
Relationship between Changes in the Protein Folding Pathway and the Process of Amyloid Formation: The Case of Bovine Carbonic Anhydrase II
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. How Mutations of a Protein Affect Its Ability to Form Amyloids?
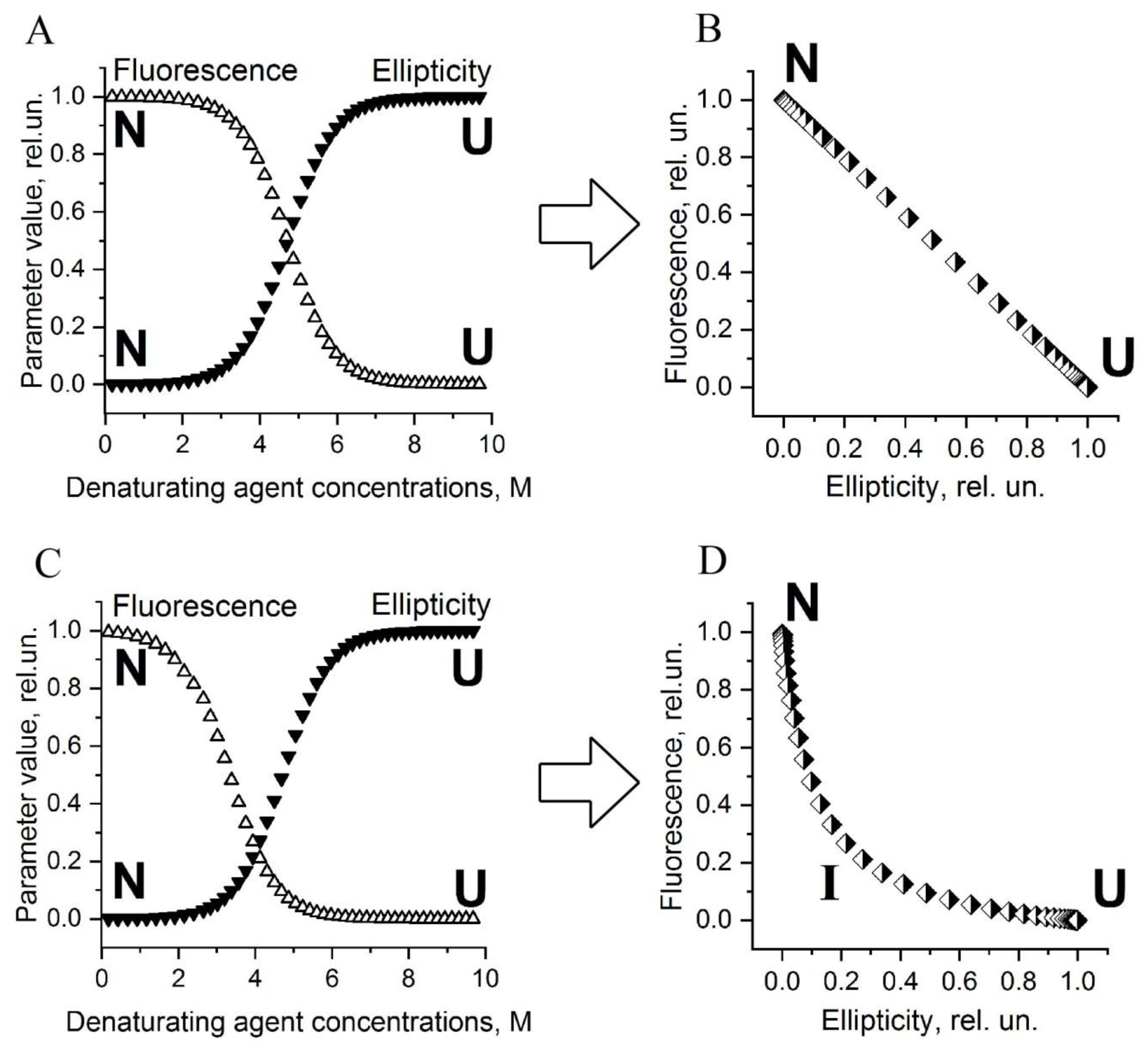
1.2. What Parametric Plots Are and How to Interpret Them
2. Results and Discussion
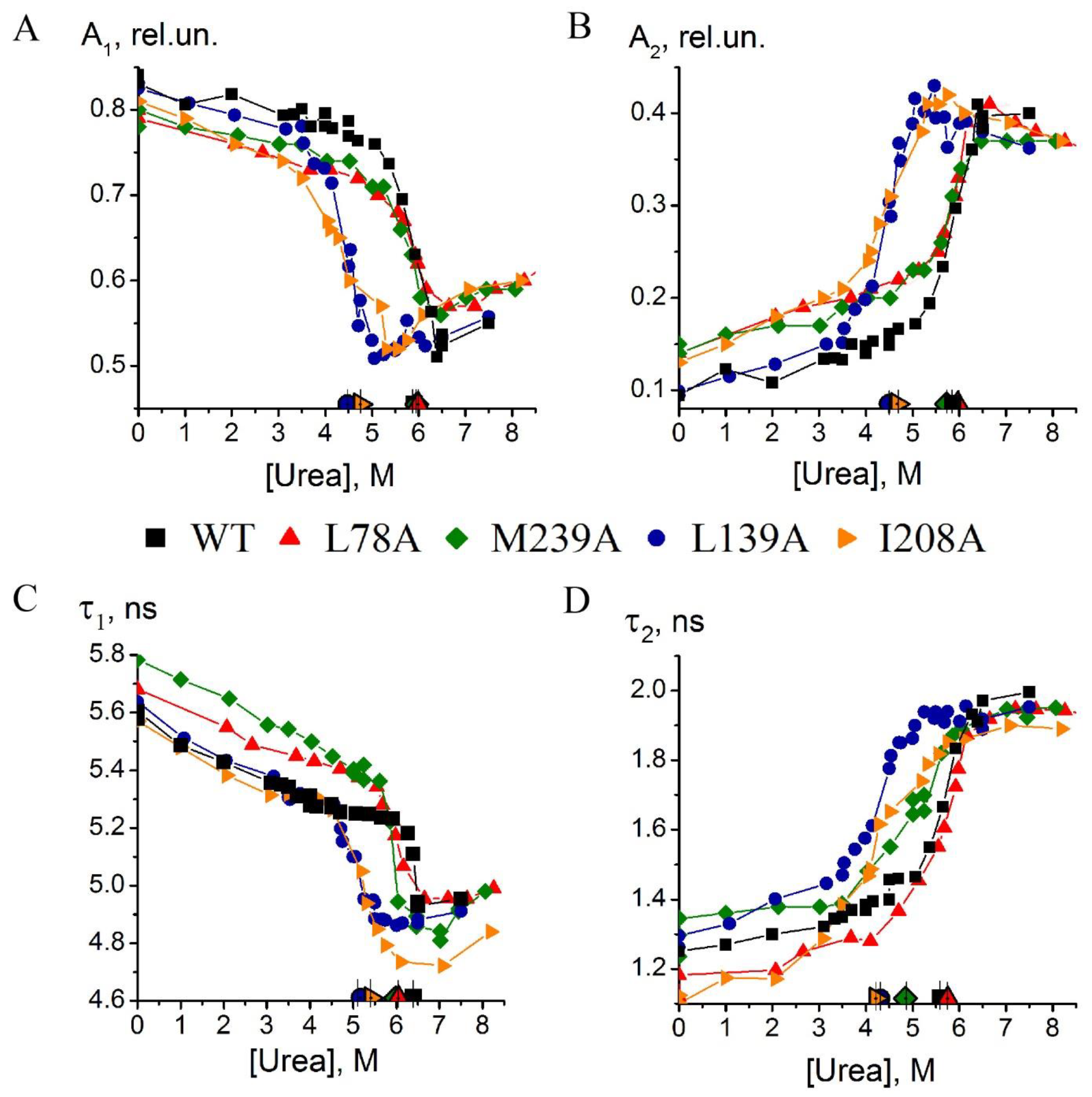
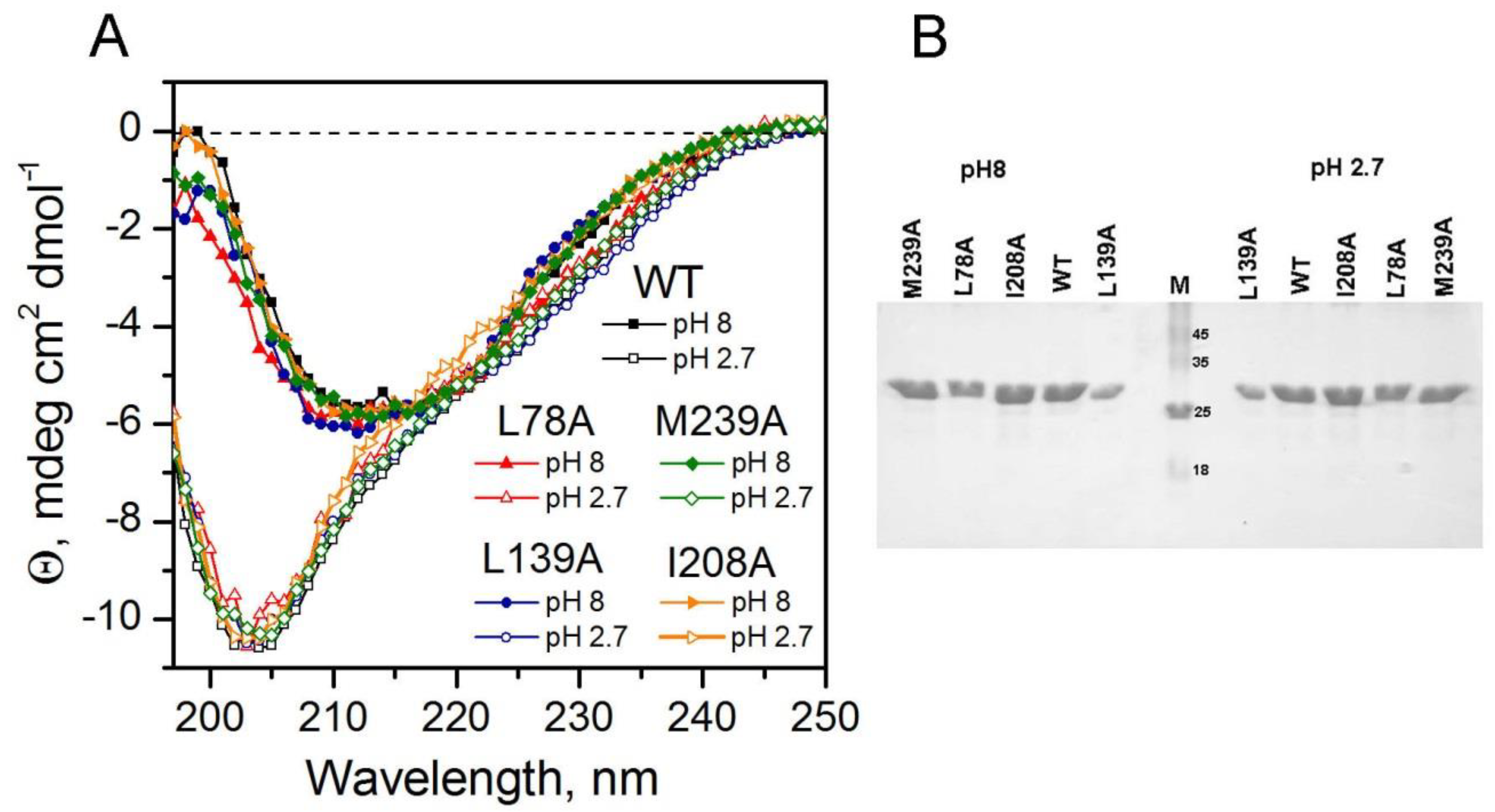
2.1. Comparison of Unfolding Pathways of Mutant Forms of Carbonic Anhydrase II Using Time-Resolved Fluorescence Technique
2.2. Growth of Amyloid Structures of the Mutant Forms of Carbonic Anhydrase II
2.3. The Relationship between Mutation Effects on Unfolding Pathway of Carbonic Anhydrase and Its Amyloid Formation
- –
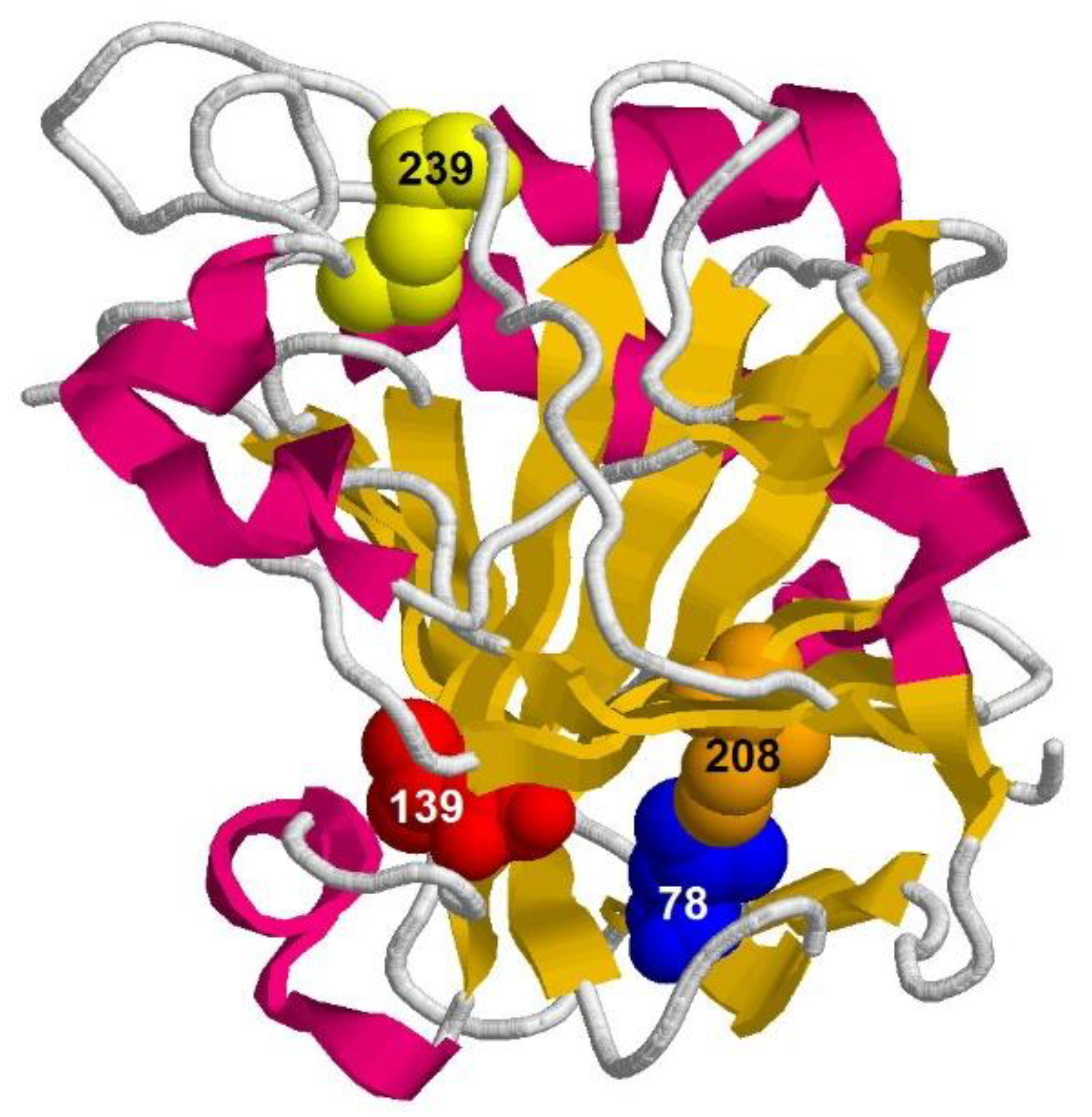
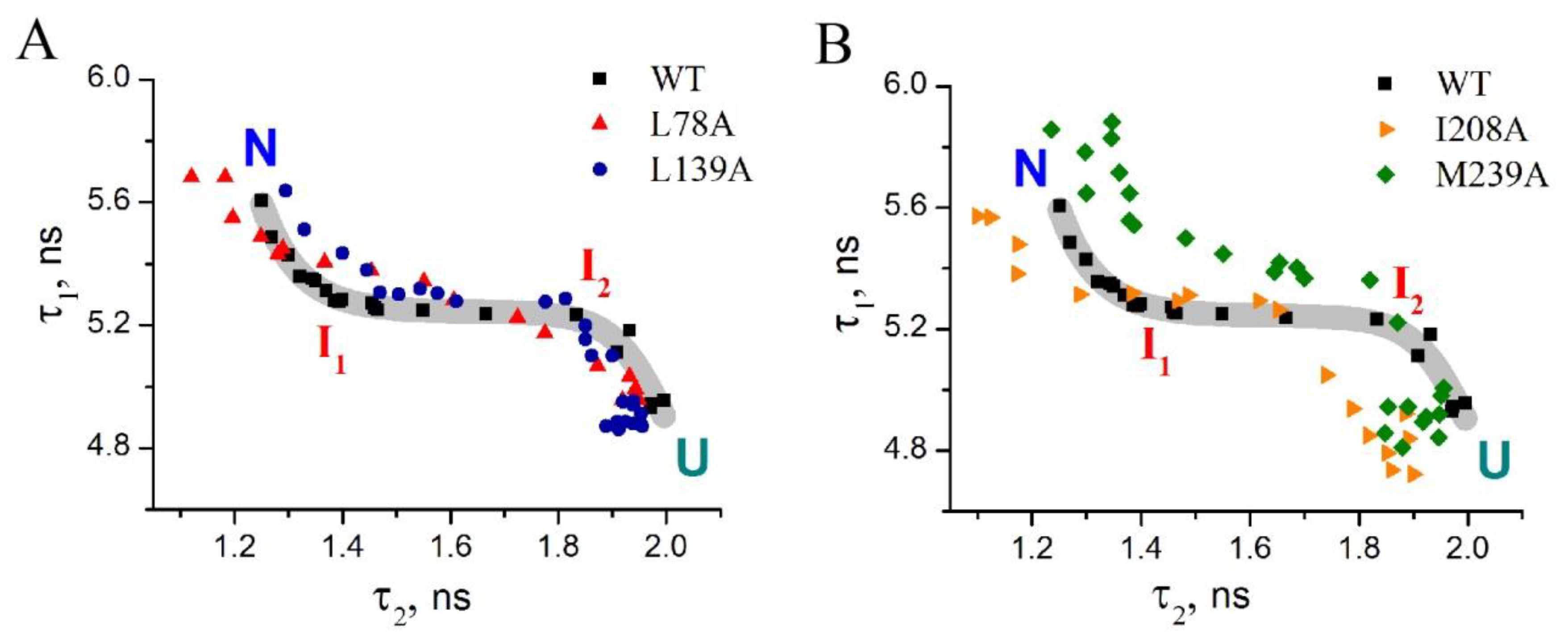
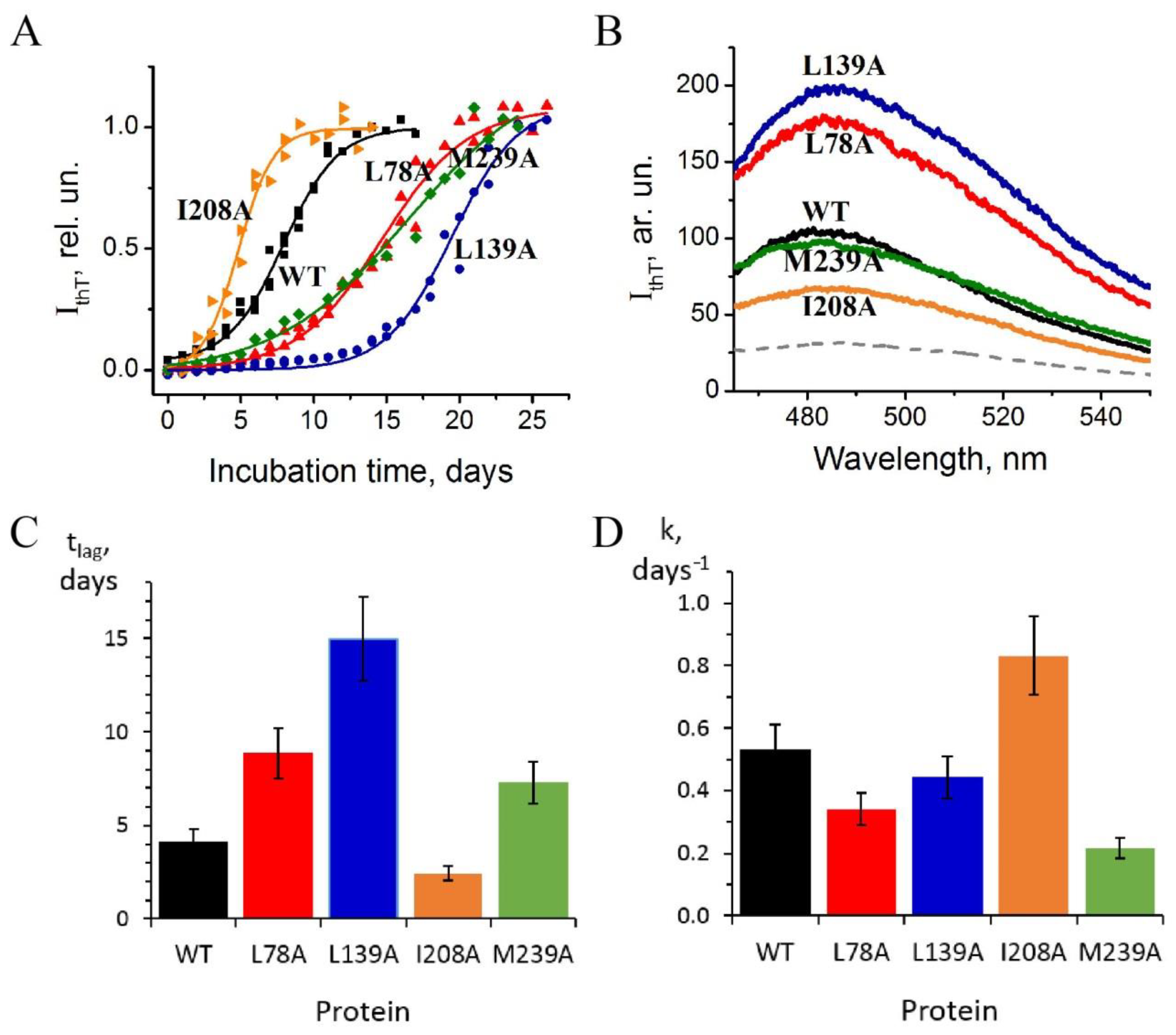
- Substitution L78A (in β-turn) slightly destabilizes both protein intermediates (Figure 3C,D) but has no effect on the structure of them (Figure 4A); this mutant form exhibits two-times-longer lag time (Figure 5C) and a slightly lower rate of amyloid growth kinetics (Figure 5D) as compared with wild-type protein;
- –
- Substitution L139A (in β-sheet) significantly decreases the intermediates’ stability (Figure 3C,D) without changing their structure (Figure 4A); for this mutant variant, a pronounced increase in the lag time (3.6-fold, Figure 5C) and a small decrease in the growth rate (Figure 5D) were found as compared with wild-type protein;
- –
- Substitution I208A (in β-turn), in addition to the destabilization of unfolding intermediates of carbonic anhydrase (Figure 3C,D), was found to disturb the structure of the late (molten globule-like, I2)-intermediate (Figure 4B); this mutant form demonstrates the fastest amyloid growth kinetics among all studied proteins (Figure 5C,D);
- –
- Substitution M239A (in unstructured C-terminal part) causes the lower stability of the protein-unfolding intermediates (Figure 3C,D) and changes the structure of the early-intermediate (I1) of carbonic anhydrase (Figure 4B); this mutant variant has a slower amyloids growth kinetics with about a two-times-longer lag time and two-times-lower growth rate (Figure 5C,D) than wild-type protein.
3. Materials and Methods
3.1. Isolation and Purification of Carbonic Anhydrase II and Its Mutant Forms
3.2. Preparation of Carbonic Anhydrase II Amyloids
3.3. SDS–Polyacrylamide Gel Electrophoresis (SDS-PAGE)
3.4. Electron Microscopy
3.5. Fluorescence Thioflavin T Measurements
3.6. Steady-State and Time-Resolved Fluorescence Measurements
4. Conclusions and Proposals
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandez-Escamilla, A.-M.; Rousseau, F.; Schymkowitz, J.; Serrano, L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat. Biotechnol. 2004, 22, 1302–1306. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, G.G.; Vendruscolo, M. The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 2008, 37, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Belli, M.; Ramazzotti, M.; Chiti, F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011, 12, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.S.; Sharpe, P.C.; Singh, Y.; Fairlie, D.P. Amyloid peptides and proteins in review. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–77. [Google Scholar]
- Goldschmidt, L.; Teng, P.K.; Riek, R.; Eisenberg, D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA 2010, 107, 3487–3492. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Chiti, F.; Taddei, N.; Bucciantini, M.; White, P.; Ramponi, G.; Dobson, C.M. Mutational analysis of the propensity for amyloid formation by a globular protein. EMBO J. 2000, 19, 1441–1449. [Google Scholar] [CrossRef]
- Katina, N.S.; Ilyina, N.B.; Kashparov, I.A.; Balobanov, V.A.; Vasiliev, V.D.; Bychkova, V.E. Apomyoglobin mutants with single point mutations at Val10 can form amyloid structures at permissive temperature. Biochemistry 2011, 76, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Langenberg, T.; Gallardo, R.; van der Kant, R.; Louros, N.; Michiels, E.; Duran-Romaña, R.; Houben, B.; Cassio, R.; Wilkinson, H.; Garcia, T.; et al. Thermodynamic and Evolutionary Coupling between the Native and Amyloid State of Globular Proteins. Cell Rep. 2020, 31, 107512. [Google Scholar] [CrossRef]
- Honda, R. Role of the Disulfide Bond in Prion Protein Amyloid Formation: A Thermodynamic and Kinetic Analysis. Biophys. J. 2018, 114, 885–892. [Google Scholar] [CrossRef]
- Choi, S.; Seong, B. A Conceptual Framework for Integrating Cellular Protein Folding, Misfolding and Aggregation. Life 2021, 11, 605. [Google Scholar] [CrossRef]
- Nemtseva, E.V.; Lashchuk, O.O.; Gerasimova, M.A.; Melnik, T.N.; Nagibina, G.S.; Melnik, B.S. Fluorescence lifetime compo-nents reveal kinetic intermediate states upon equilibrium denaturation of carbonic anhydrase II. Methods Appl. Fluoresc. 2017, 6, 015006. [Google Scholar] [CrossRef] [PubMed]
- Nemtseva, E.V.; Gerasimova, M.A.; Melnik, T.N.; Melnik, B.S. Experimental approach to study the effect of mutations on the protein folding pathway. PLoS ONE 2019, 14, e0210361. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Ptitsyn, O.B. Further Evidence on the Equilibrium “Pre-molten Globule State”: Four-state Guanidinium Chlo-ride-induced Unfolding of Carbonic Anhydrase B at Low Temperature. J. Mol. Biol. 1996, 255, 215–228. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.S.; Nagibina, G.S.; Glukhov, A.S.; Melnik, T.N.; Uversky, V.N. Substitutions of Amino Acids with Large Number of Contacts in the Native State Have no Effect on the Rates of Protein Folding. Biochim. Biophys. Acta 2016, 1864, 1809–1817. [Google Scholar] [CrossRef]
- Melnik, B.S.; Marchenkov, V.V.; Evdokimov, S.R.; Samatova, E.N.; Kotova, N.V. Multy-state protein: Determination of carbonic anhydrase free-energy landscape. Biochem. Biophys. Res. Commun. 2008, 369, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.S.; Nagibina, G.S.; Glukhov, A.S.; Melnik, T.N. An approach for the assessment of the order of disruption of the elements of protein structure upon protein unfolding: A study of carbonic anhydrase B. Biophysics 2016, 61, 860–870. [Google Scholar] [CrossRef]
- Garg, D.K.; Kundu, B. Clues for divergent, polymorphic amyloidogenesis through dissection of amyloid forming steps of bovine carbonic anhydrase and its critical amyloid forming stretch. Biochim. Biophys. Acta—Proteins Proteom. 2016, 1864, 794–804. [Google Scholar] [CrossRef]
- Es-Haghi, A.; Shariatizi, S.; Ebrahim-Habibi, A.; Nemat-Gorgani, M. Amyloid fibrillation in native and chemically-modified forms of carbonic anhydrase II: Role of surface hydrophobicity. Biochim. Biophys. Acta—Proteins Proteom. 2012, 1824, 468–477. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Stepanenko, O.V.; Turoverov, K.K.; Zhu, L.; Zhou, J.-M.; Fink, A.L.; Uversky, V.N. Unraveling multistate unfolding of rabbit muscle creatine kinase. Biochim. Biophys. Acta 2002, 1596, 138–155. [Google Scholar] [CrossRef]
- Bushmarina, N.A.; Kuznetsova, I.; Biktashev, A.G.; Turoverov, K.; Uversky, V.N. Partially Folded Conformations in the Folding Pathway of Bovine Carbonic Anhydrase II: A Fluorescence Spectroscopic Analysis. Chem. Bio. Chem. 2001, 2, 813–821. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Verkhusha, V.V.; Turoverov, K.K. Beta-barrel scaffold of fluorescent proteins: Folding, stability and role in chromophore formation. Int. Rev. Cell Mol. Biol. 2013, 302, 221–278. [Google Scholar] [PubMed]
- Kuznetsova, I.M.; Turoverov, A.K.K.; Uversky, V.N. Use of the Phase Diagram Method to Analyze the Protein Unfolding-Refolding Reactions: Fishing Out the “Invisible” Intermediates. J. Proteome Res. 2004, 3, 485–494. [Google Scholar] [CrossRef]
- Chiti, F.; Stefani, M.; Taddei, N.; Ramponi, G.; Dobson, C.M. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 2003, 424, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Marchenkov, V.; Ryabova, N.; Balobanov, V.; Glukhov, A.; Ilyina, N.; Katina, N. Under Conditions of Amyloid Formation Bovine Carbonic Anhydrase B Undergoes Fragmentation by Acid Hydrolysis. Biomolecules 2021, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.M.; Myers, D.V.; Verpoorte, J.A.; Edsall, J.T. Purification and properties of human erythrocyte carbonic anhydrases. J. Biol. Chem. 1966, 241, 5137–5149. [Google Scholar] [CrossRef]
- Dolgikh, D.A.; Kolomiets, A.P.; Bolotina, I.A.; Ptitsyn, O.B. “Molten-globule” state accumulates in carbonic anhydrase folding. FEBS Lett. 1984, 165, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melnik, B.S.; Katina, N.S.; Ryabova, N.A.; Marchenkov, V.V.; Melnik, T.N.; Karuzina, N.E.; Nemtseva, E.V. Relationship between Changes in the Protein Folding Pathway and the Process of Amyloid Formation: The Case of Bovine Carbonic Anhydrase II. Int. J. Mol. Sci. 2022, 23, 14645. https://doi.org/10.3390/ijms232314645
Melnik BS, Katina NS, Ryabova NA, Marchenkov VV, Melnik TN, Karuzina NE, Nemtseva EV. Relationship between Changes in the Protein Folding Pathway and the Process of Amyloid Formation: The Case of Bovine Carbonic Anhydrase II. International Journal of Molecular Sciences. 2022; 23(23):14645. https://doi.org/10.3390/ijms232314645
Chicago/Turabian StyleMelnik, Bogdan S., Natalya S. Katina, Natalya A. Ryabova, Victor V. Marchenkov, Tatiana N. Melnik, Natalya E. Karuzina, and Elena V. Nemtseva. 2022. "Relationship between Changes in the Protein Folding Pathway and the Process of Amyloid Formation: The Case of Bovine Carbonic Anhydrase II" International Journal of Molecular Sciences 23, no. 23: 14645. https://doi.org/10.3390/ijms232314645
APA StyleMelnik, B. S., Katina, N. S., Ryabova, N. A., Marchenkov, V. V., Melnik, T. N., Karuzina, N. E., & Nemtseva, E. V. (2022). Relationship between Changes in the Protein Folding Pathway and the Process of Amyloid Formation: The Case of Bovine Carbonic Anhydrase II. International Journal of Molecular Sciences, 23(23), 14645. https://doi.org/10.3390/ijms232314645