesis of oil bodies. Plant Mol. Biol. 1998, 38, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.-M.; Cho, K.; Lee, H.-J.; Lim, S.-H.; Kim, J.S.; Kim, Y.-M.; Lee, J.-Y. Cellular Localization of Wheat High Molecular Weight Glutenin Subunits in Transgenic Rice Grain. Int. J. Mol. Sci. 2017, 18, 2458. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Zerial, M.; Mcbride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Lawson, T.; Lycett, G.W.; Mayes, S.; Ho, W.K.; Chin, C.F. Transcriptome-wide identification and characterization of the Rab GTPase family in mango. Mol. Biol. Rep. 2020, 47, 4183–4197. [Google Scholar] [CrossRef]
- Ma, D.; Gao, H.; Du, C.; Li, L.; Sun, W.; Liu, S.; Wang, C.; Xie, Y.; Kang, G. Transcriptomic and Metabolomics Analysis of Different Endosperm Region under Nitrogen Treatments. Int. J. Mol. Sci. 2019, 20, 4212. [Google Scholar] [CrossRef]
- Määttänen, P.; Gehring, K.; Bergeron, J.J.; Thomas, D.Y. Protein quality control in the ER: The recognition of misfolded proteins. Semin. Cell Dev. Biol. 2010, 21, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.-A. The Modular Control of Cereal Endosperm Development. Trends Plant Sci. 2020, 25, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Lemmon, B.E.; Olsen, O.A. Endosperm development in barley: Microtubule involvement in the morphogenetic pathway. Plant Cell 1994, 6, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiesselbach, T.A. The Structure and Eeproduction of Corn; (Research bulletin of the Agricultural Experiment Station of Nebraska No. 161); University of Nebraska: Lincoln, NE, USA, 1949. [Google Scholar]
- Briarty, L.G.; Hughes, C.E.; Evers, A.D. The Developing Endosperm of Wheat—A Stereological Analysis. Ann. Bot. 1979, 44, 641–658. [Google Scholar] [CrossRef]
- Cao, H.; Duncan, O.; Millar, A.H. Protein turnover in the developing Tritium aestium grain. New Phytol. 2022, 233, 1188–1201. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Y.H.; Yi, H.Y.; Gong, J.M. Vacuolar membrane transporters OsVIT1 and OsVIT2 modulate iron translocation between fag leaves and seeds in rice. Plant J. 2012, 72, 400–410. [Google Scholar] [CrossRef]
- Luo, G.; Shen, L.; Song, Y.; Yu, K.; Ji, J.; Zhang, C.; Yang, W.; Li, X.; Sun, J.; Zhan, K.; et al. The MYB family transcription factor TuODORANT1 from Triticum urartu and the homolog TaODORANT1 from Triticum aestivum inhibit seed storage protein synthesis in wheat. Plant Biotechnol. J. 2021, 19, 1863–1877. [Google Scholar] [CrossRef]
- Shewry, P.R.; Halford, N.G.; Tatham, A.S. High molecular weight subunits of wheat glutenin. J. Cereal Sci. 1992, 15, 105–120. [Google Scholar] [CrossRef]
- Lawrance, G.; Moss, H.; Shepherd, K.; Wrigley, C. Dough quality of biotypes of eleven Australian wheat cultivars that differ in high-molecular-weight glutenin subunit composition. J. Cereal Sci. 1987, 6, 99–101. [Google Scholar]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Tucker, M.R.; Burton, R.A.; Shirley, N.J.; Little, A.; Morris, J.; Milne, L.; Houston, K.; Hedley, P.E.; Waugh, R.; et al. The dynamics of transcript abundance during cellularisation of developing barley endosperm. Plant Physiol. 2016, 170, 1549–1565. [Google Scholar] [CrossRef] [PubMed]
- Tabe, L.; Hagan, N.; Higgins, T.J.V. Plasticity of seed protein composition in response to nitrogen and sulfur availability. Curr. Opin. Plant Biol. 2002, 5, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Upadhyaya, N.M.; Campbell, P.M.; Akhurst, R.; Hagan, N.; Higgins, T.J.V. Decreased accumulation of glutelin types in rice grains constitutively expressing a sunflower seed albumin gene. Phytochemistry 2005, 66, 2534–2539. [Google Scholar] [CrossRef]
- Zhang, S.; Ghatak, A.; Bazargani, M.M.; Bajaj, P.; Varshney, R.K.; Chaturvedi, P.; Jiang, D.; Weckwerth, W. Spatial distribution of proteins and metabolites in developing wheat grain and their differential regulatory response during the grain filling process. Plant J. 2021, 107, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Mumolo, M.G.; Rettura, F.; Melissari, S.; Costa, F.; Ricchiuti, A.; Ceccarelli, L.; de Bortoli, N.; Marchi, S.; Bellini, M. Is Gluten the Only Culprit for Non-Celiac Gluten/Wheat Sensitivity? Nutrients 2020, 12, 3785. [Google Scholar] [CrossRef]
- Zhen, S.; Han, C.; Ma, C.; Gu, A.; Zhang, M.; Shen, X.; Li, X.; Yan, Y. Deletion of the low-molecular-weight glutenin subunit allele Glu-A3a of wheat (Triticum aestivum L.) significantly reduces dough strength and breadmaking quality. BMC Plant Biol. 2014, 14, 367. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Duncan, O.; Islam, S.; Zhang, J.; Ma, W.; Millar, A.H. Increased Wheat Protein Content via Introgression of an HMW Glutenin Selectively Reshapes the Grain Proteome. Mol. Cell Proteom. 2021, 20, 100097. [Google Scholar] [CrossRef]
- Li, P.; Chen, Y.H.; Lu, J.; Zhang, C.Q.; Liu, Q.Q.; Li, Q.F. Genes and Their Molecular Functions Determining Seed Structure, Components, and Quality of Rice. Rice 2022, 15, 18. [Google Scholar] [CrossRef]
- Gutkowska, M.; Kaus-Drobek, M.; Hoffman-Sommer, M.; Małgorzata Pamuła, M.; Daria Leja, A.; Perycz, M.; Lichocka, M.; Witek, A.; Wojtas, M.; Dadlez, M.; et al. Impact of C-terminal truncations in the Arabidopsis Rab escort protein (REP) on REP-Rab interaction and plant fertility. Plant J. 2021, 108, 1400–1421. [Google Scholar] [CrossRef]
- Pan, T.; Wang, Y.; Jing, R.; Wang, Y.; Wei, Z.; Zhang, B.; Lei, C.; Qi, Y.; Wang, F.; Bao, X.; et al. Post-Golgi trafficking of rice storage proteins requires the small GTPase Rab7 activation complex MON1-CCZ1. Plant Physiol. 2021, 187, 2174–2191. [Google Scholar] [CrossRef]
- Alhabbar, Z.; Yang, R.; Juhasz, A.; Xin, H.; She, M.; Anwar, M.; Sultana, N.; Diepeveen, D.; Ma, W.; Islam, S. NAM gene allelic composition and its relation to grain-filling duration and nitrogen utilisation efficiency of Australian wheat. PLoS One 2018, 13, e0205448. [Google Scholar] [CrossRef] [PubMed]
- El-Esawi, M.A.; Alayafi, A.A. Overexpression of Rice Rab7 Gene Improves Drought and Heat Tolerance and Increases Grain Yield in Rice (Oryza sativa L.). Genes 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buttar, Z.; Wu, S.; Arnao, M.; Wang, C.; Ullah, I.; Wang, C. Melatonin Suppressed the Heat Stress-Induced Damage in Wheat Seedlings by Modulating the Antioxidant Machinery. Plants 2020, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, L.; Li, Y.; Buttar, Z.A.; Wang, N.; Xie, Y.; Wang, C. Single Nucleotide Mutagenesis of the TaCHLI Gene Suppressed Chlorophyll and Fatty Acid Biosynthesis in Common Wheat Seedlings. Front. Plant Sci. 2020, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, D.; Xu, H.; Xie, L.; He, M.; Hou, H.; Zhang, C. A loose endosperm structure of wheat seed produced under low nitrogen level promotes early germination by accelerating water uptake. Sci. Rep. 2017, 7, 3116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Li, G.; Chu, Z.; Ru, Z.; Jiang, X.; Wen, Z.; Zhang, G.; Wang, Y.; Zhang, Y.; Wei, W. Transcriptome analysis reveals important candidate genes involved in grain-size formation at the stage of grain enlargement in common wheat cultivar “Bainong 4199”. PLoS ONE 2019, 14, e0214149. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Guan, Y.; Buttar, Z.A.; Li, G.; Zhang, G.; Li, C.; Huang, J.; Liu, M.; Wei, W. Small RNA and degradome analysis uncovering the extensive effects of miRNAs and Targets in Early developing grains of common wheat. PLoS ONE 2020. preprint. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat; Discovering splice junctions with RNA-seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 387. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Park, J.W.; Huang, J.; Dittmar, K.A.; Lu, Z.; Zhou, Q.; Carstens, R.P.; Xing, Y. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 2012, 40, e61. [Google Scholar] [CrossRef] [PubMed]
- Muntz, K. Deposition of storage proteins. Plant Mol. Biol. 1998, 38, 77–99. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variety | Flour Rate | Settlement Index | Water Absorption | Formation Time | Max. Tensile Resistance |
|---|---|---|---|---|---|
| Shannong 33 | 73.3% | 66 mL | 58.7 | 15.6 | 1038 |
| Sister line (C.K) | 71% | 27.6 mL | 63 | 3.1 | 358 |
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Error Rate (%) | GC% | Q20 (%) a | Q30 (%) b | Total Mapped |
|---|---|---|---|---|---|---|---|---|---|
| T10 | 52,242,026 | 7,888,545,926 | 51,306,098 | 7,650,889,193 | 0.0248 | 54.64 | 98.03 | 94.4 | 45,136,960 (87.98%) |
| T14 | 58,357,186 | 8,811,935,086 | 57,143,490 | 8,473,951,442 | 0.0254 | 53.14 | 97.75 | 93.89 | 45,153,078 (79.02%) |
| T20 | 49,861,908 | 7,529,148,108 | 48,289,034 | 7,121,352,201 | 0.0268 | 52.91 | 97.17 | 92.6 | 37,102,266 (76.83%) |
| T30 | 49,553,172 | 7,482,528,972 | 48,697,252 | 7,236,265,313 | 0.025 | 54.35 | 97.96 | 94.25 | 37,230,131 (76.45%) |
| CK10 | 52,674,730 | 7,953,884,230 | 51,814,526 | 7,742,996,821 | 0.0246 | 55.02 | 98.11 | 94.59 | 46,754,208 (90.23%) |
| CK14 | 52,215,230 | 7,884,499,730 | 51,232,492 | 7,611,938,755 | 0.0253 | 53.75 | 97.8 | 94 | 43,115,178 (84.16%) |
| CK20 | 48,982,074 | 7,396,293,174 | 48,113,392 | 7,143,015,218 | 0.0252 | 52.64 | 97.8 | 94.03 | 34,274,380 (71.24%) |
| Ck30 | 51,353,948 | 7,754,446,148 | 50,053,338 | 7,410,668,772 | 0.0255 | 53.85 | 97.7 | 93.8 | 35,435,811 (70.8%) |
| Pathway Type | Pathway ID | DEGs | Up/Down (Numbers of Genes) | Gene ID |
|---|---|---|---|---|
| Cell wall synthesis (ER or Golgi cellular components) | map04714 | Up | 4 | TraesCS1A02G133900, TraesCS1A02G116200 TraesCS1A02G124600, TraesCS1A02G137216 |
| structural constituent of ribosome (ER or ER cellular components) | map03010 | Up | 6 | TraesCS1A02G092900, TraesCS1A02G105400, TraesCS1A02G137294, TraesCS1A02G137403, TraesCS1A02G137409, TraesCS1A02G137505 |
| Protein folding (During ER protein processing) | map03060 | Up | 8 | TraesCS1A02G112400, TraesCS1A02G138121, TraesCS1A02G133100, TraesCS1A02G133700, TraesCS1A02G124400, TraesCS1A02G145979, TraesCS1A02G099300, TraesCS1A02G137277, TraesCS1A02G137424, TraesCS1A02G137449 |
| ATPase activity, GTPase activity (ER to Golgi trafficking) | map03060 | Up | 18 | TraesCS1A02G126900, TraesCS1A02G131500, TraesCS1A02G144411, TraesCS1A02G144222, TraesCS1A02G143755, TraesCS1A02G143642, TraesCS1A02G143622, TraesCS1A02G143335, TraesCS1A02G143297, TraesCS1A02G142230, TraesCS1A02G137242, TraesCS1A02G137259, TraesCS1A02G137367, TraesCS1A02G137536, TraesCS1A02G137607, TraesCS1A02G137612 TraesCS1A02G137736, TraesCS1A02G137782 |
| vascular transport (ER to Golgi vesicle-mediated transport) | map03060 | Up | 8 | TraesCS1A02G144928, TraesCS1A02G143916, TraesCS1A02G106000, TraesCS1A02G133100, TraesCS1A02G137356, TraesCS1A02G137364, TraesCS1A02G137405, TraesCS1A02G151679 |
| Protein quality control | Up | 4 | TraesCS1A02G137200, TraesCS1A02G150621, TraesCS1A02G151217, TraesCS1A02G108100 |
| Variety | Parentage | Year (Releasing Committee) | Developed by | Area of Adoption | Source |
|---|---|---|---|---|---|
| Shaannong 33 | Xinmai 18sp-28-14/Shannong 981sp-12-16 | 2011-Shaanxi Provincial crop variety appraisal committee | Wang Chengshe, Liu Luxiang, Zou Shufang, Xu xitang Chen, Guangdou, Xie Yangzhou | Shaanxi, and Henan Province China | NWAFU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buttar, Z.A.; Shalmani, A.; Niaz, M.; Wang, C.; Hussain, S.; Wang, C. Transcriptome Analysis Reveals Potential Mechanism in Storage Protein Trafficking within Developing Grains of Common Wheat. Int. J. Mol. Sci. 2022, 23, 14851. https://doi.org/10.3390/ijms232314851
Buttar ZA, Shalmani A, Niaz M, Wang C, Hussain S, Wang C. Transcriptome Analysis Reveals Potential Mechanism in Storage Protein Trafficking within Developing Grains of Common Wheat. International Journal of Molecular Sciences. 2022; 23(23):14851. https://doi.org/10.3390/ijms232314851
Chicago/Turabian StyleButtar, Zeeshan Ali, Abdullah Shalmani, Mohsin Niaz, Chaojie Wang, Shahid Hussain, and Chengshe Wang. 2022. "Transcriptome Analysis Reveals Potential Mechanism in Storage Protein Trafficking within Developing Grains of Common Wheat" International Journal of Molecular Sciences 23, no. 23: 14851. https://doi.org/10.3390/ijms232314851
APA StyleButtar, Z. A., Shalmani, A., Niaz, M., Wang, C., Hussain, S., & Wang, C. (2022). Transcriptome Analysis Reveals Potential Mechanism in Storage Protein Trafficking within Developing Grains of Common Wheat. International Journal of Molecular Sciences, 23(23), 14851. https://doi.org/10.3390/ijms232314851