

Distant Recurrence of a Cerebral Cavernous Malformation in the Vicinity of a Developmental Venous Anomaly: Case Report of Local Oxy-Inflammatory Events

 ,
,  , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
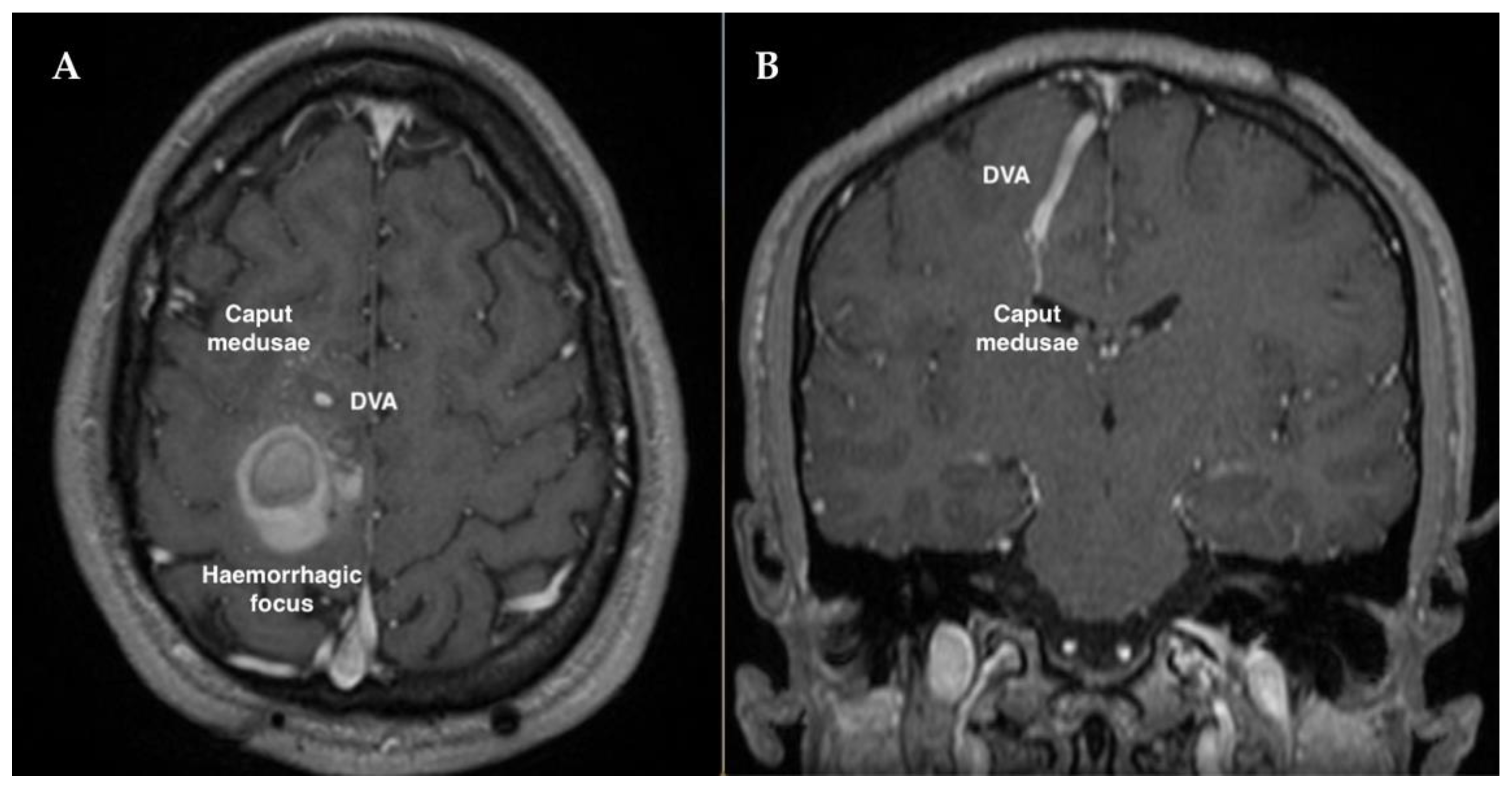
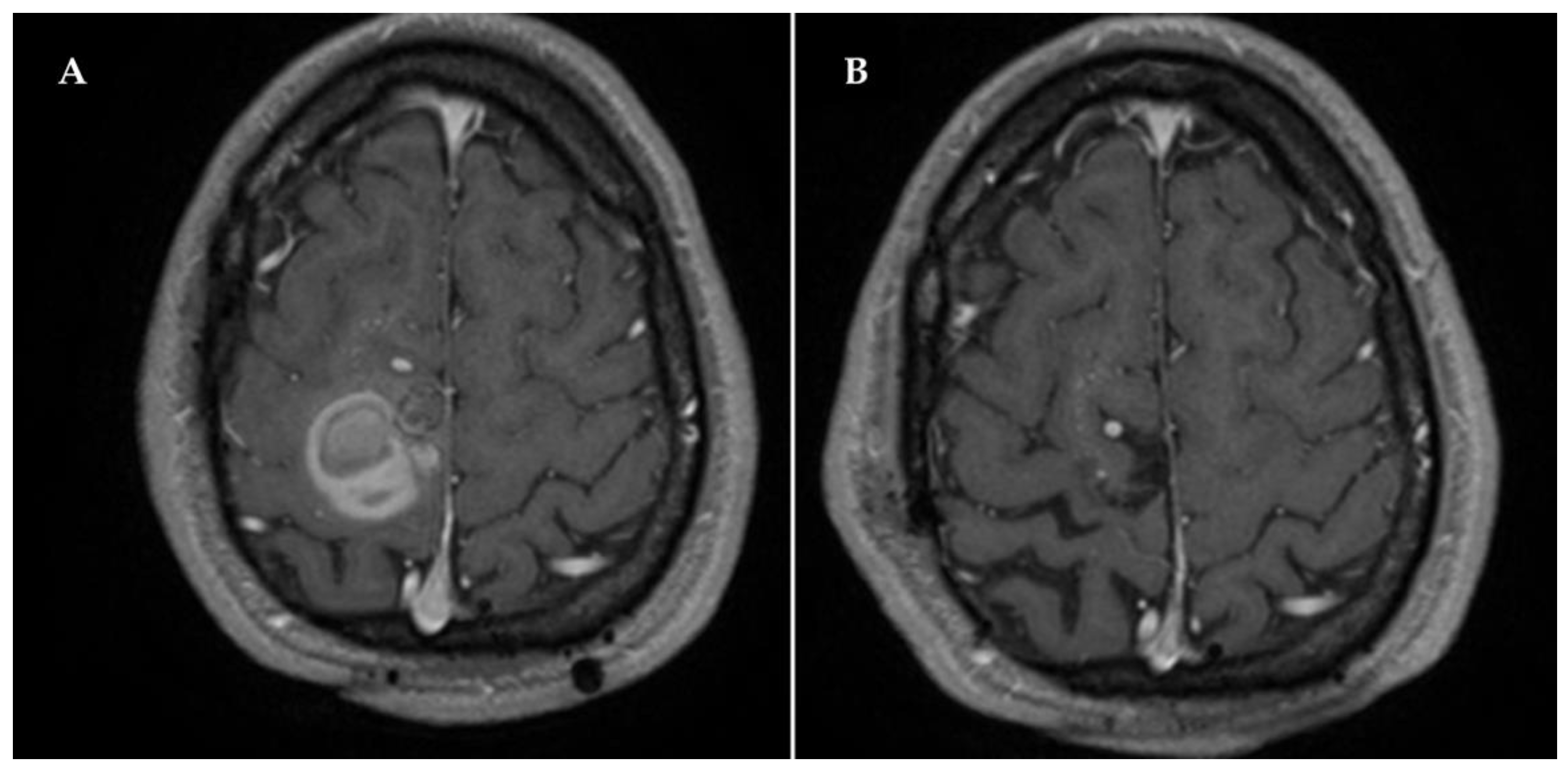
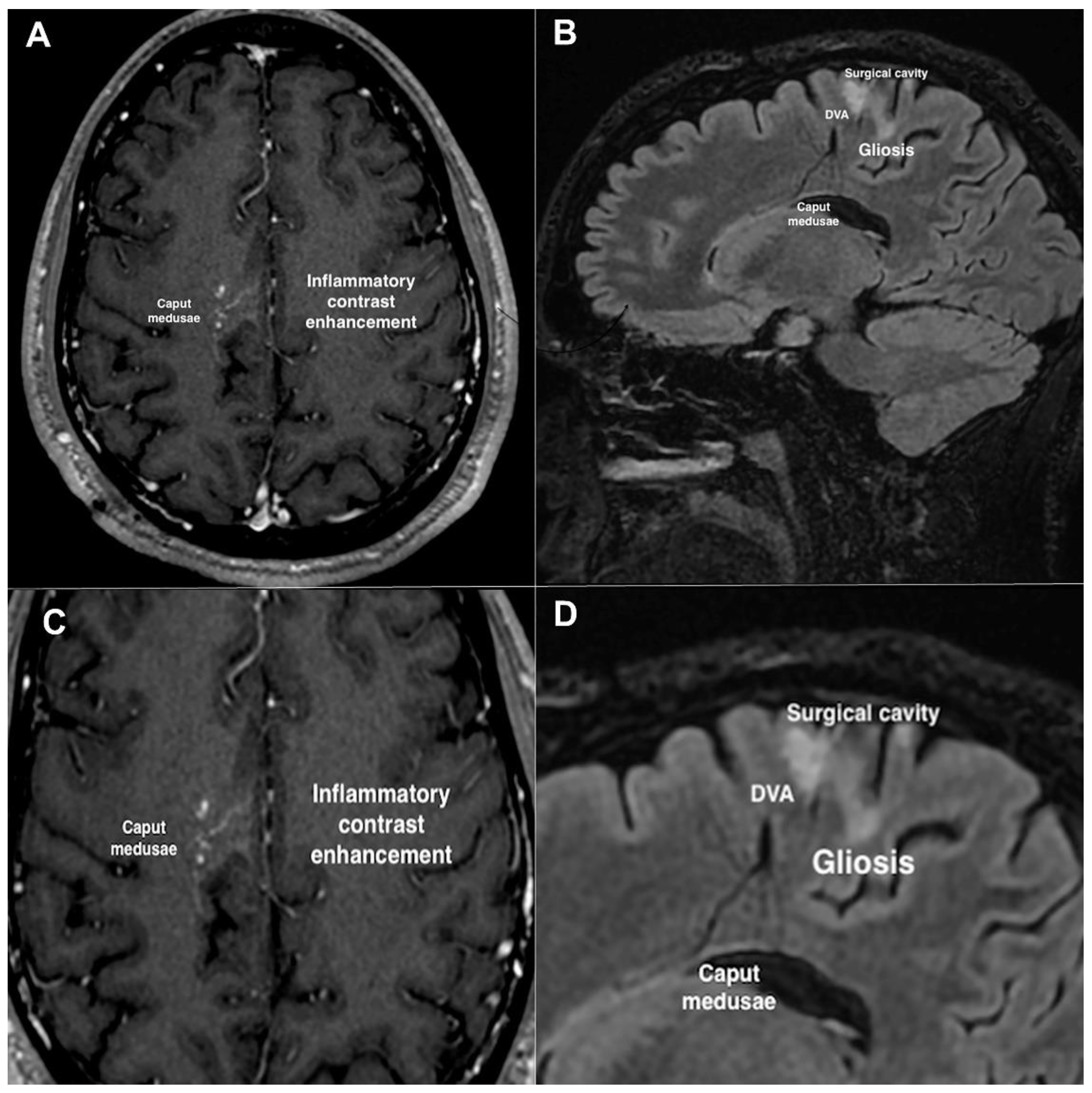
2. Case Description
3. Discussion
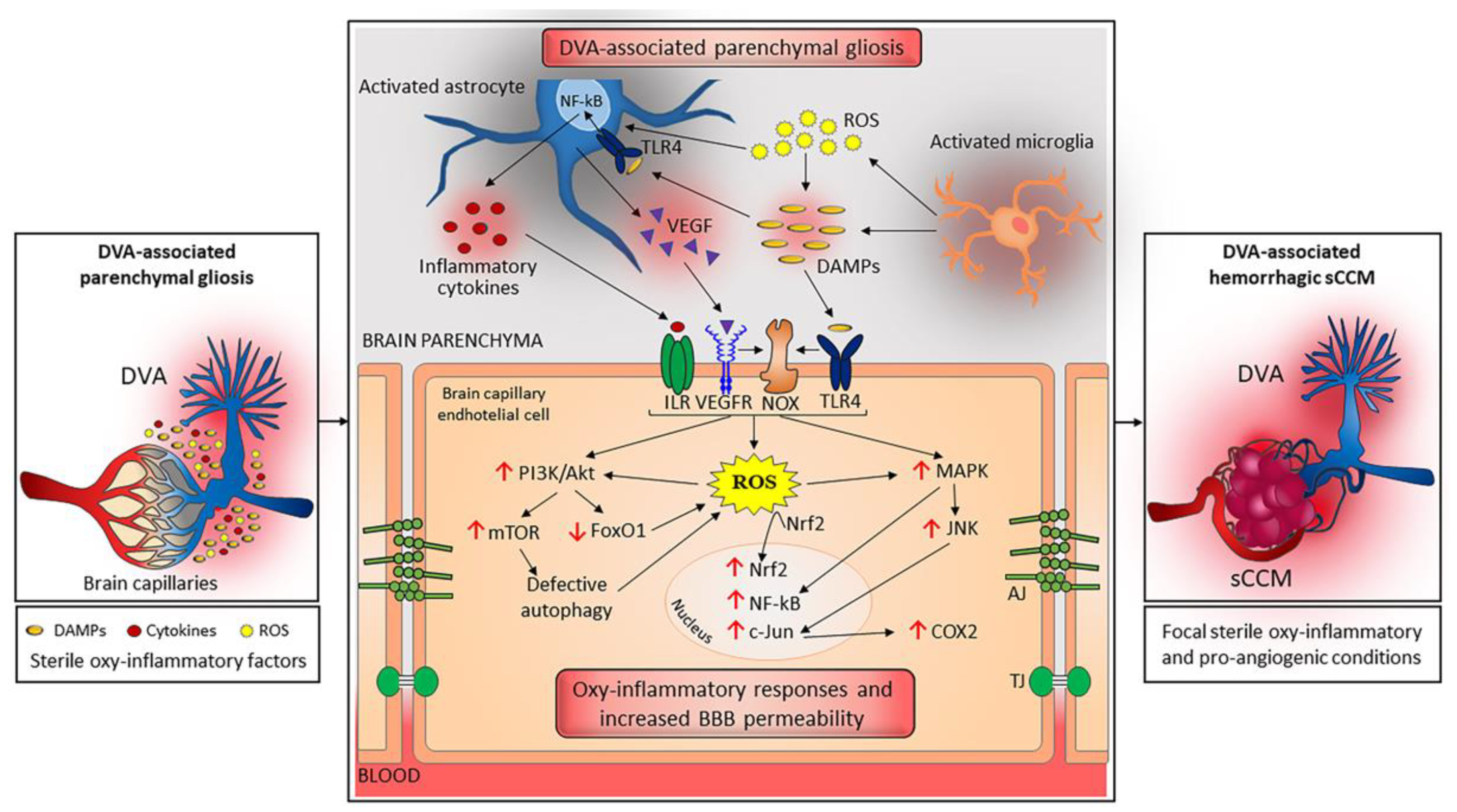
3.1. A Unifying Pathogenic Scenario for the Distinctive Features of sCCM and fCCM Lesions
3.2. DVA: A Chronic Condition That Can Trigger Sterile Inflammation
3.3. Focal Coexistence of DVA and sCCM: A Dangerous Liaison Linked to Sterile Inflammation
4. Concluding Remarks and Implications for Future Research
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Batra, S.; Lin, D.; Recinos, P.F.; Zhang, J.; Rigamonti, D. Cavernous malformations: Natural history, diagnosis and treatment. Nat. Rev. Neurol. 2009, 5, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Flemming, K.D. Incidence, Prevalence, and Clinical Presentation of Cerebral Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 27–33. [Google Scholar] [CrossRef]
- Mabray, M.; Hart, B. Clinical Imaging of Cerebral Cavernous Malformations: Computed Tomography and Magnetic Resonance Imaging. Methods Mol. Biol. 2020, 2152, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.; Kaul, A.; Kim, L. Natural history of cerebral cavernous malformations. Handb. Clin. Neurol. 2017, 143, 227–232. [Google Scholar] [CrossRef]
- Choquet, H.; Pawlikowska, L.; Lawton, M.T.; Kim, H. Genetics of cerebral cavernous malformations: Current status and future prospects. J. Neurosurg. Sci. 2015, 59, 211–220. [Google Scholar]
- Tournier-Lasserve, E. Molecular Genetic Screening of CCM Patients: An Overview. Methods Mol. Biol. 2020, 2152, 49–57. [Google Scholar] [CrossRef]
- Retta, S.F.; Perrelli, A.; Trabalzini, L.; Finetti, F. From Genes and Mechanisms to Molecular-Targeted Therapies: The Long Climb to the Cure of Cerebral Cavernous Malformation (CCM) Disease. Methods Mol. Biol. 2020, 2152, 3–25. [Google Scholar] [CrossRef]
- Benedetti, V.; Canzoneri, R.; Perrelli, A.; Arduino, C.; Zonta, A.; Brusco, A.; Retta, S.F. Next-Generation Sequencing Advances the Genetic Diagnosis of Cerebral Cavernous Malformation (CCM). Antioxidants 2022, 11, 1294. [Google Scholar] [CrossRef]
- Perrelli, A.; Ferraris, C.; Berni, E.; Glading, A.J.; Retta, S.F. KRIT1: A traffic warden at the busy crossroads between redox signaling and the pathogenesis of Cerebral Cavernous Malformation disease. Antioxid. Redox Signal. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Peyre, M.; Miyagishima, D.; Bielle, F.; Chapon, F.; Sierant, M.; Venot, Q.; Lerond, J.; Marijon, P.; Abi-Jaoude, S.; Le Van, T.; et al. Somatic PIK3CA Mutations in Sporadic Cerebral Cavernous Malformations. N. Engl. J. Med. 2021, 385, 996. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Yang, Y.; Song, D.; Huo, R.; Li, H.; Chen, Y.; Nam, Y.; Zhou, Q.; Jiao, Y.; Fu, W.; et al. Somatic MAP3K3 mutation defines a subclass of cerebral cavernous malformation. Am. J. Hum. Genet. 2021, 108, 942–950. [Google Scholar] [CrossRef]
- Hong, T.; Xiao, X.; Ren, J.; Cui, B.; Zong, Y.; Zou, J.; Kou, Z.; Jiang, N.; Meng, G.; Zeng, G.; et al. Somatic MAP3K3 and PIK3CA mutations in sporadic cerebral and spinal cord cavernous malformations. Brain 2021, 144, 2648–2658. [Google Scholar] [CrossRef]
- Petersen, T.A.; Morrison, L.A.; Schrader, R.M.; Hart, B.L. Familial versus sporadic cavernous malformations: Differences in developmental venous anomaly association and lesion phenotype. AJNR Am. J. Neuroradiol. 2010, 31, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Perrini, P.; Lanzino, G. The association of venous developmental anomalies and cavernous malformations: Pathophysiological, diagnostic, and surgical considerations. Neurosurg. Focus 2006, 21, e5. [Google Scholar] [CrossRef]
- Yu, T.; Liu, X.; Lin, X.; Bai, C.; Zhao, J.; Zhang, J.; Zhang, L.; Wu, Z.; Wang, S.; Zhao, Y.; et al. The relation between angioarchitectural factors of developmental venous anomaly and concomitant sporadic cavernous malformation. BMC Neurol. 2016, 16, 183. [Google Scholar] [CrossRef]
- Rigamonti, D.; Spetzler, R.F. The association of venous and cavernous malformations. Report of four cases and discussion of the pathophysiological, diagnostic, and therapeutic implications. Acta Neurochir. 1988, 92, 100–105. [Google Scholar] [CrossRef]
- Chen, B.; Herten, A.; Saban, D.; Rauscher, S.; Radbruch, A.; Schmidt, B.; Zhu, Y.; Jabbarli, R.; Wrede, K.H.; Kleinschnitz, C.; et al. Hemorrhage from cerebral cavernous malformations: The role of associated developmental venous anomalies. Neurology 2020, 95, e89–e96. [Google Scholar] [CrossRef]
- Mooney, M.A.; Zabramski, J.M. Developmental venous anomalies. Handb. Clin. Neurol. 2017, 143, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Brzegowy, K.; Kowalska, N.; Solewski, B.; Musiał, A.; Kasprzycki, T.; Herman-Sucharska, I.; Walocha, J.A. Prevalence and anatomical characteristics of developmental venous anomalies: An MRI study. Neuroradiology 2021, 63, 1001–1008. [Google Scholar] [CrossRef]
- Saba, P.R. The caput medusae sign. Radiology 1998, 207, 599–600. [Google Scholar] [CrossRef]
- Wurm, G.; Schnizer, M.; Fellner, F.A. Cerebral cavernous malformations associated with venous anomalies: Surgical considerations. Neurosurgery 2007, 61, 390–404; discussion 396–404. [Google Scholar] [CrossRef] [PubMed]
- Töpper, R.; Jürgens, E.; Reul, J.; Thron, A. Clinical significance of intracranial developmental venous anomalies. J. Neurol. Neurosurg. Psychiatry 1999, 67, 234–238. [Google Scholar] [CrossRef]
- Gross, B.A.; Du, R. Hemorrhage from cerebral cavernous malformations: A systematic pooled analysis. J. Neurosurg. 2017, 126, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Kondziolka, D.; Lunsford, L.D.; Kestle, J.R. The natural history of cerebral cavernous malformations. J. Neurosurg. 1995, 83, 820–824. [Google Scholar] [CrossRef]
- Horne, M.A.; Flemming, K.D.; Su, I.C.; Stapf, C.; Jeon, J.P.; Li, D.; Maxwell, S.S.; White, P.; Christianson, T.J.; Agid, R.; et al. Clinical course of untreated cerebral cavernous malformations: A meta-analysis of individual patient data. Lancet Neurol. 2016, 15, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, G.G.; Cofano, F.; Santonio, F.V.; Vincitorio, F.; Zenga, F.; Garbossa, D. Natural History, Clinical, and Surgical Management of Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 35–46. [Google Scholar] [CrossRef]
- Fontanella, M.M.; Zanin, L.; Fiorindi, A.; Spena, G.; Nicolosi, F.; Belotti, F.; Panciani, P.; Cornali, C.; Doglietto, F. Surgical Management of Brain Cavernous Malformations. Methods Mol. Biol. 2020, 2152, 109–128. [Google Scholar] [CrossRef]
- Fontanella, M.M.; Bacigaluppi, S.; Doglietto, F.; Zanin, L.; Agosti, E.; Panciani, P.; Belotti, F.; Saraceno, G.; Spena, G.; Draghi, R.; et al. An international call for a new grading system for cerebral and cerebellar cavernomas. J. Neurosurg. Sci. 2021, 65, 239–246. [Google Scholar] [CrossRef]
- Rinaldo, L.; Lanzino, G.; Flemming, K.D.; Krings, T.; Brinjikji, W. Symptomatic developmental venous anomalies. Acta Neurochir. (Wien) 2020, 162, 1115–1125. [Google Scholar] [CrossRef]
- McLaughlin, M.R.; Kondziolka, D.; Flickinger, J.C.; Lunsford, S.; Lunsford, L.D. The prospective natural history of cerebral venous malformations. Neurosurgery 1998, 43, 195–200; discussion 191–200. [Google Scholar] [CrossRef]
- Brinjikji, W.; Flemming, K.D.; Lanzino, G. De novo formation of a large cavernoma associated with a congenital torcular dural arteriovenous fistula: Case report. J. Neurosurg. Pediatr. 2017, 19, 567–570. [Google Scholar] [CrossRef]
- Kumar, S.; Lanzino, G.; Brinjikji, W.; Hocquard, K.W.; Flemming, K.D. Infratentorial Developmental Venous Abnormalities and Inflammation Increase Odds of Sporadic Cavernous Malformation. J. Stroke Cerebrovasc. Dis. 2019, 28, 1662–1667. [Google Scholar] [CrossRef]
- Idiculla, P.S.; Gurala, D.; Philipose, J.; Rajdev, K.; Patibandla, P. Cerebral Cavernous Malformations, Developmental Venous Anomaly, and Its Coexistence: A Review. Eur. Neurol. 2020, 83, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Bai, C.; Yu, T.; Wu, Z.; Liu, X.; Zhang, J.; Zhao, J. The association between cerebral developmental venous anomaly and concomitant cavernous malformation: An observational study using magnetic resonance imaging. BMC Neurol. 2014, 14, 50. [Google Scholar] [CrossRef]
- Nakase, K.; Motoyama, Y.; Nakai, T.; Takeshima, Y.; Nakagawa, I.; Park, Y.S.; Ohbayashi, C.; Nakase, H. Cavernous Malformation Associated With Arterialized Developmental Venous Anomaly: A Case Report. Neurosurgery 2017, 80, E257–E262. [Google Scholar] [CrossRef] [PubMed]
- Maeder, P.; Gudinchet, F.; Meuli, R.; de Tribolet, N. Development of a cavernous malformation of the brain. AJNR Am. J. Neuroradiol. 1998, 19, 1141–1143. [Google Scholar] [PubMed]
- Hong, Y.J.; Chung, T.S.; Suh, S.H.; Park, C.H.; Tomar, G.; Seo, K.D.; Kim, K.S.; Park, I.K. The angioarchitectural factors of the cerebral developmental venous anomaly; can they be the causes of concurrent sporadic cavernous malformation? Neuroradiology 2010, 52, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Zipfel, G.J.; Hildebolt, C.; Derdeyn, C.P. Hemodynamic effects of developmental venous anomalies with and without cavernous malformations. AJNR Am. J. Neuroradiol. 2013, 34, 1746–1751. [Google Scholar] [CrossRef] [PubMed]
- Maish, W.N. Developmental venous anomalies and brainstem cavernous malformations: A proposed physiological mechanism for haemorrhage. Neurosurg. Rev. 2019, 42, 663–670. [Google Scholar] [CrossRef]
- Brinjikji, W.; El-Masri, A.E.; Wald, J.T.; Flemming, K.D.; Lanzino, G. Prevalence of cerebral cavernous malformations associated with developmental venous anomalies increases with age. Childs Nerv. Syst. 2017, 33, 1539–1543. [Google Scholar] [CrossRef]
- Santucci, G.M.; Leach, J.L.; Ying, J.; Leach, S.D.; Tomsick, T.A. Brain parenchymal signal abnormalities associated with developmental venous anomalies: Detailed MR imaging assessment. AJNR Am. J. Neuroradiol. 2008, 29, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Akers, A.L.; Johnson, E.; Steinberg, G.K.; Zabramski, J.M.; Marchuk, D.A. Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two-hit mechanism of CCM pathogenesis. Hum. Mol. Genet. 2009, 18, 919–930. [Google Scholar] [CrossRef]
- McDonald, D.A.; Shenkar, R.; Shi, C.; Stockton, R.A.; Akers, A.L.; Kucherlapati, M.H.; Kucherlapati, R.; Brainer, J.; Ginsberg, M.H.; Awad, I.A.; et al. A novel mouse model of cerebral cavernous malformations based on the two-hit mutation hypothesis recapitulates the human disease. Hum. Mol. Genet. 2011, 20, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Trapani, E.; Retta, S.F. Cerebral cavernous malformation (CCM) disease: From monogenic forms to genetic susceptibility factors. J. Neurosurg. Sci. 2015, 59, 201–209. [Google Scholar] [PubMed]
- Retta, S.F.; Glading, A.J. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int. J. Biochem. Cell Biol. 2016, 81, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief 2018, 16, 929–938. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Antognelli, C.; Perrelli, A.; Armeni, T.; Nicola Talesa, V.; Retta, S.F. Dicarbonyl Stress and S-Glutathionylation in Cerebrovascular Diseases: A Focus on Cerebral Cavernous Malformations. Antioxidants 2020, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, A.; Retta, S.F. Polymorphisms in genes related to oxidative stress and inflammation: Emerging links with the pathogenesis and severity of Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2021, 172, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Pozzati, E.; Acciarri, N.; Tognetti, F.; Marliani, F.; Giangaspero, F. Growth, subsequent bleeding, and de novo appearance of cerebral cavernous angiomas. Neurosurgery 1996, 38, 662–669; discussion 669–670. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.J.; Ball, W.S.; Bove, K.E.; Crone, K.R.; Tew, J.M. Formation of intracerebral cavernous malformations after radiation treatment for central nervous system neoplasia in children. J. Neurosurg. 1998, 88, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Kondziolka, D.; Dempsey, P.K.; Lunsford, L.D. The case for conservative management of venous angiomas. Can. J. Neurol. Sci. 1991, 18, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Biller, J.; Toffol, G.J.; Shea, J.F.; Fine, M.; Azar-Kia, B. Cerebellar venous angiomas. A continuing controversy. Arch. Neurol. 1985, 42, 367–370. [Google Scholar] [CrossRef]
- Rigamonti, D.; Spetzler, R.F.; Medina, M.; Rigamonti, K.; Geckle, D.S.; Pappas, C. Cerebral venous malformations. J. Neurosurg. 1990, 73, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Rigamonti, D.; Vivas-Buitrago, T. A brief history of cerebral cavernous malformations: A personal perspective. Vessel. Plus 2021, 5, 47. [Google Scholar] [CrossRef]
- Rigamonti, D.; Hadley, M.N.; Drayer, B.P.; Johnson, P.C.; Hoenig-Rigamonti, K.; Knight, J.T.; Spetzler, R.F. Cerebral cavernous malformations. Incidence and familial occurrence. N. Engl. J. Med. 1988, 319, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Rabinstein, A.A.; Flemming, K.D. Cavernous malformations with DVA: Hold those knives. Neurology 2020, 95, 13–14. [Google Scholar] [CrossRef]
- Bacigaluppi, S.; Retta, S.F.; Pileggi, S.; Fontanella, M.; Goitre, L.; Tassi, L.; La Camera, A.; Citterio, A.; Patrosso, M.C.; Tredici, G.; et al. Genetic and cellular basis of cerebral cavernous malformations: Implications for clinical management. Clin. Genet. 2013, 83, 7–14. [Google Scholar] [CrossRef]
- Clatterbuck, R.E.; Eberhart, C.G.; Crain, B.J.; Rigamonti, D. Ultrastructural and immunocytochemical evidence that an incompetent blood-brain barrier is related to the pathophysiology of cavernous malformations. J. Neurol. Neurosurg. Psychiatry 2001, 71, 188–192. [Google Scholar] [CrossRef]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Delle Monache, S.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced S-Glutathionylation of Distinct Structural and Regulatory Proteins. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef]
- De Luca, E.; Perrelli, A.; Swamy, H.; Nitti, M.; Passalacqua, M.; Furfaro, A.L.; Salzano, A.M.; Scaloni, A.; Glading, A.J.; Retta, S.F. Protein kinase Cα regulates the nucleocytoplasmic shuttling of KRIT1. J. Cell Sci. 2021, 134, jcs250217. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Retta, S.F.; Pinton, P. Cellular processes underlying cerebral cavernous malformations: Autophagy as another point of view. Autophagy 2016, 12, 424–425. [Google Scholar] [CrossRef]
- De Luca, E.; Pedone, D.; Moglianetti, M.; Pulcini, D.; Perrelli, A.; Retta, S.F.; Pompa, P.P. Multifunctional Platinum@BSA-Rapamycin Nanocarriers for the Combinatorial Therapy of Cerebral Cavernous Malformation. ACS Omega 2018, 3, 15389–15398. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ramirez, M.A.; Lai, C.C.; Soliman, S.I.; Hale, P.; Pham, A.; Estrada, E.J.; McCurdy, S.; Girard, R.; Verma, R.; Moore, T.; et al. Astrocytes propel neurovascular dysfunction during cerebral cavernous malformation lesion formation. J. Clin. Invest. 2021, 131, e139570. [Google Scholar] [CrossRef]
- Hulsmans, M.; Holvoet, P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J. Cell Mol. Med. 2010, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, T.; Patel, M. Crosstalk between neuroinflammation and oxidative stress in epilepsy. Front. Cell Dev. Biol. 2022, 10, 976953. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Miller, A.A.; Drummond, G.R.; Kemp-Harper, B.K.; Sobey, C.G. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front. Biosci. 2011, 16, 1733–1745. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.; Coleman, P.; Chen, J.; Ting, K.K.; Choi, J.P.; Zheng, X.; Vadas, M.A.; Gamble, J.R. Low fluid shear stress conditions contribute to activation of cerebral cavernous malformation signalling pathways. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165519. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Banjara, M.; Ghosh, C. Sterile Neuroinflammation and Strategies for Therapeutic Intervention. Int. J. Inflam. 2017, 2017, 8385961. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Aimaretti, E.; Ferreira Alves, G.; Cento, A.S.; Fornelli, C.; Dal Bello, F.; Ferraris, C.; Goitre, L.; Perrelli, A.; Retta, S.F. Heterozygous Loss of KRIT1 in Mice Affects Metabolic Functions of the Liver, Promoting Hepatic Oxidative and Glycative Stress. Int. J. Mol. Sci. 2022, 23, 1151. [Google Scholar] [CrossRef] [PubMed]
- Ruíz, D.S.; Yilmaz, H.; Gailloud, P. Cerebral developmental venous anomalies: Current concepts. Ann. Neurol. 2009, 66, 271–283. [Google Scholar] [CrossRef]
- San Millán Ruíz, D.; Gailloud, P. Cerebral developmental venous anomalies. Childs Nerv. Syst. 2010, 26, 1395–1406. [Google Scholar] [CrossRef]
- Jung, H.N.; Kim, S.T.; Cha, J.; Kim, H.J.; Byun, H.S.; Jeon, P.; Kim, K.H.; Kim, B.J. Diffusion and perfusion MRI findings of the signal-intensity abnormalities of brain associated with developmental venous anomaly. AJNR Am. J. Neuroradiol. 2014, 35, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Iv, M.; Fischbein, N.J.; Zaharchuk, G. Association of developmental venous anomalies with perfusion abnormalities on arterial spin labeling and bolus perfusion-weighted imaging. J. Neuroimaging 2015, 25, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Umino, M.; Maeda, M.; Matsushima, N.; Matsuura, K.; Yamada, T.; Sakuma, H. High-signal-intensity abnormalities evaluated by 3D fluid-attenuated inversion recovery imaging within the drainage territory of developmental venous anomalies identified by susceptibility-weighted imaging at 3 T. Jpn. J. Radiol. 2014, 32, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Larvie, M.; Timerman, D.; Thum, J.A. Brain metabolic abnormalities associated with developmental venous anomalies. AJNR Am. J. Neuroradiol. 2015, 36, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Lazor, J.W.; Schmitt, J.E.; Loevner, L.A.; Nabavizadeh, S.A. Metabolic Changes of Brain Developmental Venous Anomalies on. Acad. Radiol. 2019, 26, 443–449. [Google Scholar] [CrossRef]
- Condon, L.; Blazak, J. Focal 18F-DOPA Uptake in Brain Parenchyma Surrounding Developmental Venous Anomalies. Clin. Nucl. Med. 2018, 43, e37–e38. [Google Scholar] [CrossRef]
- Qu, D.; Wang, L.; Huo, M.; Song, W.; Lau, C.W.; Xu, J.; Xu, A.; Yao, X.; Chiu, J.J.; Tian, X.Y.; et al. Focal TLR4 activation mediates disturbed flow-induced endothelial inflammation. Cardiovasc. Res. 2020, 116, 226–236. [Google Scholar] [CrossRef]
- Colombo, P.C.; Onat, D.; Harxhi, A.; Demmer, R.T.; Hayashi, Y.; Jelic, S.; LeJemtel, T.H.; Bucciarelli, L.; Kebschull, M.; Papapanou, P.; et al. Peripheral venous congestion causes inflammation, neurohormonal, and endothelial cell activation. Eur. Heart J. 2014, 35, 448–454. [Google Scholar] [CrossRef]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Zietek, T.; Rath, E. Inflammation Meets Metabolic Disease: Gut Feeling Mediated by GLP-1. Front. Immunol. 2016, 7, 154. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.M.; Peckham, M.E.; Shah, L.M.; Wiggins, R.H. Association of Developmental Venous Anomalies with Demyelinating Lesions in Patients with Multiple Sclerosis. AJNR Am. J. Neuroradiol. 2018, 39, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Haacke, E.M.; Ge, Y.; Sethi, S.K.; Buch, S.; Zamboni, P. An Overview of Venous Abnormalities Related to the Development of Lesions in Multiple Sclerosis. Front. Neurol. 2021, 12, 561458. [Google Scholar] [CrossRef] [PubMed]
- Dammann, P.; Wrede, K.; Zhu, Y.; Matsushige, T.; Maderwald, S.; Umutlu, L.; Quick, H.H.; Hehr, U.; Rath, M.; Ladd, M.E.; et al. Correlation of the venous angioarchitecture of multiple cerebral cavernous malformations with familial or sporadic disease: A susceptibility-weighted imaging study with 7-Tesla MRI. J. Neurosurg. 2017, 126, 570–577. [Google Scholar] [CrossRef]
- Frantz, S.; Vincent, K.A.; Feron, O.; Kelly, R.A. Innate immunity and angiogenesis. Circ. Res. 2005, 96, 15–26. [Google Scholar] [CrossRef]
- Wu, C.; Li, F.; Niu, G.; Chen, X. PET imaging of inflammation biomarkers. Theranostics 2013, 3, 448–466. [Google Scholar] [CrossRef]
- Sala, Q.; Metellus, P.; Taieb, D.; Kaphan, E.; Figarella-Branger, D.; Guedj, E. 18F-DOPA, a clinically available PET tracer to study brain inflammation? Clin. Nucl. Med. 2014, 39, e283–e285. [Google Scholar] [CrossRef]
- Kang, Y.; Mozley, P.D.; Verma, A.; Schlyer, D.; Henchcliffe, C.; Gauthier, S.A.; Chiao, P.C.; He, B.; Nikolopoulou, A.; Logan, J.; et al. Noninvasive PK11195-PET Image Analysis Techniques Can Detect Abnormal Cerebral Microglial Activation in Parkinson’s Disease. J. Neuroimaging 2018, 28, 496–505. [Google Scholar] [CrossRef]
- Tronel, C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Dupont, A.C.; Arlicot, N. Molecular Targets for PET Imaging of Activated Microglia: The Current Situation and Future Expectations. Int. J. Mol. Sci. 2017, 18, 802. [Google Scholar] [CrossRef]
- Zhu, F.D.; Hu, Y.J.; Yu, L.; Zhou, X.G.; Wu, J.M.; Tang, Y.; Qin, D.L.; Fan, Q.Z.; Wu, A.G. Nanoparticles: A Hope for the Treatment of Inflammation in CNS. Front. Pharmacol. 2021, 12, 683935. [Google Scholar] [CrossRef]
- Perrelli, A.; Fatehbasharzad, P.; Benedetti, V.; Ferraris, C.; Fontanella, M.; De Luca, E.; Moglianetti, M.; Battaglia, L.; Retta, S.F. Towards precision nanomedicine for cerebrovascular diseases with emphasis on Cerebral Cavernous Malformation (CCM). Expert. Opin. Drug Deliv. 2021, 18, 849–876. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bianconi, A.; Salvati, L.F.; Perrelli, A.; Ferraris, C.; Massara, A.; Minardi, M.; Aruta, G.; Rosso, M.; Massa Micon, B.; Garbossa, D.; et al. Distant Recurrence of a Cerebral Cavernous Malformation in the Vicinity of a Developmental Venous Anomaly: Case Report of Local Oxy-Inflammatory Events. Int. J. Mol. Sci. 2022, 23, 14643. https://doi.org/10.3390/ijms232314643
Bianconi A, Salvati LF, Perrelli A, Ferraris C, Massara A, Minardi M, Aruta G, Rosso M, Massa Micon B, Garbossa D, et al. Distant Recurrence of a Cerebral Cavernous Malformation in the Vicinity of a Developmental Venous Anomaly: Case Report of Local Oxy-Inflammatory Events. International Journal of Molecular Sciences. 2022; 23(23):14643. https://doi.org/10.3390/ijms232314643
Chicago/Turabian StyleBianconi, Andrea, Luca Francesco Salvati, Andrea Perrelli, Chiara Ferraris, Armando Massara, Massimiliano Minardi, Gelsomina Aruta, Miriam Rosso, Barbara Massa Micon, Diego Garbossa, and et al. 2022. "Distant Recurrence of a Cerebral Cavernous Malformation in the Vicinity of a Developmental Venous Anomaly: Case Report of Local Oxy-Inflammatory Events" International Journal of Molecular Sciences 23, no. 23: 14643. https://doi.org/10.3390/ijms232314643
APA StyleBianconi, A., Salvati, L. F., Perrelli, A., Ferraris, C., Massara, A., Minardi, M., Aruta, G., Rosso, M., Massa Micon, B., Garbossa, D., & Retta, S. F. (2022). Distant Recurrence of a Cerebral Cavernous Malformation in the Vicinity of a Developmental Venous Anomaly: Case Report of Local Oxy-Inflammatory Events. International Journal of Molecular Sciences, 23(23), 14643. https://doi.org/10.3390/ijms232314643