
An Overview of the Molecular Cues and Their Intracellular Signaling Shared by Cancer and the Nervous System: From Neurotransmitters to Synaptic Proteins, Anatomy of an All-Inclusive Cooperation


Abstract
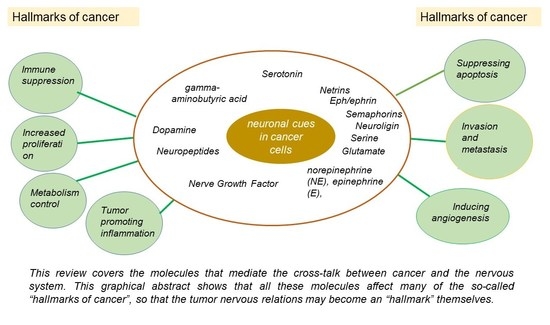
1. Introduction
2. Perineural Invasion and the Perineural Niche
3. The Sympathetic Nervous System and Adrenergic Signaling
4. The Parasympathetic Nervous System and Acetylcholine
5. The Tumor/Nervous Interface in the Central Nervous System Tumors and Electrochemical Communication
6. Molecules with a Widespread Activity in Various Types of Cancer: Neurotransmitters, Amino Acids, Growth Factors, Axon Guidance, and Synaptic Proteins
6.1. Dopamine
6.2. Gamma-Aminobutyric Acid
6.3. Serotonin
6.4. Glutamate
6.5. Serine
6.6. Neuropeptides
6.7. Nerve Growth Factor
6.8. Axon Guidance Molecules and Neuroligin
6.8.1. Axon Guidance Molecules
6.8.2. Neuroligins
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lacina, L.; Coma, M.; Dvovránková, B.; Kodet, O.; Melegova, N.; Gal, P.; Smetana, K. Evolution of Cancer progression in the context of Darwinism. Anticancer Res. 2019, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gal, P.; Varinska, L.; Faber, L.; Novákv, S.; Szabo, P.; Mitrengova, P.; Mirossay, A.; Muvcaji, P.; Smetana, K. How signaling molecules regulate tumor microenvironment: Parallels to wound repair. Molecules 2017, 22, 1818. [Google Scholar] [CrossRef] [PubMed]
- Fife, A.; Beasley, P.J.; Fertig, D.L. Psychoneuroimmunology and cancer: Historical perspectives and current research. Adv. Neuroimmunol. 1996, 6, 179–190. [Google Scholar] [CrossRef]
- Kiecolt-Glaser, J.K.; Robles, T.; Heffner, K.; Loving, T.; Glaser, R. Psycho-oncology and cancer: Psychoneuroimmunology and cancer. Ann. Oncol. 2002, 13, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Zhu, H.; Wang, Y.; Li, P.; Wang, D. The dark side of synaptic proteins in tumours. Br. J. Cancer 2022, 3, 1184–1192. [Google Scholar] [CrossRef]
- Shi, D.D.; Guo, J.A.; Hoffman, H.I.; Su, J.; Mino-Kenudson, M.; Barth, J.L.; Schenkel, J.M.; Loeffler, J.S.; Shih, H.A.; Hong, T.S.; et al. Therapeutic avenues for cancer neuroscience: Translational frontiers and clinical opportunities. Lancet Oncol. 2022, 23, e62–e74. [Google Scholar] [CrossRef]
- Hernandez, S.; Serrano, A.G.; Solis Soto, L.M. The Role of Nerve Fibers in the Tumor Immune Microenvironment of Solid Tumors. Adv. Biol. 2022, 6, 2200046. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Winkler, F. Insights and opportunities at the crossroads of cancer and neuroscience. Nat. Cell Biol 2022, 24, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Amit, M. Cancer Neuroscience: Evidence for a New Hallmark of Cancer. Adv. Biol. 2022, 6, 2200192. [Google Scholar] [CrossRef] [PubMed]
- Silverman, D.A.; Martinez, V.K.; Dougherty, P.M.; Myers, J.N.; Calin, G.A.; Amit, M. Cancer-Associated Neurogenesis and Nerve-Cancer Cross-talk. Cancer Res. 2021, 81, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Kayahara, M.; Nakagawara, H.; Kitagawa, H.; Ohta, T. The nature of neural invasion by pancreatic cancer. Pancreas 2007, 35, 218. [Google Scholar] [CrossRef]
- Liebig, C.; Ayala, G.; Wilks, J.A.; Berger, D.H.; Albo, D. Perineural invasion in cancer. Cancer 2009, 115, 3379–3391. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Amit, M.; Na’ara, S.; Gil, Z. Mechanisms of cancer dissemination along nerves. Nat. Rev. 2016, 16, 399. [Google Scholar] [CrossRef]
- Ayala, G.E.; Wheeler, T.M.; Shine, H.D.; Schmelz, M.; Frolov, A.; Chakraborty, S.; Rowley, D. In vitro dorsal root ganglia and human prostate cell line interaction: Redefining perineural invasion in prostate cancer. Prostate 2001, 49, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Förster, S.; Muders, M. The Role of Perineural Invasion in Prostate Cancer and Its Prognostic Significance. Cancers 2022, 14, 4065. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Romeo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Edwards, G.; Campbell, T.; Henderson, V.; Danaher, A.; Wu, D.; Srinivasan, R.; Rezvani, K.; Odero-Marah, V.A. SNAIL Transctiption factor in prostate cancer cells promotes neurite outgrowth. Biochimie 2021, 180, 1–9. [Google Scholar] [CrossRef]
- Eksi, S.E.; Chitsazan, A.; Sayar, Z.; Thomas, G.V.; Fields, A.J.; Kopp, R.P.; Spellman, P.T.; Adey, A.C. Epigenetic loss of heterogeneity from low to high grade localized prostate tumours. Nat. Commun. 2021, 12, 7292. [Google Scholar] [CrossRef] [PubMed]
- Frunza, A.; Slavescu, D.; Lascar, I. Perineural invasion in head and neck cancers-a review. J. Med. Life 2014, 7, 121. [Google Scholar]
- Li, R.; Wheeler, T.; Dai, H.; Ayala, G. Neural cell adhesion molecule is upregulated in nerves with prostate cancer invasion. Hum. Pathol. 2003, 34, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; He, D.; Florentin, D.; Frolov, A.; Hilsenbeck, S.; Ittmann, M.; Kadmon, D.; Miles, B.; Rowley, D.; Ayala, G. Semaphorin 4F as a Critical Regulator of Neuroepithelial Interactions and a Biomarker of Aggressive Prostate CancerSemaphorin 4F and Nerves. Clin. Cancer Res. 2013, 19, 6101–6111. [Google Scholar] [CrossRef]
- Yin, L.; Li, J.; Wang, J.; Pu, T.; Wei, J.; Li, Q.; Wu, B.J. MAOA promotes prostate cancer cell perineural invasion through SEMA3C/PlexinA2/NRP1-cMET signaling. Oncogene 2021, 40, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Adekoya, T.O.; Richardson, R.M. Cytokines and chemokines as mediators of prostate cancer metastasis. Int. J. Mol. Sci. 2020, 21, 4449. [Google Scholar] [CrossRef]
- Na’ara, S.; Amit, M.; Gil, Z. L1CAM induces perineural invasion of pancreas cancer cells by upregulation of metalloproteinase expression. Oncogene 2019, 38, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, C.S.; Banerjee, R.; Inglehart, R.C.; Liu, M.; Russo, N.; Hariharan, A.; Van Tubergen, E.A.; Corson, S.L.; Asangani, I.A.; Mistretta, C.M.; et al. Galanin modulates the neural niche to favour perineural invasion in head and neck cancer. Nat. Commun 2015, 6, 6885. [Google Scholar] [CrossRef]
- Lin, C.; Ren, Z.; Yang, X.; Yang, R.; Chen, Y.; Liu, Z.; Dai, Z.; Zhang, Y.; He, Y.; Zhang, C.; et al. Nerve growth factor (NGF)-TrkA axis in head and neck squamous cell carcinoma triggers EMT and confers resistance to the EGFR inhibitor erlotinib. Cancer Lett. 2020, 472, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Ban, K.; Feng, S.; Shao, L.; Ittmann, M. RET signaling in prostate cancer. Clin. Cancer Res. 2017, 23, 4885–4896. [Google Scholar] [CrossRef] [PubMed]
- Jobling, P.; Pundavela, J.; Oliveira, S.M.; Roselli, S.; Walker, M.M.; Hondermarck, H. Nerve-Cancer Cell Cross-talk: A Novel Promoter of Tumor Progression. Cancer Res. 2015, 75, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Mo, R.-J.; Han, Z.-D.; Liang, Y.-K.; Ye, J.-H.; Wu, S.-L.; Lin, S.X.; Zhang, Y.-Q.; Song, S.-D.; Jiang, F.-N.; Zhong, W.-D.; et al. Expression of PD-L1 in tumor-associated nerves correlates with reduced CD8+ tumor-associated lymphocytes and poor prognosis in prostate cancer. Int. J. Cancer 2019, 144, 3099–3110. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Mizushima, N. Diverse cellular roles of autophagy. Annu. Rev. Cell Dev. Biol. 2019, 35, 453–475. [Google Scholar] [CrossRef]
- Cai, Q.; Ganesan, D. Regulation of neuronal autophagy and the implications in neurodegenerative diseases. Neurobiol. Dis. 2021, 4, 105582. [Google Scholar] [CrossRef] [PubMed]
- Yazdankhah, M.; Ghosh, S.; Shang, P.; Stepicheva, N.; Hose, S.; Liu, H.; Chamling, X.; Tian, S.; Sullivan, M.L.; Calderon, M.J.; et al. BNIP3L-mediated mitophagy is required for mitochondrial remodeling during the differentiation of optic nerve oligodendrocytes. Autophagy 2021, 17, 3140–3159. [Google Scholar] [CrossRef] [PubMed]
- Griffey, C.J.; Yamamoto, A. Macroautophagy in CNS health and disease. Nat. Rev. Neurosci. 2022, 23, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, X.; Wang, Y.; Jia, C.; Liu, X.; Wang, Y.; Wu, H.; Cai, H.; Shen, H.-M.; Le, W. Essential role for autophagy protein VMP1 in maintaining neuronal homeostasis and preventing axonal degeneration. Cell Death Dis. 2021, 12, 116. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Regulska, K.; Regulski, M.; Karolak, B.; Murias, M.; Stanisz, B. Can cardiovascular drugs support cancer treatment? The rationale for drug repurposing. Drug Discov. Today 2019, 24, 1059–1065. [Google Scholar] [CrossRef]
- Demir, I.E.; Reyes, C.M.; Alrawashdeh, W.; Ceyhan, G.O.; Deborde, S.; Friess, H.; Görgülü, K.; Istvanffy, R.; Jungwirth, D.; Kuner, R.; et al. Future directions in preclinical and translational cancer neuroscience research. Nature 2020, 1, 1027–1031. [Google Scholar] [CrossRef]
- Shaw, V.; Srivastava, S.; Srivastava, S.K. Repurposing antipsychotics of the diphenylbutylpiperidine class for cancer therapy. Semin. Cancer Biol. 2021, 1, 75–83. [Google Scholar] [CrossRef]
- Shi, J.; Xu, J.; Li, Y.; Li, B.; Ming, H.; Nice, E.C.; Huang, C.; Li, Q.; Wang, C. Drug repurposing in cancer neuroscience: From the viewpoint of the autophagy-mediated innervated niche. Front. Pharmacol. 2022, 13, 990665. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Garc’ia-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma ablates pericytes antitumor immune function through aberrant up-regulation of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Zhang, B.-Y.; Zhou, B.; Zhu, C.-Z.; Sun, L.-Q.; Feng, Y.-J. Perineural invasion of cancer: A complex crosstalk between cells and molecules in the perineural niche. Am. J. Cancer Res. 2019, 9, 1–21. [Google Scholar] [PubMed]
- William Tank, A.; Lee Wong, D. Peripheral and central effects of circulating catecholamines. Compr. Physiol. 2011, 5, 1–15. [Google Scholar]
- Lillberg, K.; Verkasalo, P.K.; Kaprio, J.; Teppo, L.; Helenius, H.; Koskenvuo, M. Stressful life events and risk of breast cancer in 10,808 women: A cohort study. Am. J. Epidemiol. 2003, 157, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Chida, Y.; Hamer, M.; Wardle, J.; Steptoe, A. Do stress-related psychosocial factors contribute to cancer incidence and survival? Nat. Clin. Pract. Oncol. 2008, 5, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Partecke, L.I.; Speerforck, S.; Käding, A.; Seubert, F.; Kühn, S.; Lorenz, E.; Schwandke, S.; Sendler, M.; Keßler, W.; Trung, D.N.; et al. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology 2016, 16, 423–433. [Google Scholar] [CrossRef]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R.; Danial, N.; et al. Behavioral stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 2972. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Al-Wadei, H.A.; Ullah, M.F.; Plummer, H.K. Regulation of pancreatic cancer by neuropsychological stress responses: A novel target for intervention. Carcinogenesis 2012, 33, 2871. [Google Scholar] [CrossRef] [PubMed]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 6142. [Google Scholar] [CrossRef]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Investig. 2010, 120, 24579. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018, 33, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, X.M.; Wang, Y.H.; Feng, M.X.; Liu, X.J.; Zhang, Y.L. Monoamine oxidase A suppresses hepatocellular carcinoma metastasis by inhibiting the adrenergic system and its transactivation of EGFR signaling. J. Hepatol. 2014, 60, 246. [Google Scholar] [CrossRef]
- Han, J.; Jiang, Q.; Ma, R.; Zhang, H.; Tong, D.; Tang, K.; Wang, X.; Ni, L.; Miao, J.; Duan, B.; et al. Norepinephrine-CREB1-miR-373 axis promotes progression of colon cancer. Mol. Oncol. 2020, 14, 1059–1073. [Google Scholar] [CrossRef]
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Lv, S.; Li, J.; Chen, K.; Jiang, Z.; Cheng, L.; Zhou, C.; Yan, B.; Cao, J.; Ma, Q.; et al. Norepinephrine enhances cell viability and invasion, and inhibits apoptosis of pancreatic cancer cells in a Notch-1-dependent manner. Oncol. Rep. 2018, 40, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Decker, A.M.; Jung, Y.; Cackowski, F.C.; Yumoto, K.; Wang, J.; Taichman, R.S. Sympathetic signaling reactivates quiescent disseminated prostate cancer cells in the bone marrow. Mol. Cancer Res. 2017, 15, 1644–1655. [Google Scholar] [CrossRef]
- Jang, H.-J.; Boo, H.-J.; Lee, H.J.; Min, H.-Y.; Lee, H.-Y. Chronic stress facilitates lung tumorigenesis by promoting exocytosis of IGF2 in lung epithelial cells. Cancer Res. 2016, 76, 6607–6619. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Hondermarck, H.; Jobling, P. The sympathetic nervous system drives tumor angiogenesis. Trends Cancer 2018, 4, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hansen, L.S.; Lillelund, C.; Andersen, C.; Gehl, J.; Christensen, J.F.; Pedersen, B.K.; Hojman, P. Exercise-induced catecholamines activate the hippo tumor suppressor pathway to reduce risks of breast cancer development. Cancer Res. 2017, 77, 4894–4904. [Google Scholar] [CrossRef]
- Song, Y.; Gan, Y.; Wang, Q.; Meng, Z.; Li, G.; Shen, Y.; Wu, Y.; Li, P.; Yao, M.; Gu, J.; et al. Enriching the housing environment for mice enhances their NK cell antitumor immunity via sympathetic nerve-dependent regulation of NKG2D and CCR5. Cancer Res. 2017, 77, 1611–1622. [Google Scholar] [CrossRef]
- Kim, T.-H.; Ly, C.; Christodoulides, A.; Nowell, C.J.; Gunning, P.W.; Sloan, E.K.; Rowat, C.A. Stress hormone signaling through β-adrenergic receptors regulates macrophage mechanotype and function. FASEB J. 2019, 33, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, S.; Lange, T.; Gouttefangeas, C.; Jensen, A.T.; Szczepanski, M.; Lehnnolz, J.; Soekadar, S.; Rammensee, H.-G.; Born, J.; Besedovsky, L. Gαs-coupled receptor signaling and sleep regulate integrin activation of human antigen-specific T cells. J. Exp. Med. 2019, 216, 517–526. [Google Scholar] [CrossRef]
- Sloan, E.K.; Priceman, S.J.; Cox, B.F.; Yu, S.; Pimentel, M.A.; Tangkanangnukul, V.; Arevalo, J.M.; Morizono, K.; Karanikolas, B.D.; Wu, L.; et al. The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Res. 2010, 70, 7042–7052. [Google Scholar] [CrossRef]
- Mohammadpour, H.; MacDonald, C.R.; Qiao, G.; Chen, M.; Dong, B.; Hylander, B.L.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A.; Priceman, S.J.; et al. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Investig. 2019, 129, 5537–5552. [Google Scholar] [CrossRef] [PubMed]
- Dubeykovskaya, Z.; Si, Y.; Chen, X.; Worthley, D.L.; Renz, B.W.; Urbanska, A.M.; Hayakawa, Y.; Xu, T.; Westphalen, C.B.; Dubeykovskiy, A.; et al. Neural innervation stimulates splenic TFF2 to arrest myeloid cell expansion and cancer. Nat. Commun. 2016, 7, 10517. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Seshacharyulu, P.; Lakshmanan, I.; Rachagani, S.; Chugh, S.; Karmakar, S. Cigarette smoke induces stem cell features of pancreatic cancer cells via PAF1. Gastroenterology 2018, 155, 2862. [Google Scholar] [CrossRef]
- Elisia, I.; Cho, B.; Hay, M.; Li, M.Y.; Hofs, E.; Lam, V.; Dyer, R.A.; Lum, J.; Krystal, G. The effect of diet and exercise on tobacco carcinogen-induced lung cancer. Carcinogenesis 2019, 40, 448–460. [Google Scholar] [CrossRef]
- Shimizu, R.; Ibaragi, S.; Eguchi, T.; Kuwajima, D.; Kodama, S.; Nishioka, T.; Okui, T.; Obata, K.; Takabatake, K.; Kawai, H.; et al. Nicotine promotes lymph node metastasis and cetuximab resistance in head and neck squamous cell carcinoma. Int. J. Oncol. 2019, 54, 283–294. [Google Scholar] [CrossRef]
- Schaal, C.M.; Bora-Singhal, N.; Kumar, D.M.; Chellappan, S.P. Regulation of Sox2 and stemness by nicotine and electronic-cigarettes in non-small cell lung cancer. Mol. Cancer 2018, 17, 149. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Cho, C.H. Neurotransmitters, more than meets the eye–neurotransmitters and their perspectives in cancer development and therapy. Eur. J. Pharm. 2011, 2, 667. [Google Scholar] [CrossRef] [PubMed]
- Patane, S. M3 muscarinic acetylcholine receptor in cardiology and oncology. Int. J. Cardiol. 2014, 4, 177. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xia, H.; Tang, Q.; Xu, H.; Wei, G.; Chen, Y.; Dai, X.; Gong, Q.; Bi, F. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci. Rep. 2017, 7, 40802. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, Y.; Sakitani, K.; Konishi, M.; Asfaha, S.; Niikura, R.; Tomita, H.; Renz, B.W.; Tailor, Y.; Macchini, M.; Middelhoff, M.; et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 2017, 31, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Partecke, L.I.; Käding, A.; Trung, D.N.; Diedrich, S.; Sendler, M.; Weiss, F.; Kühn, J.-P.; Mayerle, J.; Beyer, K.; von Bernstorff, W.; et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget 2017, 8, 22501. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, A.; Hayama, Y.; Kato, S.; Shimomura, A.; Shimomura, T.; Irie, K.; Kaneko, R.; Yanagawa, Y.; Kobayashi, K.; Ochiya, T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat. Neurosci. 2019, 22, 1289–1305. [Google Scholar] [CrossRef]
- Fujii, T.; Mashimo, M.; Moriwaki, Y.; Misawa, H.; Ono, S.; Horiguchi, K. Physiological functions of the cholinergic system in immune cells. J. Pharm. Sci. 2017, 1, 134. [Google Scholar] [CrossRef]
- Zila, I.; Mokra, D.; Kopincova, J.; Kolomaznik, M.; Javorka, M.; Calkovska, A. Vagal-immune interactions involved in cholinergic anti-inflammatory pathway. Physiol. Res. 2017, 1, 66. [Google Scholar] [CrossRef]
- Yang, M.-W.; Tao, L.-Y.; Jiang, Y.-S.; Yang, J.-Y.; Huo, Y.-M.; Liu, D.-J.; Li, J.; Fu, X.-L.; He, R.; Lin, C.; et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef] [PubMed]
- John Lin, C.-C.; Yu, K.; Hatcher, A.; Huang, T.-W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Chaichana, K.L.; Parker, S.L.; Olivi, A.; Quiñones-Hinojosa, A. Long-term seizure outcomes in adult patients undergoing primary resection of malignant brain astrocytomas. J. Neurosurg. 2009, 111, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Rub’i, B.; Maechler, P. Minireview: New roles for peripheral dopamine on metabolic control and tumor growth: Let’s seek the balance. Endocrinology 2010, 151, 5570–5581. [Google Scholar] [CrossRef]
- Roney, M.S.I.; Park, S.-K. Antipsychotic dopamine receptor antagonists, cancer, and cancer stem cells. Arch. Pharm. Res. 2018, 41, 384–408. [Google Scholar] [CrossRef]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [PubMed]
- Jandaghi, P.; Najafabadi, H.S.; Bauer, A.S.; Papadakis, A.I.; Fassan, M.; Hall, A.; Monast, A.; von Knebel Doeberitz, M.; Neoptolemos, J.P.; Costello, E.; et al. Expression of DRD2 is increased in human pancreatic ductal adenocarcinoma and inhibitors slow tumor growth in mice. Gastroenterology 2016, 151, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, C.; Chakroborty, D.; Chowdhury, U.R.; Dasgupta, P.S.; Basu, S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 2008, 14, 2502–2510. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, D.; Sarkar, C.; Mitra, R.B.; Banerjee, S.; Dasgupta, P.S.; Basu, S. Depleted dopamine in gastric cancer tissues: Dopamine treatment retards growth of gastric cancer by inhibiting angiogenesis. Clin. Cancer Res. 2004, 10, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Chakroborty, D.; Chowdhury, U.R.; Sarkar, C.; Baral, R.; Dasgupta, P.S.; Basu, S. Dopamine regulates endothelial progenitor cell mobilization from mouse bone marrow in tumor vascularization. J. Clin. Investig. 2008, 118, 1380–1389. [Google Scholar] [CrossRef]
- Pinoli, M.; Marino, F.; Cosentino, M. Dopaminergic regulation of innate immunity: A review. J. Neuroimmune Pharmacol. 2017, 12, 602–623. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, R.; Tang, N.; Gong, Z.; Zhou, J.; Chen, Y.; Chen, K.; Cai, W. Dopamine inhibits the function of Gr-1+ CD115+ myeloid-derived suppressor cells through D1-like receptors and enhances anti-tumor immunity. J. Leukoc. Biol. 2015, 97, 191–200. [Google Scholar] [CrossRef]
- Cosentino, M.; Fietta, A.M.; Ferrari, M.; Rasini, E.; Bombelli, R.; Carcano, E.; Saporiti, F.; Meloni, F.; Marino, F.; Lecchini, S. Human CD4+ CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 2007, 109, 632–642. [Google Scholar] [CrossRef]
- Maemura, K.; Shiraishi, N.; Sakagami, K.; Kawakami, K.; Inoue, T.; Murano, M. Proliferative effects of gamma-aminobutyric acid on the gastric cancer cell line are associated with extracellular signal-regulated kinase 1/2 activation. J. Gastroenterol. Hepatol. 2009, 2, 24. [Google Scholar]
- Hujber, Z.; Horváth, G.; PetHovári, G.; Krencz, I.; Dankó, T.; Mészáros, K.; Rajnai, H.; Szoboszlai, N.; Leenders, W.P.; Jeney, A.; et al. GABA, glutamine, glutamate oxidation and succinic semialdehyde dehydrogenase expression in human gliomas. J. Exp. Clin. Cancer Res. 2018, 37, 271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, X.; Yao, Z.; Wei, C.; Ning, N.; Li, J. GABAergic signaling facilitates breast cancer metastasis by promoting ERK1/2-dependent phosphorylation. Cancer Lett. 2014, 348, 100–108. [Google Scholar] [CrossRef]
- Kanbara, K.; Otsuki, Y.; Watanabe, M.; Yokoe, S.; Mori, Y.; Asahi, M.; Neo, M. GABA B receptor regulates proliferation in the high-grade chondrosarcoma cell line OUMS-27 via apoptotic pathways. BMC Cancer 2018, 18, 263. [Google Scholar] [CrossRef]
- Sung, H.Y.; Yang, S.-D.; Ju, W.; Ahn, J.-H. Aberrant epigenetic regulation of GABRP associates with aggressive phenotype of ovarian cancer. Exp. Mol. Med. 2017, 49, e335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Du, Z.; Liu, J.; He, J. Gamma-aminobutyric acid receptors affect the progression and migration of tumor cells. J. Recept. Signal Transduct. Res. 2014, 1, 34. [Google Scholar]
- Gumireddy, K.; Li, A.; Kossenkov, A.V.; Sakurai, M.; Yan, J.; Li, Y.; Xu, H.; Wang, J.; Zhang, P.J.; Zhang, L.; et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat. Commun. 2016, 7, 10715. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.H.; Zhu, L.L.; Zhang, M.; Li, R.K.; Yang, Q.; Yan, J.Y.; Zhang, C.; Yang, J.Y.; Dong, F.Y.; Dai, M.; et al. GABRP regulates chemokine signalling, macrophage recruitment and tumour progression in pancreatic cancer through tuning KCNN4-mediated Ca2+ signalling in a GABA-independent manner. Gut 2019, 68, 1551. [Google Scholar] [CrossRef] [PubMed]
- Abdul, M.; Mccray, S.D.; Hoosein, N.M. Expression of gamma-aminobutyric acid receptor (subtype A) in prostate cancer. Acta Oncol. 2008, 47, 1546–1550. [Google Scholar] [CrossRef]
- Takehara, A.; Hosokawa, M.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Nakamura, Y.; Nakagawa, H. γ-aminobutyric acid (GABA) stimulates pancreatic cancer growth through overexpressing GABAA receptor π subunit. Cancer Res. 2007, 67, 9704–9712. [Google Scholar] [CrossRef]
- Wang, T.; Huang, W.; Chen, F. Baclofen, a GABAB receptor agonist, inhibits human hepatocellular carcinoma cell growth in vitro and in vivo. Life Sci. 2008, 82, 536–541. [Google Scholar] [CrossRef]
- Schuller, H.M. Regulatory role of G protein-coupled receptors in pancreatic cancer development and progression. Curr. Med. Chem. 2018, 25, 2566–2575. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, M.; Iishi, H.; Baba, M.; Nakaizumi, A.; Ichii, M.; Taniguchi, H. Inhibition by γ-Amino-n-butyric Acid and Baclofen of Gastric Carcinogenesis Induced by N′-Methyl-N′-nitro-N′-nitrosoguanidine in Wistar Rats. Cancer Res. 1990, 50, 4931–4934. [Google Scholar] [PubMed]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. The neurotransmitter γ-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res. 2002, 62, 6467–6469. [Google Scholar]
- Thaker, P.H.; Yokoi, K.; Jennings, N.B.; Li, Y.; Rebhun, R.B.; Rousseau Jr, D.L.; Fan, D.; Sood, A.K. Inhibition of experimental colon cancer metastasis by the GABA-receptor agonist nembutal. Cancer Biol. Ther. 2005, 4, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Neurotransmission and cancer: Implications for prevention and therapy. Anti-Cancer Drugs 2008, 19, 655–671. [Google Scholar] [CrossRef]
- Azuma, H.; Inamoto, T.; Sakamoto, T.; Kiyama, S.; Ubai, T.; Shinohara, Y.; Maemura, K.; Tsuji, M.; Segawa, N.; Masuda, H.; et al. γ-Aminobutyric acid as a promoting factor of cancer metastasis; induction of matrix metalloproteinase production is potentially its underlying mechanism. Cancer Res. 2003, 63, 8090–8096. [Google Scholar]
- Xia, S.; He, C.; Zhu, Y.; Wang, S.; Li, H.; Zhang, Z.; Jiang, X.; Liu, J. GABABR-induced EGFR transactivation promotes migration of human prostate cancer cells. Mol. Pharmacol. 2017, 92, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ren, L.; Wan, Y.; Prud’homme, G.J. GABAergic regulation of pancreatic islet cells: Physiology and antidiabetic effects. J. Cell. Physiol. 2019, 234, 14432–14444. [Google Scholar] [CrossRef]
- Mohammad-Zadeh, L.F.; Moses, L.; Gwaltney-Brant, S.M. Serotonin: A review. J. Vet. Pharmacol. Ther. 2008, 31, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat. Med. 2010, 16, 804. [Google Scholar] [CrossRef] [PubMed]
- Chabbi-Achengli, Y.; Coudert, A.E.; Callebert, J.; Geoffroy, V.; Côté, F.; Collet, C.; de Vernejoul, M.-C. Decreased osteoclastogenesis in serotonin-deficient mice. Proc. Natl. Acad. Sci. USA 2012, 109, 2567–2572. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, E.J.; Shabbir, M.; Mikhailidis, D.P.; Thompson, C.S.; Mumtaz, F.H. The Role of Serotonin (5-Hydroxytryptamine1A and 1B) Receptors in Prostate Cancer Cell Proliferation. J. Urol. 2006, 176, 1648–1653. [Google Scholar] [CrossRef]
- Siddiqui, E.J.; Shabbir, M.A.; Mikhailidis, D.P.; Mumtaz, F.H.; Thompson, C.S. The effect of serotonin and serotonin antagonists on bladder cancer cell proliferation. BJU Int. 2006, 97, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, H.; Wang, Z.; Wu, P.; Gong, W. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur. J. Cancer 2019, 1, 114. [Google Scholar] [CrossRef] [PubMed]
- Del Bello, F.; Bonifazi, A.; Giorgioni, G.; Quaglia, W.; Amantini, C.; Morelli, M.B.; Santoni, G.; Battiti, F.O.; Vistoli, G.; Cilia, A.; et al. Chemical manipulations on the 1, 4-dioxane ring of 5-HT1A receptor agonists lead to antagonists endowed with antitumor activity in prostate cancer cells. Eur. J. Med. Chem. 2019, 168, 461–473. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Regmi, S.C.; Ahn, S.; Jeon, Y.H.; Jeong, H.; Kim, S.J.; Nam, T.; Jeong, B.-S.; Kim, J.-A. Tryptophan hydroxylase 1 and 5-HT 7 receptor preferentially expressed in triple-negative breast cancer promote cancer progression through autocrine serotonin signaling. Mol. Cancer 2016, 15, 75. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Mesnil, M. Serotonin and human cancer: A critical view. Biochimie 2019, 161, 46–50. [Google Scholar] [CrossRef]
- Dizeyi, N.; Bjartell, A.; Nilsson, E.; Hansson, J.; Gadaleanu, V.; Cross, N.; Abrahamsson, P.-A. Expression of Serotonin Receptors and Role of Serotonin in Human Prostate Cancer Tissue and Cell Lines. Prostate 2004, 59, 328–336. [Google Scholar] [CrossRef]
- Sarrouilhe, D.; Clarhaut, J.; Defamie, N.; Mesnil, M. Serotonin and cancer: What is the link? Curr. Mol. Med. 2015, 15, 62–77. [Google Scholar] [CrossRef]
- Jose, J.; Tavares, C.D.; Ebelt, N.D.; Lodi, A.; Edupuganti, R.; Xie, X.; Devkota, A.K.; Kaoud, T.S.; Van Den Berg, C.L.; Anslyn, E.V.; et al. Serotonin analogues as inhibitors of breast cancer cell growth. ACS Med. Chem. Lett. 2017, 8, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-H.; Li, J.; Dong, F.-Y.; Yang, J.-Y.; Liu, D.-J.; Yang, X.-M.; Wang, Y.-H.; Yang, M.-W.; Fu, X.-L.; Zhang, X.-X.; et al. Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterology 2017, 153, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Mammadova-Bach, E.; Mauler, M.; Braun, A.; Duerschmied, D. Autocrine and paracrine regulatory functions of platelet serotonin. Platelets 2018, 29, 541–548. [Google Scholar] [CrossRef]
- Asada, M.; Ebihara, S.; Yamanda, S.; Niu, K.; Okazaki, T.; Sora, I.; Arai, H. Depletion of serotonin and selective inhibition of 2B receptor suppressed tumor angiogenesis by inhibiting endothelial nitric oxide synthase and extracellular signal-regulated kinase 1/2 phosphorylation. Neoplasia 2009, 11, 408–410. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.P.; Custódio, J.B.; Santos, A.E. Ionotropic glutamate receptor antagonists and cancer therapy: Time to think out of the box? Cancer Chemother. Pharmacol. 2017, 79, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, J.Y.; Wall, B.A.; Wangari-Talbot, J.; Chen, S. Metabotropic glutamate receptors in cancer. Neuropharmacology 2017, 115, 193–202. [Google Scholar]
- Yoo, B.C.; Jeon, E.; Hong, S.-H.; Shin, Y.-K.; Chang, H.J.; Park, J.-G. Metabotropic glutamate receptor 4-mediated 5-fluorouracil resistance in a human colon cancer cell line. Clin. Cancer Res. 2004, 10, 4176–4184. [Google Scholar] [CrossRef]
- Groot, J.F.; Piao, Y.; Lu, L.; Fuller, G.N.; Yung, W.K.A. Knockdown of GluR1 expression by RNA interference inhibits glioma proliferation. J. Neuro-Oncol. 2008, 88, 121–133. [Google Scholar] [CrossRef]
- Liu, J.-W.; Myoung, S.K.; Nagpal, J.; Yamashita, K.; Poeta, L.; Chang, X.; Lee, J.; Park, H.L.; Jeronimo, C.; Westra, W.H.; et al. Quantitative hypermethylation of NMDAR2B in human gastric cancer. Int. J. Cancer 2007, 121, 1994–2000. [Google Scholar] [CrossRef]
- Park, S.-Y.; Lee, S.-A.; Han, I.-H.; Yoo, B.-C.; Lee, S.-H.; Park, J.-Y.; Cha, I.-H.; Kim, J.; Choi, S.-W. Clinical significance of metabotropic glutamate receptor 5 expression in oral squamous cell carcinoma. Oncol. Rep. 2007, 17, 81–87. [Google Scholar] [CrossRef][Green Version]
- Pollock, P.M.; Cohen-Solal, K.; Sood, R.; Namkoong, J.; Martino, J.J.; Koganti, A.; Zhu, H.; Robbins, C.; Makalowska, I.; Shin, S.-S.; et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat. Genet. 2003, 34, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Park, S.-Y.; Hong, S.-P.; Pai, H.; Choi, J.-Y.; Kim, S.-G. The expression of NMDA receptor 1 is associated with clinicopathological parameters and prognosis in the oral squamos cell carcinoma. J. Oral Pathol. Med. 2004, 33, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.J.; Yoo, B.C.; Lim, S.-B.; Jeong, S.-Y.; Kim, W.H.; Park, J.-G. Metabotropic glutamate receptor 4 expression in colorectal carcinoma and its prognostic significance. Clin. Cancer Res. 2005, 11, 3288–3295. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, L.; Arcella, A.; Battaglia, G.; Pazzaglia, S.; Aronica, E.; Spinsanti, P.; Caruso, A.; De Smaele, E.; Saran, A.; Gulino, A.; et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J. Neurosci. 2006, 26, 8388–8397. [Google Scholar] [CrossRef] [PubMed]
- Palamiuc, L.; Emerling, B.M. PSMA brings new flavors to PI3K signaling: A role for glutamate in prostate cancer. J. Exp. Med. 2018, 215, 17. [Google Scholar] [CrossRef] [PubMed]
- Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 2020, 183, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Kaczy’nska, K.; Zajkac, D.; Wojciechowski, P.; Kogut, E.; Szereda-Przestaszewska, M. Neuropeptides and breathing in health and disease. Pulm. Pharmacol. Ther. 2018, 48, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Covenas, R.; Munoz, M. Cancer progression and substance P. Histol. Histopathol. 2014, 1, 29. [Google Scholar]
- Tilan, J.; Kitlinska, J. Neuropeptide Y (NPY) in tumor growth and progression: Lessons learned from pediatric oncology. Neuropeptides 2016, 10, 55. [Google Scholar] [CrossRef]
- Munoz, M.; Rosso, M.; Covenas, R. The NK-1 receptor: A new target in cancer therapy. Curr. Drug Targets 2011, 1, 12. [Google Scholar] [CrossRef]
- Munoz, M.; Rosso, M.; Covenas, R. The NK-1 receptor antagonist L-732,138 induces apoptosis in human gastrointestinal cancer cell lines. Pharm. Rep. 2017, 6, 69. [Google Scholar]
- Zhang, L.; Wang, L.; Dong, D.; Wang, Z.; Ji, W.; Yu, M.; Zhang, F.; Niu, R.; Zhou, Y. MiR-34b/c-5p and the neurokinin-1 receptor regulate breast cancer cell proliferation and apoptosis. Cell Prolif. 2019, 52, e12527. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Glioma and neurokinin-1 receptor antagonists: A new therapeutic approach. Anti-Cancer Agents Med. Chem. 2019, 19, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tian, Y.; Wu, A. Neuropeptide Y receptors: A promising target for cancer imaging and therapy. Regen. Biomater. 2015, 2, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.; Naranjo, A.; Van Ryn, C.; Tilan, J.U.; Trinh, E.; Yang, C.; Tsuei, J.; Hong, S.-H.; Wang, H.; Izycka-Swieszewska, E.; et al. Neuropeptide Y as a biomarker and therapeutic target for neuroblastoma. Am. J. Pathol. 2016, 186, 3040–3053. [Google Scholar] [CrossRef]
- Czarnecka, M.; Trinh, E.; Lu, C.; Kuan-Celarier, A.; Galli, S.; Hong, S.-H.; Tilan, J.U.; Talisman, N.; Izycka-Swieszewska, E.; Tsuei, J.; et al. Neuropeptide Y receptor Y5 as an inducible pro-survival factor in neuroblastoma: Implications for tumor chemoresistance. Oncogene 2015, 34, 3131–3143. [Google Scholar] [CrossRef] [PubMed]
- Jeppsson, S.; Srinivasan, S.; Chandrasekharan, B. Neuropeptide Y (NPY) promotes inflammation-induced tumorigenesis by enhancing epithelial cell proliferation. Am. J. Physiol. -Gastrointest. Liver Physiol. 2017, 312, G103–G111. [Google Scholar] [CrossRef] [PubMed]
- Molloy, N.H.; Read, D.E.; Gorman, A.M. Nerve growth factor in cancer cell death and survival. Cancers 2011, 3, 510–530. [Google Scholar] [CrossRef] [PubMed]
- Pundavela, J.; Roselli, S.; Faulkner, S.; Attia, J.; Scott, R.J.; Thorne, R.F.; Forbes, J.F.; Bradshaw, R.A.; Walker, M.M.; Jobling, P.; et al. Nerve fibers infiltrate the tumor microenvironment and are associated with nerve growth factor production and lymph node invasion in breast cancer. Mol. Oncol. 2015, 9, 1626–1635. [Google Scholar] [CrossRef]
- Ylivinkka, I.; Keski-Oja, J.; Hyytiäinen, M. Netrin-1: A regulator of cancer cell motility? Eur. J. Cell Biol. 2016, 95, 513–520. [Google Scholar] [CrossRef]
- Bruikman, C.S.; Zhang, H.; Kemper, A.M.; van Gils, J.M. Netrin family: Role for protein isoforms in cancer. J. Nucleic Acids 2019, 2019, 3947123. [Google Scholar] [CrossRef] [PubMed]
- Rehman, M.; Tamagnone, L. Semaphorins in cancer: Biological mechanisms and therapeutic approaches. Semin. Cell Dev. Biol. 2013, 1, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Kumar, D.; Tomar, D.; Chakraborty, G.; Kumar, S.; Kundu, G.C. The potential of class 3 semaphorins as both targets and therapeutics in cancer. Expert Opin. Ther. Targets 2015, 19, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Killeen, M.T.; Sybingco, S.S. Netrin, Slit and Wnt receptors allow axons to choose the axis of migration. Dev. Biol. 2008, 323, 143–151. [Google Scholar] [CrossRef][Green Version]
- Jiang, S.; Richaud, M.; Vieugué, P.; Rama, N.; Delcros, J.-G.; Siouda, M.; Sanada, M.; Redavid, A.; Ducarouge, B.; Hervieu, M.; et al. Targeting netrin-3 in small cell lung cancer and neuroblastoma. EMBO Mol. Med. 2021, 13, e12878. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H. Netrin-1 and its receptors in tumorigenesis. Nat. Rev. 2004, 4, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Meyerhardt, J.A.; Caca, K.; Eckstrand, B.C.; Hu, G.; Lengauer, C.; Banavali, S.; Look, A.T.; Fearon, E.R. Netrin-1: Interaction with deleted in colorectal cancer (DCC) and alterations in brain tumors and neuroblastomas. Cell Growth Differ. 1999, 10, 35–42. [Google Scholar]
- Latil, A.; Chêne, L.; Cochant-Priollet, B.; Mangin, P.; Fournier, G.; Berthon, P.; Cussenot, O. Quantification of expression of netrins, slits and their receptors in human prostate tumors. Int. J. Cancer 2003, 103, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Fearon, E.R. Role of the dependence receptor DCC in colorectal cancer pathogenesis. J. Clin. Oncol. Oncol. 2004, 22, 3420–3428. [Google Scholar] [CrossRef] [PubMed]
- Boussouar, A.; Tortereau, A.; Manceau, A.; Paradisi, A.; Gadot, N.; Vial, J.; Neves, D.; Larue, L.; Battistella, M.; Leboeuf, C.; et al. Netrin-1 and Its Receptor DCC Are Causally Implicated in Melanoma ProgressionDCC and Netrin-1 in Melanoma. Cancer Res. 2020, 80, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.-J.; Rama, N.; Imbach, J.; Fiore, S.; Ducarouge, B.; Neves, D.; Chen, H.-W.; Bernard, D.; Yang, P.-C.; Bernet, A.; et al. Cancer-associated fibroblasts produce netrin-1 to control cancer cell plasticity. Cancer Res. 2019, 79, 3651–3661. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, A.; Mehlen, P. Netrin-1, a missing link between chronic inflammation and tumor progression. Cell Cycle 2010, 9, 1253–1262. [Google Scholar] [CrossRef]
- Qi, Q.; Li, D.Y.; Luo, H.R.; Guan, K.-L.; Ye, K. Netrin-1 exerts oncogenic activities through enhancing Yes-associated protein stability. Proc. Natl. Acad. Sci. USA 2015, 112, 7255–7260. [Google Scholar] [CrossRef]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Cutforth, T.; Moring, L.; Mendelsohn, M.; Nemes, A.; Shah, N.M.; Kim, M.M.; Frisén, J.; Axel, R. Axonal ephrin-As and odorant receptors: Coordinate determination of the olfactory sensory map. Cell 2003, 114, 311–322. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Petrogiannopoulos, L.; Pergaris, A.; Theocharis, S. The EPH/Ephrin System in Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 2761. [Google Scholar] [CrossRef]
- Kakarla, M.; ChallaSivaKanaka, S.; Dufficy, M.F.; Gil, V.; Filipovich, Y.; Vickman, R.; Crawford, S.E.; Hayward, S.W.; Franco, O.E. Ephrin B Activate Src Family Kinases in Fibroblasts Inducing Stromal Remodeling in Prostate Cancer. Cancers 2022, 14, 2336. [Google Scholar] [CrossRef] [PubMed]
- Shiuan, E.; Chen, J. Eph receptor tyrosine kinases in tumor immunity. Cancer Res. 2016, 76, 6452–6457. [Google Scholar] [CrossRef]
- Broggini, T.; Piffko, A.; Hoffmann, C.J.; Ghori, A.; Harms, C.; Adams, R.H.; Vajkoczy, P.; Czabanka, M. Ephrin-B2-EphB4 communication mediates tumor-endothelial cell interactions during hematogenous spread to spinal bone in a melanoma metastasis model. Oncogene 2020, 39, 7063–7075. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.P.; Kandpal, R.P. Invasiveness of breast carcinoma cells and transcript profile: Eph receptors and ephrin ligands as molecular markers of potential diagnostic and prognostic application. Biochem. Biophys. Res. Commun. 2004, 318, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Hu, Z.; Kinch, M.S.; Pan, C.-X.; Flockhart, D.A.; Kao, C.; Gardner, T.A.; Zhang, S.; Li, L.; Baldridge, L.A.; et al. High-level expression of EphA2 receptor tyrosine kinase in prostatic intraepithelial neoplasia. Am. J. Pathol. 2003, 163, 2271–2276. [Google Scholar] [CrossRef]
- Nikas, I.; Giaginis, C.; Petrouska, K.; Alexandrou, P.; Michail, A.; Sarantis, P.; Tsourouflis, G.; Danas, E.; Pergaris, A.; Politis, P.K.; et al. EPHA2, EPHA4, and EPHA7 Expression in Triple-Negative Breast Cancer. Diagnostics 2022, 12, 366. [Google Scholar] [CrossRef] [PubMed]
- Venkitachalam, S.; Babu, D.; Ravillah, D.; Katabathula, R.M.; Joseph, P.; Singh, S.; Udhayakumar, B.; Miao, Y.; Martinez-Uribe, O.; Hogue, J.A.; et al. The Ephrin B2 Receptor Tyrosine Kinase is a Regulator of Proto-oncogene MYC and Molecular Programs Central to Barrett’s Neoplasia. Gastroenterology 2022, 163, 1228–1241. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rivera, E.; Ran, S.; Brekken, R.A.; Minna, J.D. Semaphorin 3B inhibits the phosphatidylinositol 3-kinase/Akt pathway through neuropilin-1 in lung and breast cancer cells. Cancer Res. 2008, 68, 8295–8303. [Google Scholar] [CrossRef]
- Caunt, M.; Mak, J.; Liang, W.-C.; Stawicki, S.; Pan, Q.; Tong, R.K.; Kowalski, J.; Ho, C.; Reslan, H.B.; Ross, J.; et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell 2008, 13, 331–342. [Google Scholar] [CrossRef]
- Leffers, N.; Gooden, M.J.; de Jong, R.A.; Hoogeboom, B.-N.; ten Hoor, K.A.; Hollema, H.; Boezen, H.M.; van der Zee, A.G.; Daemen, T.; Nijman, H.W. Prognostic significance of tumor-infiltrating T-lymphocytes in primary and metastatic lesions of advanced stage ovarian cancer. Cancer Immunol. Immunother. 2009, 58, 449–459. [Google Scholar] [CrossRef]
- Fard, D.; Tamagnone, L. Semaphorins in health and disease. CytokineGrowth Factor Rev. 2021, 57, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Franzolin, G.; Tamagnone, L. Semaphorin signaling in cancer-associated inflammation. Int. J. Mol. Sci. 2019, 20, 377. [Google Scholar] [CrossRef] [PubMed]
- Butti, R.; Kumar, T.V.; Nimma, R.; Kundu, G.C. Impact of semaphorin expression on prognostic characteristics in breast cancer. Breast Cancer Targets Ther. 2018, 10, 79. [Google Scholar] [CrossRef]
- Toledano, S.; Nir-Zvi, I.; Engelman, R.; Kessler, O.; Neufeld, G. Class-3 semaphorins and their receptors: Potent multifunctional modulators of tumor progression. Int. J. Mol. Sci. 2019, 20, 556. [Google Scholar] [CrossRef]
- Hagel, C.; Stavrou, D. Neuronal markers in non-neuronal tissues. Neuronal Act. Tumor Tissue 2007, 39, 64–77. [Google Scholar]
- Samarelli, A.V.; Riccitelli, E.; Bizzozero, L.; Silveira, T.N.; Seano, G.; Pergolizzi, M.; Vitagliano, G.; Cascone, I.; Carpentier, G.; Bottos, A.; et al. Neuroligin 1 induces blood vessel maturation by cooperating with the α6 integrin. J. Biol. Chem. 2014, 289, 19466–19476. [Google Scholar] [CrossRef] [PubMed]
- Rissone, A.; Foglia, E.; Sangiorgio, L.; Cermenati, S.; Nicoli, S.; Cimbro, S.; Beltrame, M.; Bussolino, F.; Cotelli, F.; Arese, M. The Synaptic Proteins β-Neurexin and Neuroligin Synergize With Extracellular Matrix-Binding Vascular Endothelial Growth Factor A During Zebrafish Vascular Development. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1563–1572. [Google Scholar] [CrossRef][Green Version]
- Bottos, A.; Rissone, A.; Bussolino, F.; Arese, M. Neurexins and neuroligins: Synapses look out of the nervous system. Cell. Mol. Life Sci. 2011, 68, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Destro, E.; Rissone, A.; Graziano, S.; Cordara, G.; Assenzio, B.; Cera, M.R.; Mascia, L.; Bussolino, F.; Arese, M. The synaptic proteins neurexins and neuroligins are widely expressed in the vascular system and contribute to its functions. Proc. Natl. Acad. Sci. USA 2009, 106, 20782–20787. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, X.; Yang, Y.; Chi, P.; Huang, J.; Qiu, S.; Zheng, X.; Chen, X. Upregulated NLGN1 predicts poor survival in colorectal cancer. BMC Cancer 2021, 21, 884. [Google Scholar] [CrossRef] [PubMed]
- Pergolizzi, M.; Bizzozero, L.; Maione, F.; Maldi, E.; Isella, C.; Macagno, M.; Mariella, E.; Bardelli, A.; Medico, E.; Marchiò, C.; et al. The neuronal protein Neuroligin 1 promotes colorectal cancer progression by modulating the APC/β-catenin pathway. J. Exp. Clin. Cancer Res. 2022, 41, 266. [Google Scholar] [CrossRef] [PubMed]
- Bizzozero, L.; Pergolizzi, M.; Pascal, D.; Maldi, E.; Villari, G.; Erriquez, J.; Volante, M.; Serini, G.; Marchiò, C.; Bussolino, F.; et al. Tumoral Neuroligin 1 Promotes Cancer-Nerve Interactions and Synergizes with the Glial Cell Line-Derived Neurotrophic Factor. Cells 2022, 11, 280. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Suzuki, K.; Hayashi, Y.; Nakahara, S.; Kumazaki, H.; Prox, J.; Horiuchi, K.; Zeng, M.; Tanimura, S.; Nishiyama, Y.; Osawa, S.; et al. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron 2012, 76, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.J.; Kabotyanski, K.E.; Calin, G.A.; Xie, T.; Myers, J.N.; Amit, M. Interrupting Neuron—Tumor Interactions to Overcome Treatment Resistance. Cancers 2020, 12, 3741. [Google Scholar] [CrossRef]
- Jung, E.; Alfonso, J.; Monyer, H.; Wick, W.; Winkler, F. Neuronal signatures in cancer. Int. J. Cancer 2020, 147, 3281–3291. [Google Scholar] [CrossRef] [PubMed]
- Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L.; Pascal, D. Tumor progression: The neuronal input. Ann. Transl. Med. 2018, 6, 2972. [Google Scholar] [CrossRef] [PubMed]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2011, 1816, 105–118. [Google Scholar]
- Jiang, S.-H.; Hu, L.-P.; Wang, X.; Li, J.; Zhang, Z.-G. Neurotransmitters: Emerging targets in cancer. Oncogene 2020, 39, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Mendler, L.; Pinter, S.; Kiricsi, M.; Baka, Z.; Dux, L. Regeneration of reinnervated rat soleus muscle is accompanied by fiber transition toward a faster phenotype. J. Histochem. Cytochem. 2008, 56, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, G.F.; Burke, R.E.; Lowey, S.; Hobbs, A.W. Myosin isozymes in normal and cross-reinnervated cat skeletal muscle fibers. J. Cell Biol. 1983, 97, 756–771. [Google Scholar] [CrossRef] [PubMed]




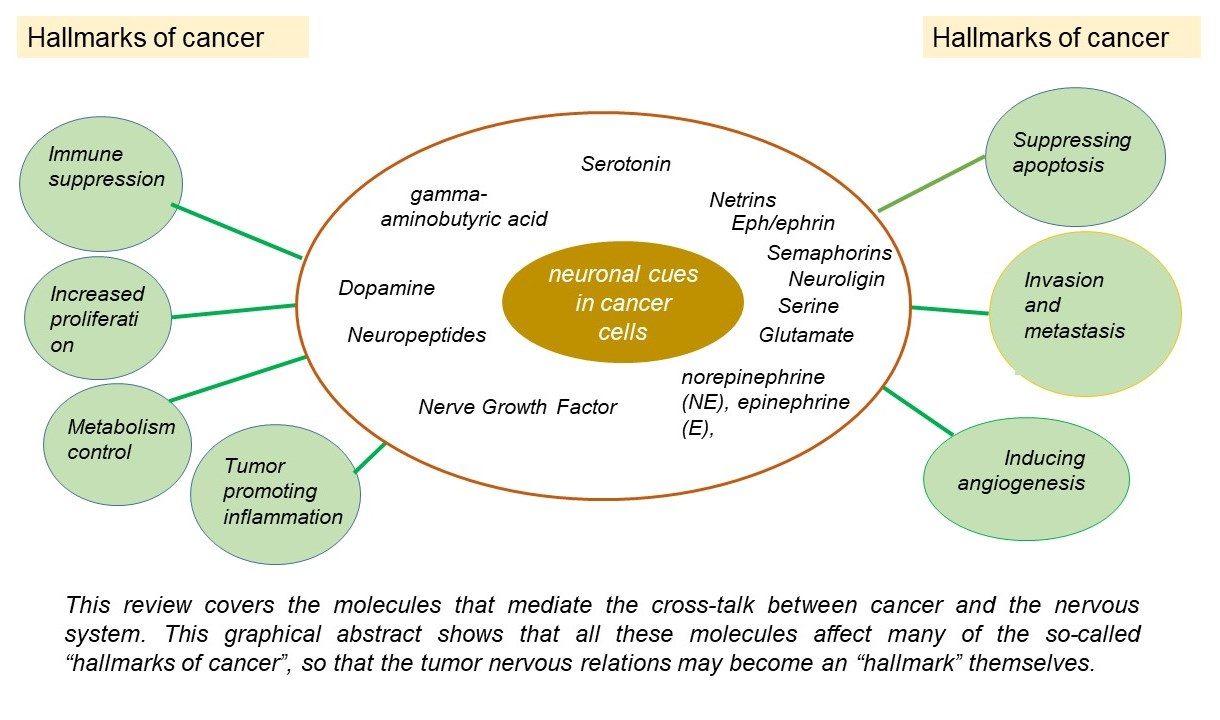{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Member | Cellular Receptor | Affected Intracellular Pathways | Refs |
|---|---|---|---|---|
| Neurotransmitters/amino acids | Norepinephrine (noradrenaline) | α-β adrenergic receptors | Gαs–cAMP, MAPK, PKA, EPAC ERK/cyclooxygenase 2 CREB1-miR-373 axis, Src PKA, VDCC, Hippo | [50,56,58,61,64] |
| Dopamine | Dopamine 1–5 (D1–5) receptors | VEGFA-mediated ERK1/2 phosphorylation | [95,96] | |
| Serotonin (5-hydroxytryptamine) | 5-HT1-7 receptors | Akt/Mtor, glycolysis, endothelial nitric oxide synthase, p-ERK1/2 | [130,131,133] | |
| Acetylcholine | Muscarinic acetylcholine receptor, nicotinic acetylcholine receptor | Ca2+ flux, PKC/ERK1/2, COX2, PGE2, CREB, SRC, AKT, Ras-RAF1, MAPK, survivin, BCL-2, NF-κB, PI3K/AKT | [2,5,73,75,76,79] | |
| Glutamate | Metabotropic glutamate receptor, NMDA receptor, Kainate receptor, AMPA receptor | PI3K (p110) | [144] | |
| Gamma-aminobutyric acid | GABAB GABAA GABAc receptors | Ca2+ flux MAPK/ERK | [103,109] | |
| Serine | Amino acid transporters | mRNA translation | [145] | |
| Neuropeptides | Neuropeptide Y | NPY1R, NPY2R, PPYR1, NPY5R | PI3-K/pAkt microRNA-375 | [156] |
| Substance P | NK1, NNK2, NNK3 | inositol 1,4,5-triphosphate (IP3) DAG, c-myc, mitogen-activated protein kinases, activator protein 1, extracellular signal-regulated kinases 1 and 2, glycogen breakdown | [149,152] | |
| Growth factors | NGF | TrkA, p75NTR | PI3K, MAPK c-Jun N-terminal kinase | [157] |
| Axon guidance | Netrin 1 | DCC, UNC5 | Netrin 1 is a target of NFkB and induces YAP. | [171,172] |
| Ephrins | Ephs | Forward signaling: Src family kinases, Rho GTPases, Ephexins (GEFs that can activate Rho GTPases), ERK/MAPK pathway, which promote cell proliferation. Additionally, FAK and the JAK/STAT pathway interact with EPHs, which modifies cell adhesion. Reverse signaling: interactions between ephrins and Src, Erk, Rac, T, paxillin, p75, and integrin-dependent cell adhesion as well as with Src, Grb4, PTP-BL, and PDZ-RGS3, which controls a variety of actions, including cell adhesion, migration, and proliferation. | [175] | |
| Semaphorins | Neuropilins, plexins | Sema3A: nrp1 PTEN/FOXO 3a-dependent MelCAM (CD146) expression. Sema3B binds to NRP1 and inhibits PI3K/Akt signaling. Full-length Sema3C NRP2 inhibits VEGF-C-dependent ERK1/2 and Akt signaling and suppresses lymphangiogenesis and metastasis. Cleaved Sema3C promotes cancer cell survival. Sema4D binds to plexin-B1 and activates ErbB2, phosphorylating plexin-B1. By activating RhoA GTPase, phosphorylated plexin-B1 promotes migration. Through ErbB2-dependent MAPK signaling, cleaved p61-Sema3E binds to plexin-D1 to promote metastasis. Sema3E binds to plexin-D1 and disrupts the interactions between plexin-D1 and NR4A, which is known to induce caspase-9-mediated apoptosis. | [188] | |
| Synaptic proteins | Neuroligin 1 | - | Neuroligin 1 recruits APC to the plasma membrane, blocking beta catenin degradation and causing its transfer to the nucleus, where it promotes EMT-linked gene transcription. | [196] |
| Neuroligin 1 | - | Neuroligin 1 synergizes with GDNF to induce cancer cell invasion of nerves and to activate the cytoskeleton-regulating protein cofilin. | [199] | |
| Neuroligin 3 | - | NLGN3 extracellular domain shedding by ADAM10 into the tumor microenvironment promotes glioma growth. | [198] | |
| For all other synaptic proteins, see [6]. | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arese, M.; Bussolino, F.; Pergolizzi, M.; Bizzozero, L. An Overview of the Molecular Cues and Their Intracellular Signaling Shared by Cancer and the Nervous System: From Neurotransmitters to Synaptic Proteins, Anatomy of an All-Inclusive Cooperation. Int. J. Mol. Sci. 2022, 23, 14695. https://doi.org/10.3390/ijms232314695
Arese M, Bussolino F, Pergolizzi M, Bizzozero L. An Overview of the Molecular Cues and Their Intracellular Signaling Shared by Cancer and the Nervous System: From Neurotransmitters to Synaptic Proteins, Anatomy of an All-Inclusive Cooperation. International Journal of Molecular Sciences. 2022; 23(23):14695. https://doi.org/10.3390/ijms232314695
Chicago/Turabian StyleArese, Marco, Federico Bussolino, Margherita Pergolizzi, and Laura Bizzozero. 2022. "An Overview of the Molecular Cues and Their Intracellular Signaling Shared by Cancer and the Nervous System: From Neurotransmitters to Synaptic Proteins, Anatomy of an All-Inclusive Cooperation" International Journal of Molecular Sciences 23, no. 23: 14695. https://doi.org/10.3390/ijms232314695
APA StyleArese, M., Bussolino, F., Pergolizzi, M., & Bizzozero, L. (2022). An Overview of the Molecular Cues and Their Intracellular Signaling Shared by Cancer and the Nervous System: From Neurotransmitters to Synaptic Proteins, Anatomy of an All-Inclusive Cooperation. International Journal of Molecular Sciences, 23(23), 14695. https://doi.org/10.3390/ijms232314695