
Understanding In Vitro Pathways to Drug Discovery for TDP-43 Proteinopathies

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
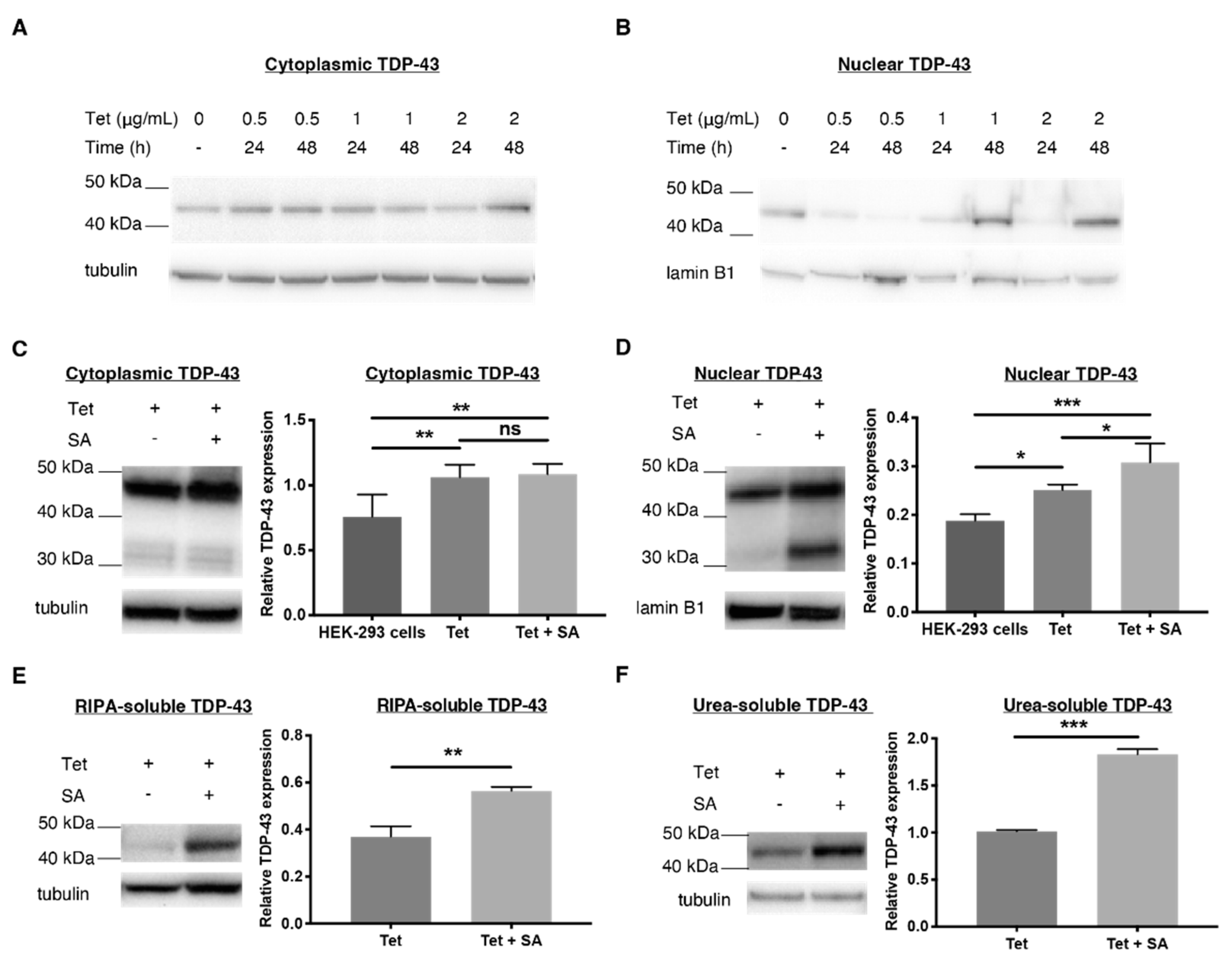
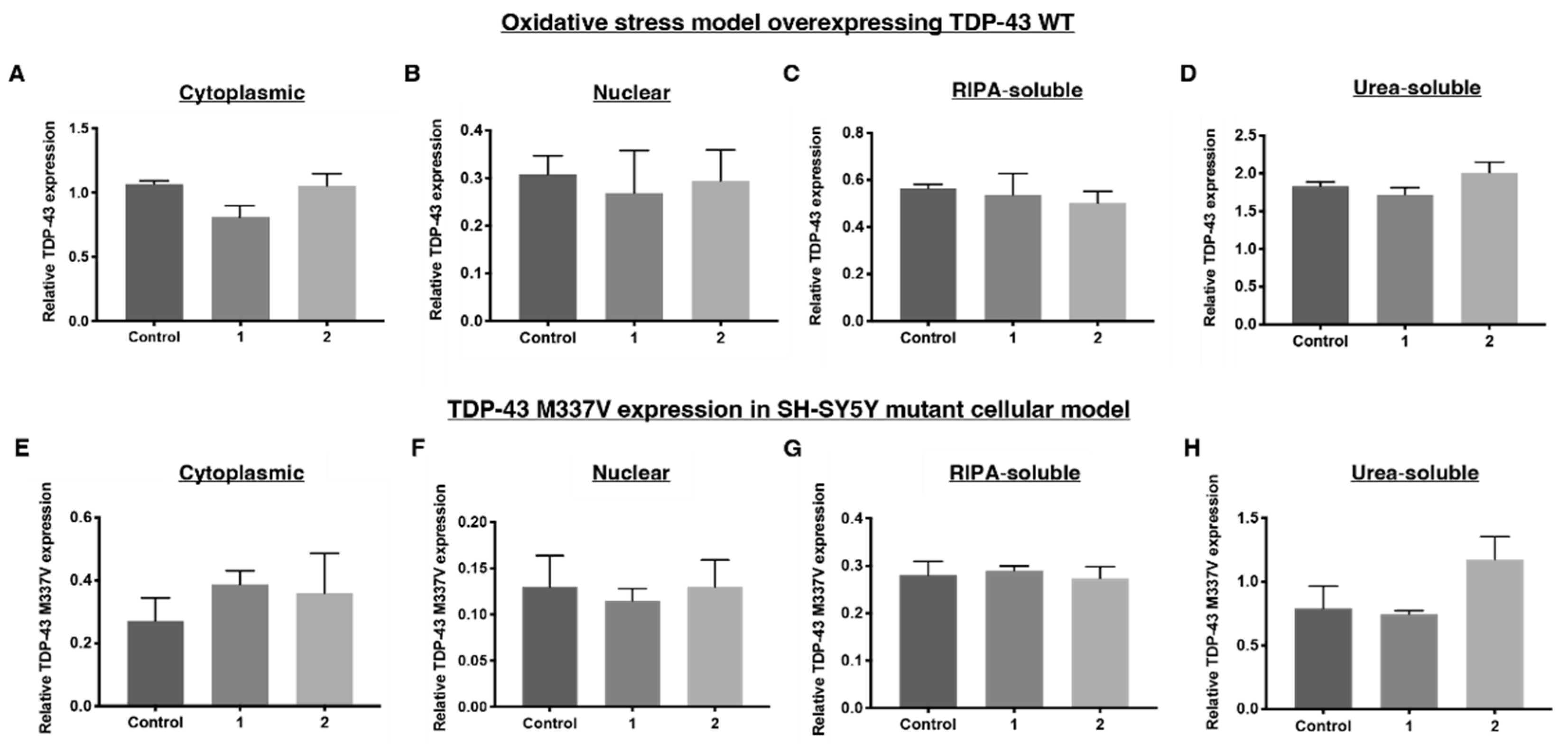
2.1. Establishing a TDP-43 WT-Overexpressing HEK-293 Cell Line—An Oxidative Stress Model
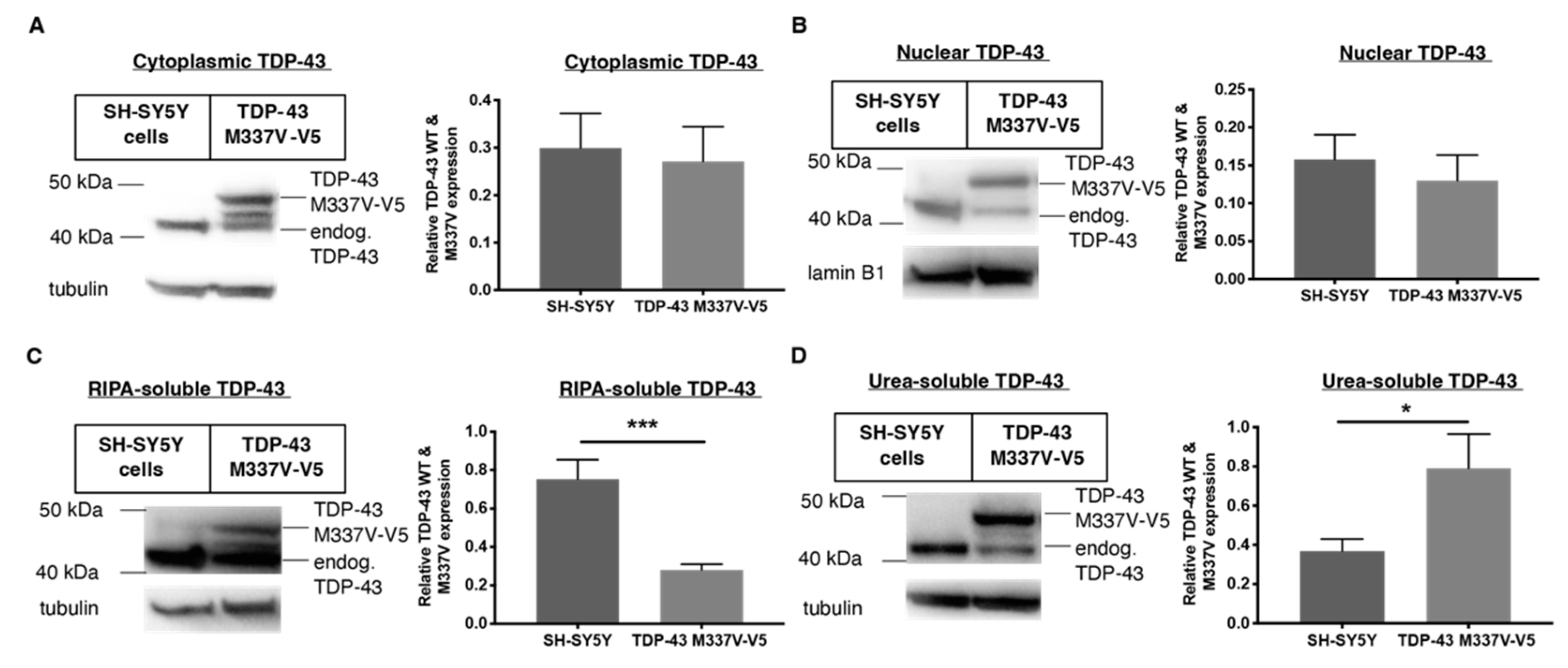
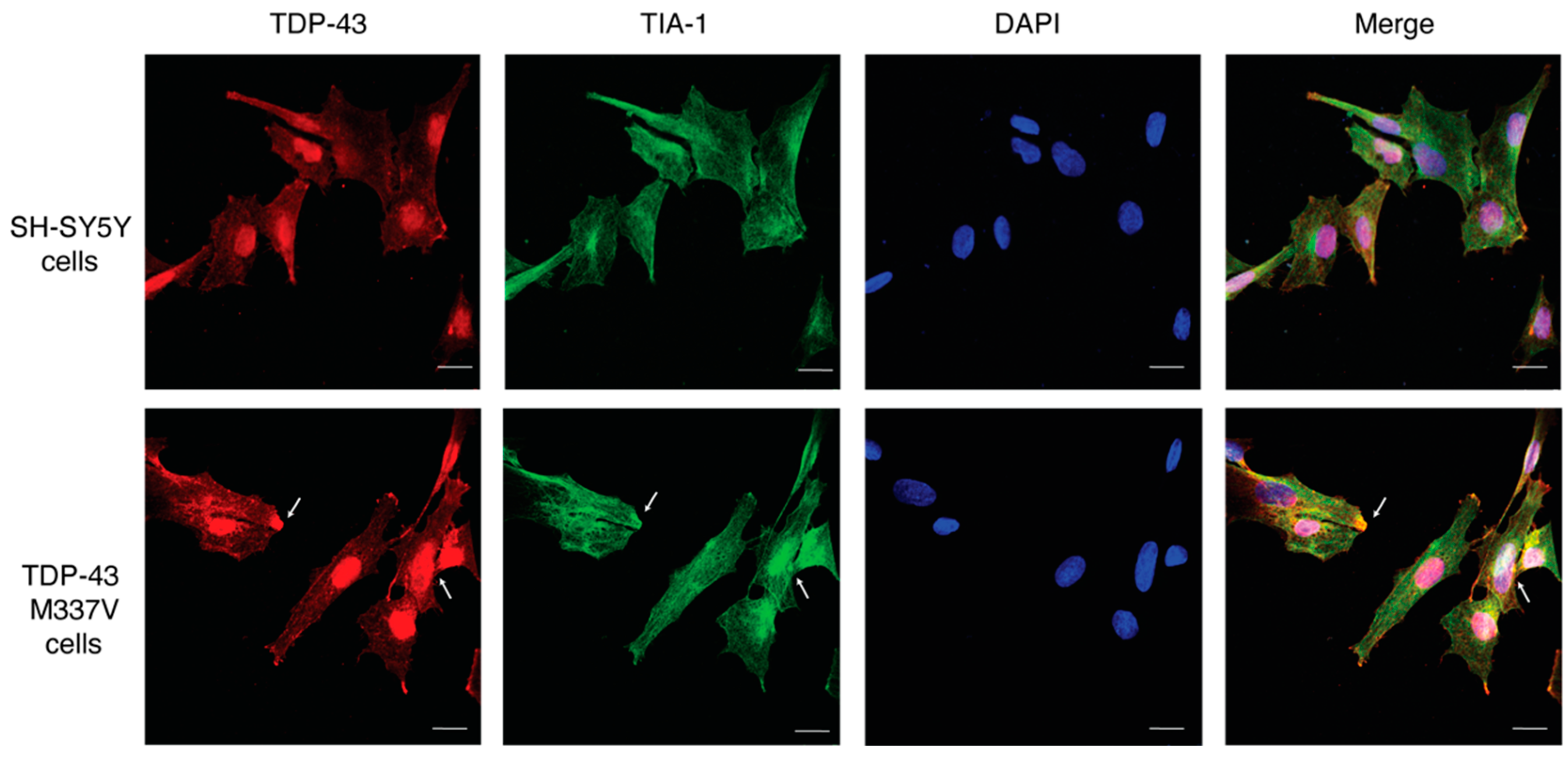
2.2. Establishing a SH-SY5Y TDP-43 M337V Mutant Cellular Model
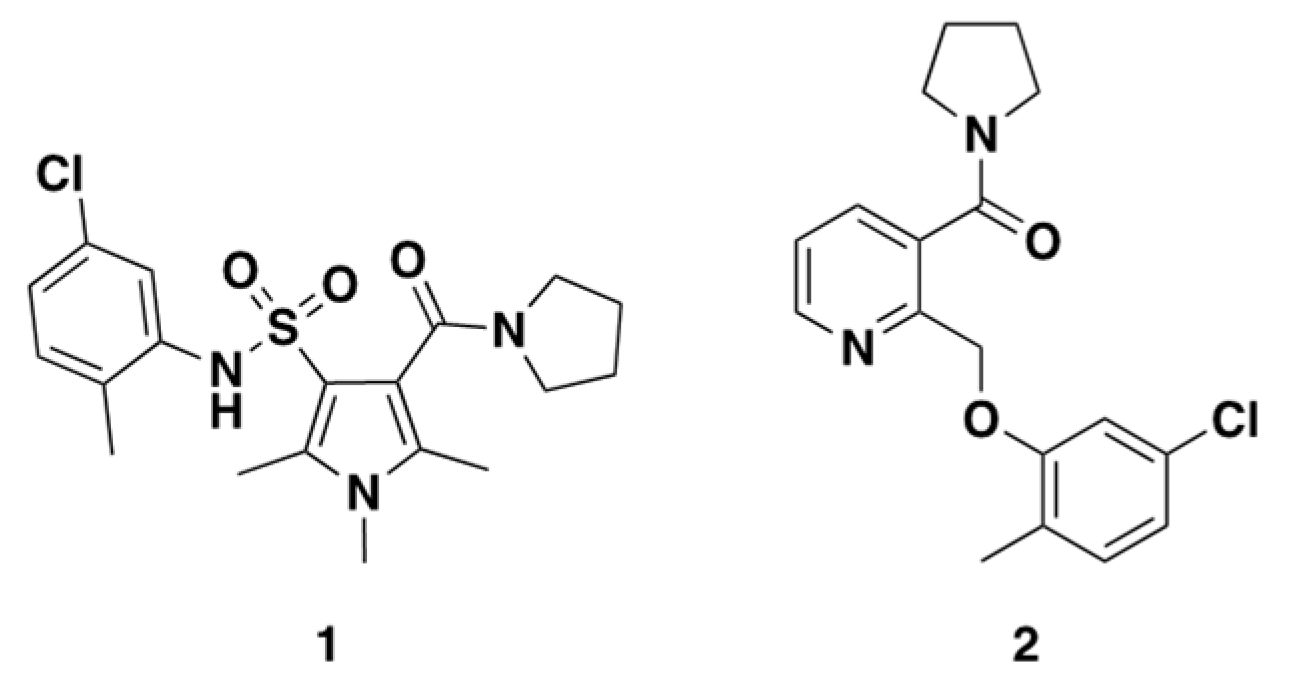
2.3. Rationale of Compound 1 and 2
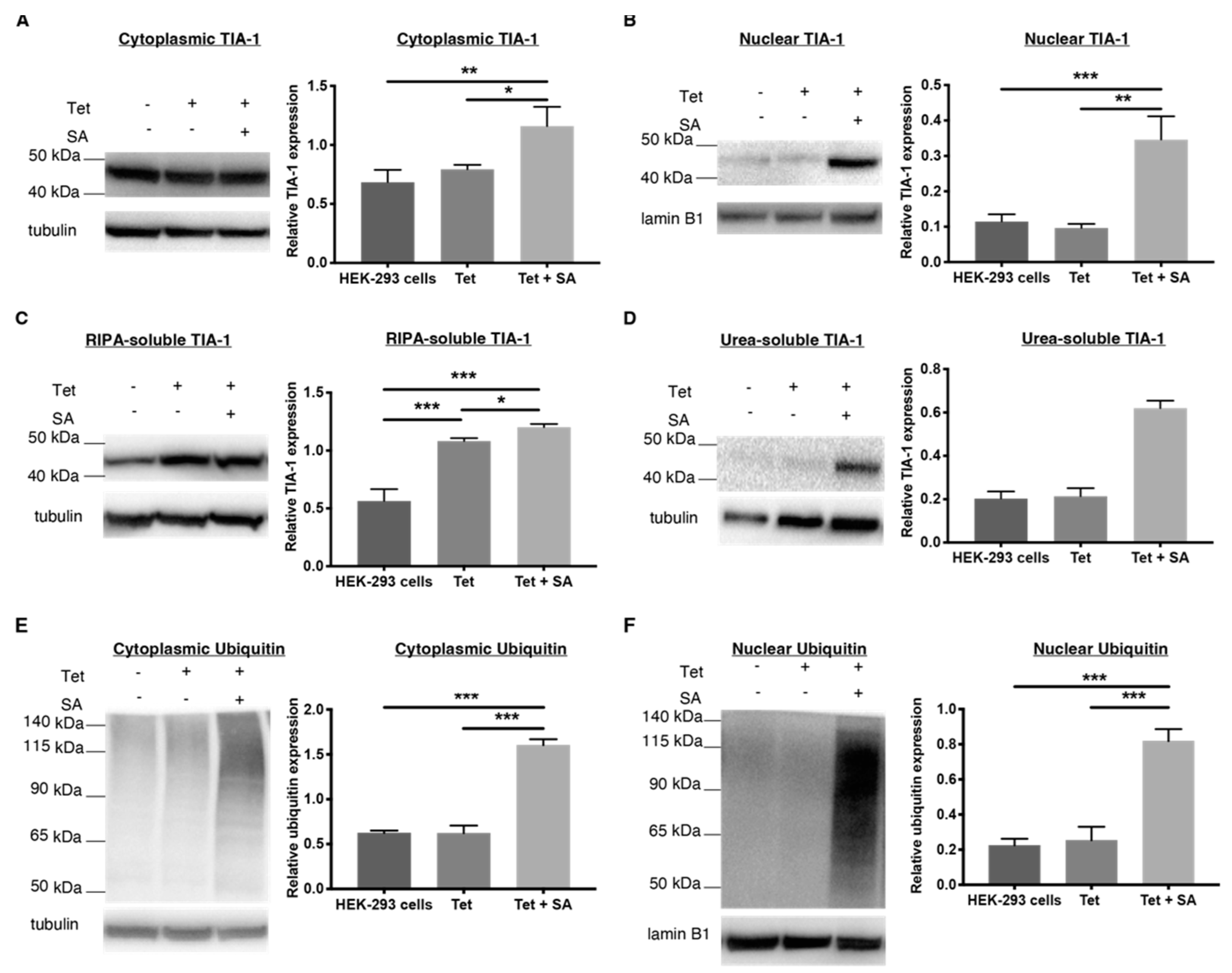
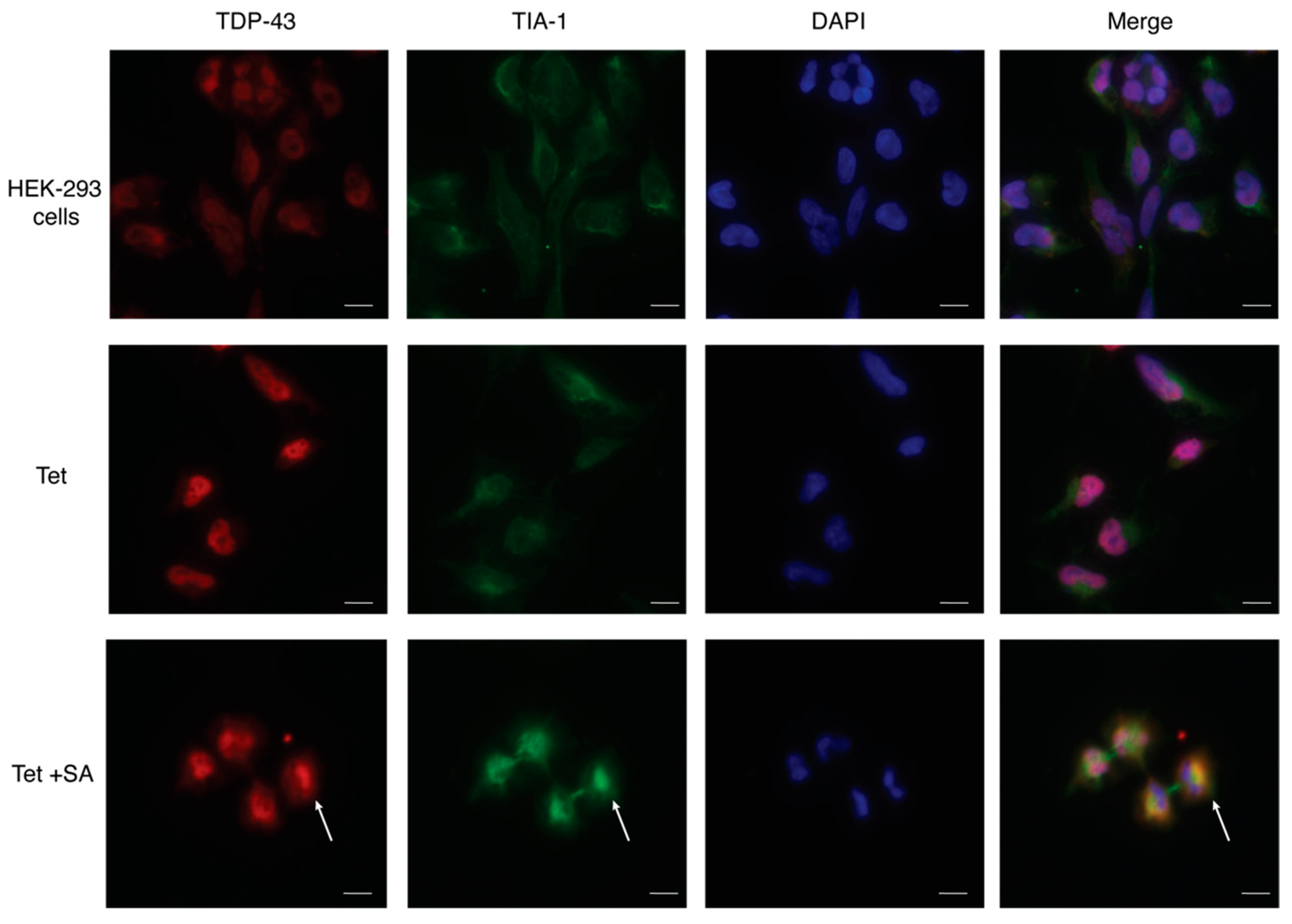
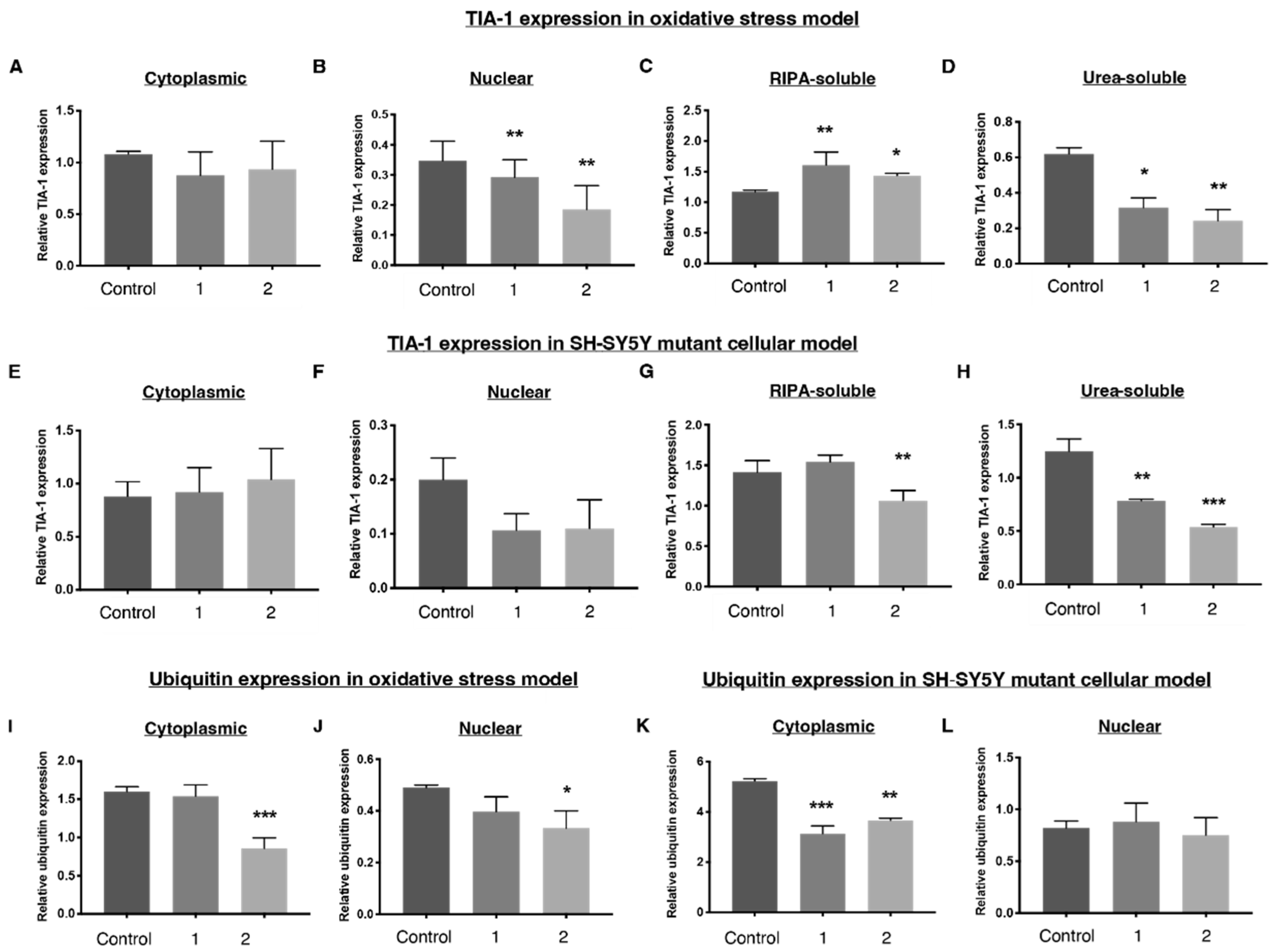
2.4. 1 and 2 Alter Stress Granule Solubility in Both Cellular Models
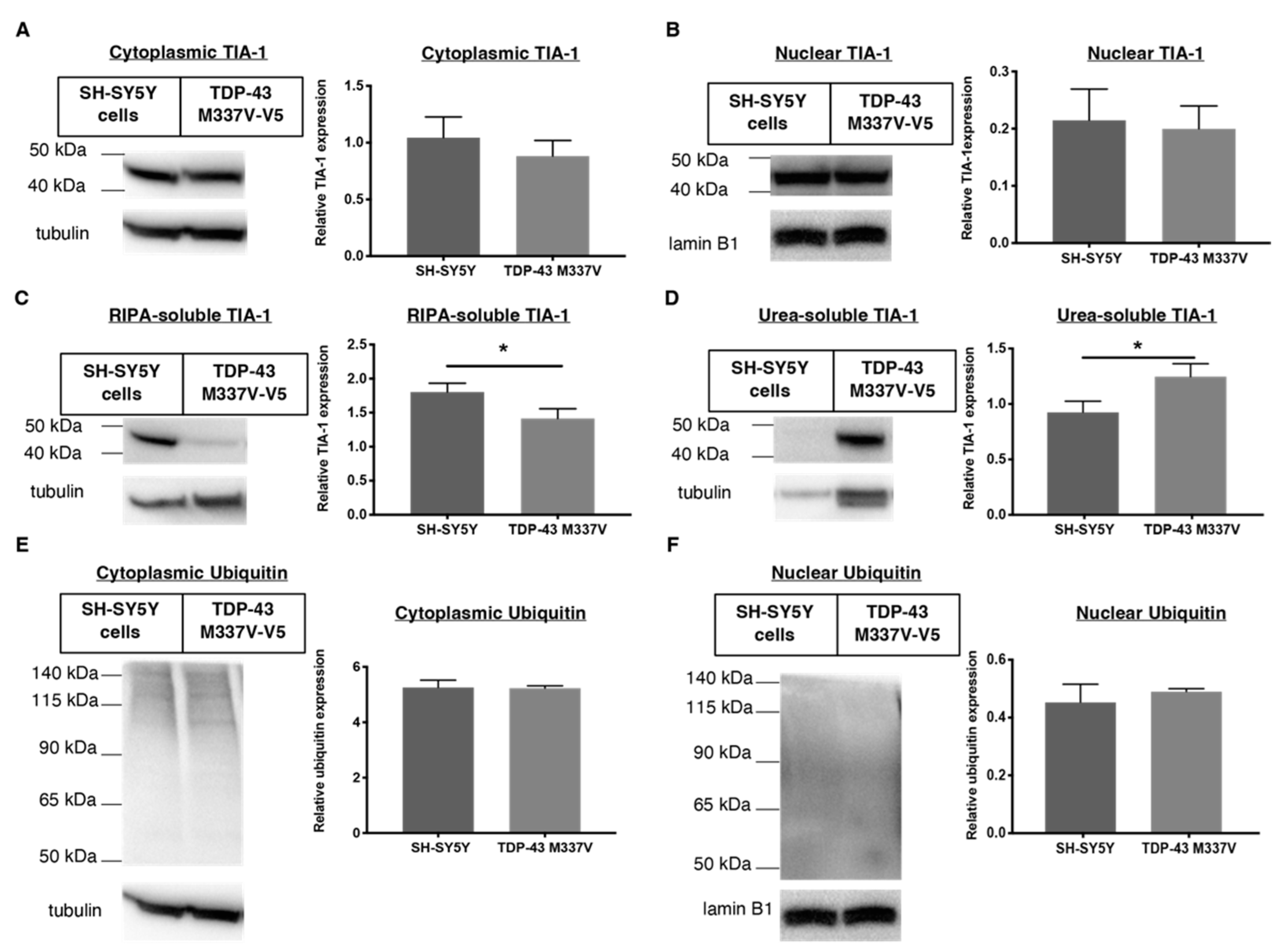
2.5. Downregulation of Ubiquitin in Both Cellular Models
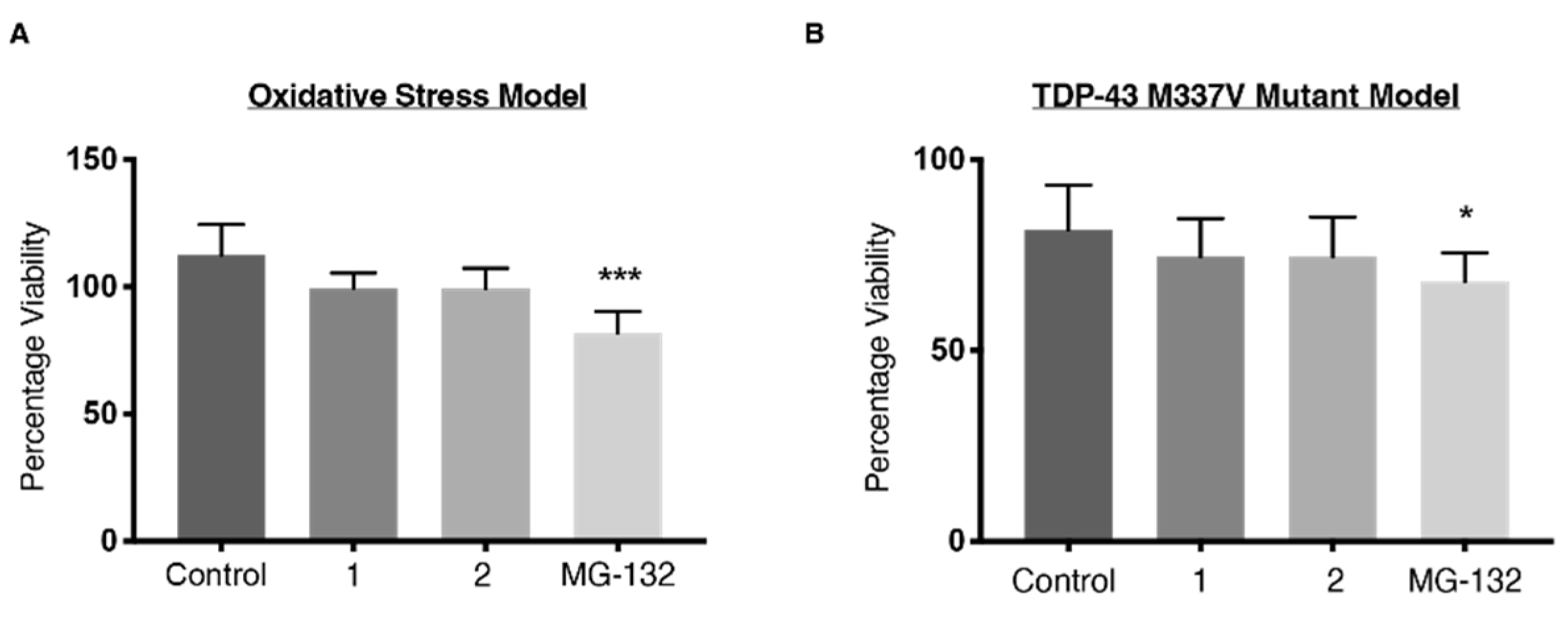
2.6. Viability
3. Materials and Methods
3.1. General Procedures
3.2. Reagents
3.3. SH-SY5Y Cell Culture
3.4. Generation of Wild-Type TDP-43-Expressing HEK-293 Cells
3.5. Cellular Treatments
3.6. Nucleocytoplasmic Fractionation
3.7. Solubility Fractionation
3.8. Protein Quantification and Western Blot
3.9. Immunofluorescence
3.10. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly (ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 2018, 71, 703–717. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Visibly stressed: The role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 2002, 7, 213. [Google Scholar] [CrossRef]
- Ding, Q.; Chaplin, J.; Morris, M.J.; Hilliard, M.A.; Wolvetang, E.; Ng, D.C.H.; Noakes, P.G. TDP-43 Mutation Affects Stress Granule Dynamics in Differentiated NSC-34 Motoneuron-Like Cells. Front. Cell Neurosci. 2021, 9, 611601. [Google Scholar] [CrossRef]
- Bentmann, E.; Neumann, M.; Tahirovic, S.; Rodde, R.; Dormann, D.; Haass, C. Requirements for stress granule recruitment of fused in Sarcoma (FUS) and TAR DNA binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2012, 287, 23079–23094. [Google Scholar] [CrossRef]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Meyerowitz, J.; Parker, S.J.; Vella, L.J.; Ng, D.; Price, K.A.; Liddell, J.R.; Caragounis, A.; Li, Q.X.; Masters, C.L.; Nonaka, T.; et al. N-terminal kinase controls TDP-43 accumulation in stress granules induced by oxidative stress. Mol. Neurodegener. 2011, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y. Small-molecule modulation of TDP-43 recruitment to stress granules prevents persistent TDP-43 accumulation in ALS/FTD. Neuron 2019, 103, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Izumi, Y.; Watanabe, A.; Tsukita, K.; Woltjen, K.; Yamamoto, T.; Hotta, A.; Kondo, T.; Kitaoka, S.; Ohta, A.; et al. The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, eaaf3962. [Google Scholar] [CrossRef]
- Lanznaster, D.; Bourgeais, J.; Bruno, C.; Hergesheimer, R.C.; Thepault, R.A.; Vourc’h, P.; Corcia, P.; Andres, C.R.; Herault, O.; Blasco, H. TDP-43-Mediated Toxicity in HEK293T Cells: A Fast and Reproducible Protocol to Be Employed in the Search of New Therapeutic Options against Amyotrophic Lateral Sclerosis. Cells 2019, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Human Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 aggregation in neurodegeneration: Are stress granules the key? Brain Res. 2012, 1462, 16–25. [Google Scholar] [CrossRef]
- Emara, M.M.; Fujimura, K.; Sciaranghella, D.; Ivanova, V.; Ivanov, P.; Anderson, P. Hydrogen peroxide induces stress granule formation independent of eIF2α phosphorylation. Biochem. Biophys. Res. Commun. 2012, 423, 763–769. [Google Scholar] [CrossRef]
- Palangi, F.; Samuel, S.M.; Thompson, I.R.; Triggle, C.R.; Emara, M.M. Effects of oxidative and thermal stresses on stress granule formation in human induced pluripotent stem cells. PLoS ONE 2017, 12, e0182059. [Google Scholar] [CrossRef]
- Sauer, B.; Henderson, N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc. Natl. Acad. Sci. USA 1988, 85, 5166–5170. [Google Scholar] [CrossRef]
- Liu, S.X.; Athar, M.; Lippai, I.; Waldren, C.; Hei, T.K. Induction of oxyradicals by arsenic: Implication for mechanism of genotoxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 1643–1648. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250. [Google Scholar] [CrossRef] [PubMed]
- Mihevc, S.P.; Baralle, M.; Buratti, E.; Rogelj, B. TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 2016, 6, 33996. [Google Scholar] [CrossRef] [PubMed]
- Davidson, Y.; Kelley, T.; Mackenzie, I.R.; Pickering-Brown, S.; Du Plessis, D.; Neary, D.; Snowden, J.S.; Mann, D.M. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta. Neuropathol. 2007, 113, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Neumann, M.; Bigio, E.H.; Holm, I.E.; Troost, D.; Hatanpaa, K.J.; Foong, C.; White III, C.L.; Schneider, J.A.; Kretzschmar, H.A. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am. J. Pathol. 2007, 171, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-Y.; Jeon, G.S.; Sung, J.-J. ALS-linked mutant SOD1 associates with TIA-1 and alters stress granule dynamics. Neurochem. Res. 2020, 45, 2884–2893. [Google Scholar] [CrossRef]
- Fujita, K.; Ito, H.; Nakano, S.; Kinoshita, Y.; Wate, R.; Kusaka, H. Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta. Neuropathol. 2008, 116, 439–445. [Google Scholar] [CrossRef]
- Bigio, E.H.; Johnson, N.A.; Rademaker, A.W.; Fung, B.B.; Mesulam, M.-M.; Siddique, N.; Dellefave, L.; Caliendo, J.; Freeman, S.; Siddique, T. Neuronal ubiquitinated intranuclear inclusions in familial and non-familial frontotemporal dementia of the motor neuron disease type associated with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2004, 63, 801–811. [Google Scholar] [CrossRef]
- López de Silanes, I.; Galbán, S.; Martindale, J.L.; Yang, X.; Mazan-Mamczarz, K.; Indig, F.E.; Falco, G.; Zhan, M.; Gorospe, M. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol. Cell. Biol. 2005, 25, 9520–9531. [Google Scholar] [CrossRef]
- Ling, S.-C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Velde, C.V. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2009, 19, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.D.; Lee-Armandt, J.P.; Feiler, M.S.; Zaarur, N.; Liu, M.; Kraemer, B.; Concannon, J.B.; Ebata, A.; Wolozin, B.; Glicksman, M.A. A high-content screen identifies novel compounds that inhibit stress-induced TDP-43 cellular aggregation and associated cytotoxicity. J. Biomol. Screen 2014, 19, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Polymenidou, M. Mammalian Prion Biology: One Century of Evolving Concepts. Cell 2004, 116, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.-E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 2007, 18, 2603–2618. [Google Scholar] [CrossRef] [PubMed]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.-M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 2021, 372, eabf6548. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. eLife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Nicholson, A.M.; Sarkar, M.; Messing, J.; Purice, M.D.; Pottier, C.; Annu, K.; Baker, M.; Perkerson, R.B.; Kurti, A. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 2017, 95, 808–816. [Google Scholar] [CrossRef] [PubMed]
- van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE 2011, 6, e22850. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Kawakami, E.; Endo, K.; Misawa, H.; Watabe, K. Formation and spreading of TDP-43 aggregates in cultured neuronal and glial cells demonstrated by time-lapse imaging. PLoS ONE 2017, 12, e0179375. [Google Scholar] [CrossRef] [PubMed]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Smith, A.S.; Chun, C.; Hesson, J.; Mathieu, J.; Valdmanis, P.N.; Mack, D.L.; Choi, B.-O.; Kim, D.-H.; Bothwell, M. Human Induced Pluripotent Stem Cell-Derived TDP-43 Mutant Neurons Exhibit Consistent Functional Phenotypes Across Multiple Gene Edited Lines Despite Transcriptomic and Splicing Discrepancies. Front. Cell Neurosci. 2021, 9, 728707. [Google Scholar] [CrossRef]
- Lee, S.; Huang, E.J. Modeling ALS and FTD with iPSC-derived neurons. Brain Res. 2017, 1656, 88–97. [Google Scholar] [CrossRef]
- Estévez, V.; Sridharan, V.; Sabaté, S.; Villacampa, M.; Menéndez, J.C. Three-Component Synthesis of Pyrrole-Related Nitrogen Heterocycles by a Hantzsch-Type Process: Comparison between Conventional and High-Speed Vibration Milling Conditions. Asian J. Org. Chem. 2016, 5, 652–662. [Google Scholar] [CrossRef]
- Larsen, G.R.; Weigele, M.; Vacca, J.P. Preparation of Pyrrolidinecarbonyl-Pyrrole- Sulfonamides Useful in Modulation of the Inclusion Formation Treatment of Diseases. WO2016090313A1, 9 June 2016. [Google Scholar]
- He, G.; Chen, G. A Practical Strategy for the Structural Diversification of Aliphatic Scaffolds through the Palladium-Catalyzed Picolinamide-Directed Remote Functionalization of Unactivated C(sp3) H Bonds. Angew. Chem. Int. Ed. 2011, 50, 5192–5196. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, J.; Thaker, K. Ring–chain tautomerism in the acid esters of pyridine-2: 3-dicarboxylic acid. J. Chem. Soc. 1957, 2531–2536. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, H.W.A.; Callis, T.B.; Montgomery, A.P.; Danon, J.J.; Jorgensen, W.T.; Ke, Y.D.; Ittner, L.M.; Werry, E.L.; Kassiou, M. Understanding In Vitro Pathways to Drug Discovery for TDP-43 Proteinopathies. Int. J. Mol. Sci. 2022, 23, 14769. https://doi.org/10.3390/ijms232314769
Cheng HWA, Callis TB, Montgomery AP, Danon JJ, Jorgensen WT, Ke YD, Ittner LM, Werry EL, Kassiou M. Understanding In Vitro Pathways to Drug Discovery for TDP-43 Proteinopathies. International Journal of Molecular Sciences. 2022; 23(23):14769. https://doi.org/10.3390/ijms232314769
Chicago/Turabian StyleCheng, Hei W. A., Timothy B. Callis, Andrew P. Montgomery, Jonathan J. Danon, William T. Jorgensen, Yazi D. Ke, Lars M. Ittner, Eryn L. Werry, and Michael Kassiou. 2022. "Understanding In Vitro Pathways to Drug Discovery for TDP-43 Proteinopathies" International Journal of Molecular Sciences 23, no. 23: 14769. https://doi.org/10.3390/ijms232314769
APA StyleCheng, H. W. A., Callis, T. B., Montgomery, A. P., Danon, J. J., Jorgensen, W. T., Ke, Y. D., Ittner, L. M., Werry, E. L., & Kassiou, M. (2022). Understanding In Vitro Pathways to Drug Discovery for TDP-43 Proteinopathies. International Journal of Molecular Sciences, 23(23), 14769. https://doi.org/10.3390/ijms232314769