
cNTnC and fYTnC2, Genetically Encoded Green Calcium Indicators Based on Troponin C from Fast Animals

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
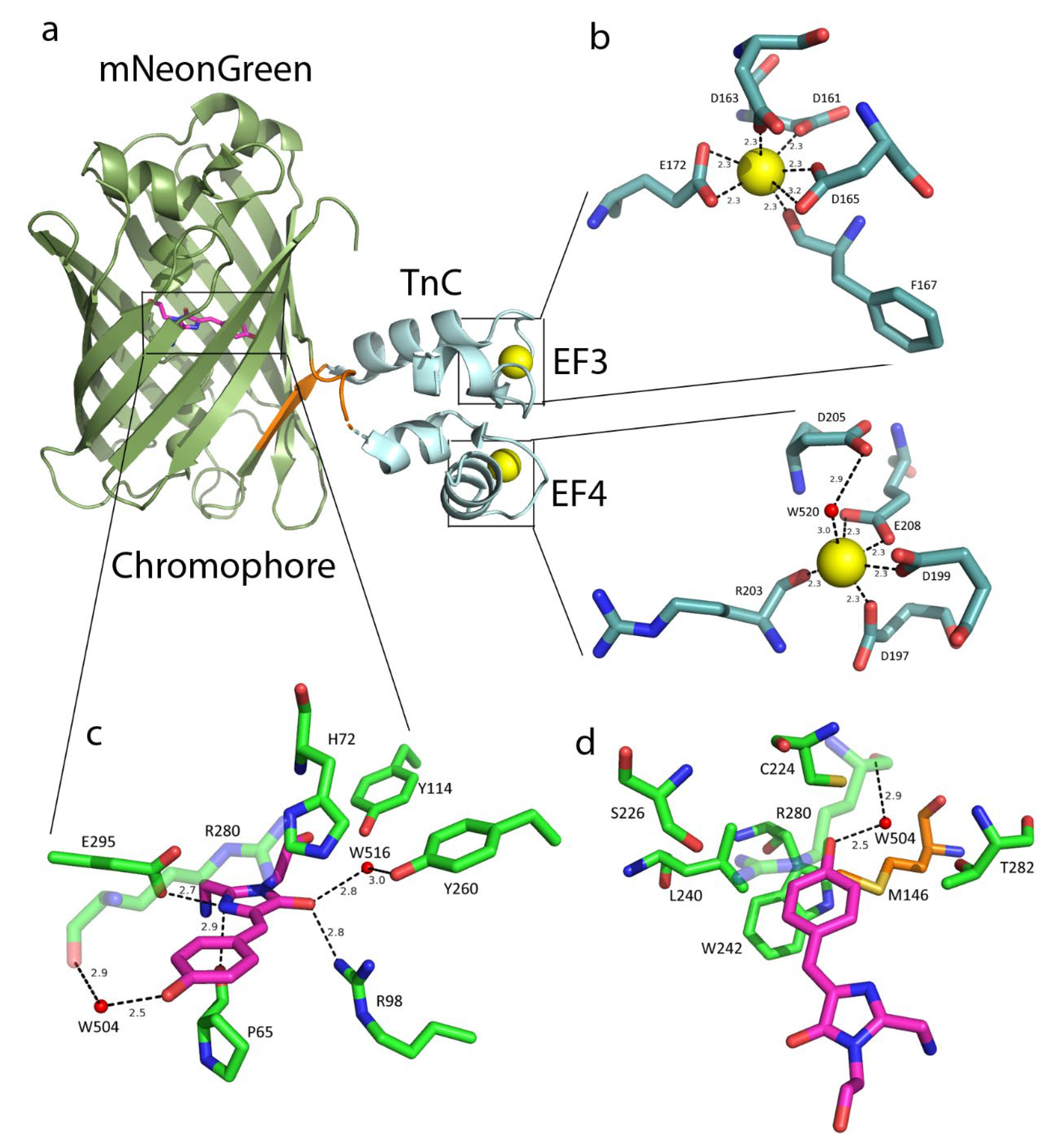
2.1. NTnC Crystal Structure
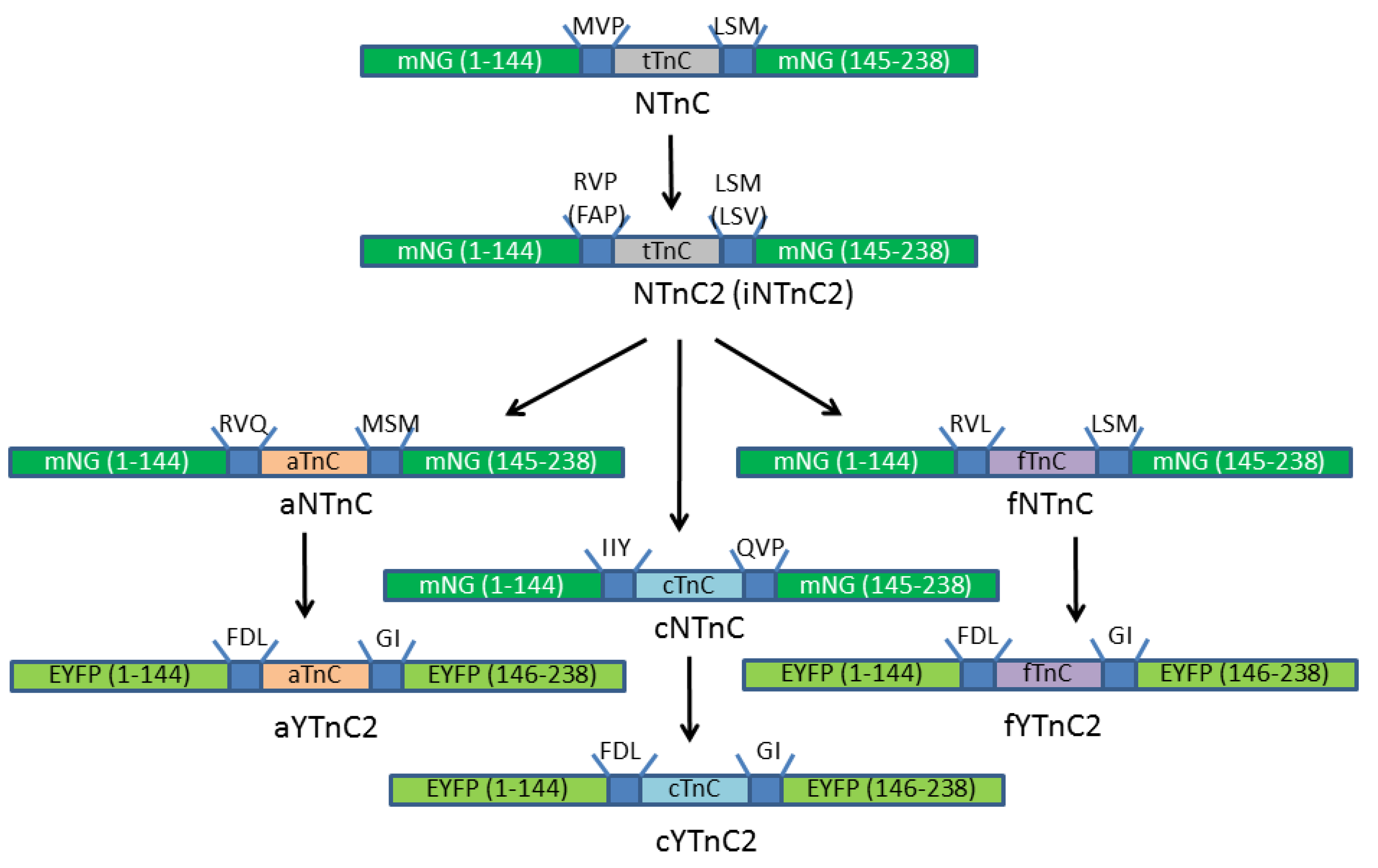
2.2. Development and Characterization of NTnC2 with Troponin C from Toadfish
2.3. Development and Characterization of Calcium Indicators Based on mNeonGreen and Troponin C from Fasting Animals
2.4. Engineering of Calcium Indicators Composed of EYFP and Truncated Troponins C from Fast Animals
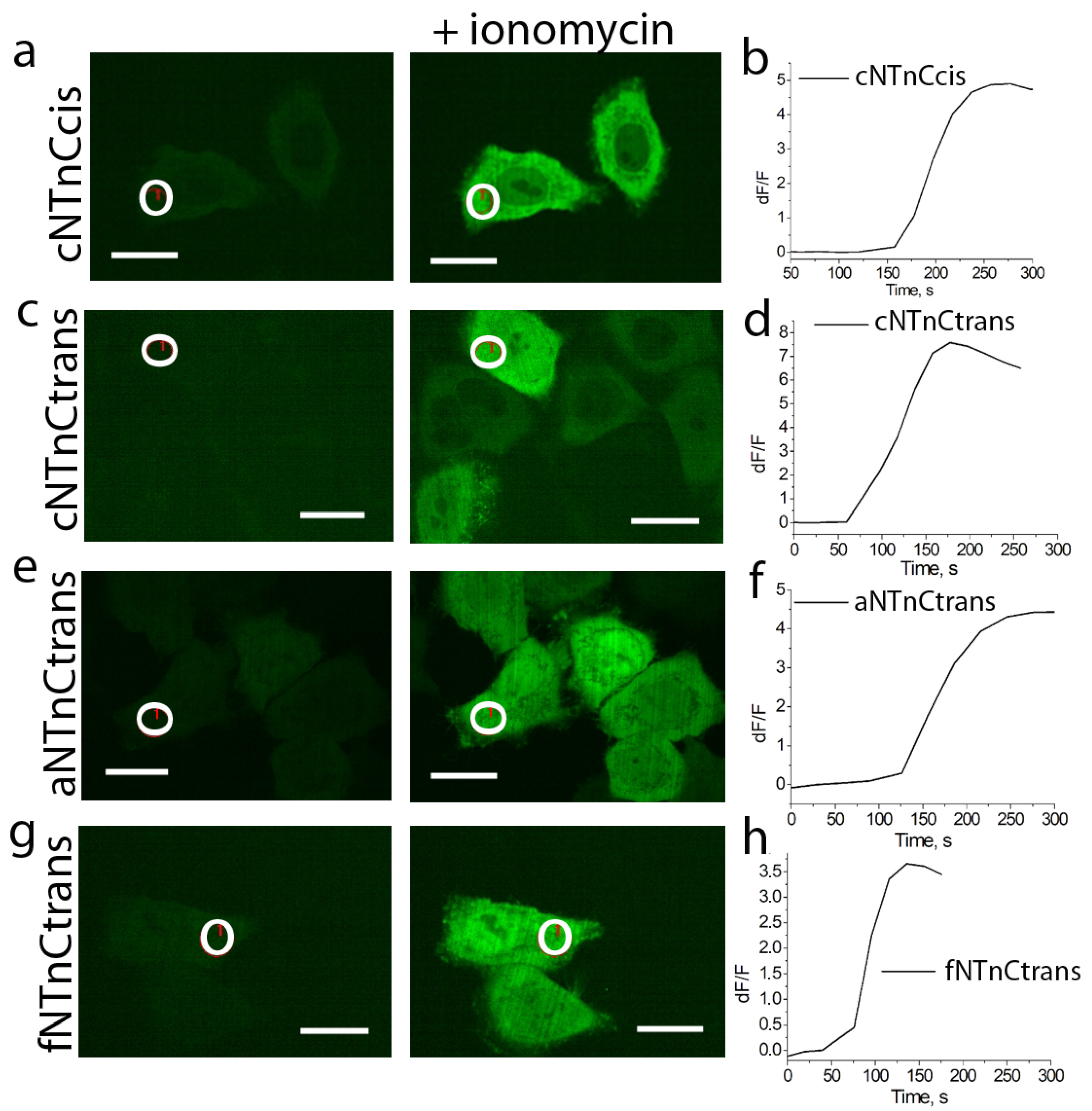
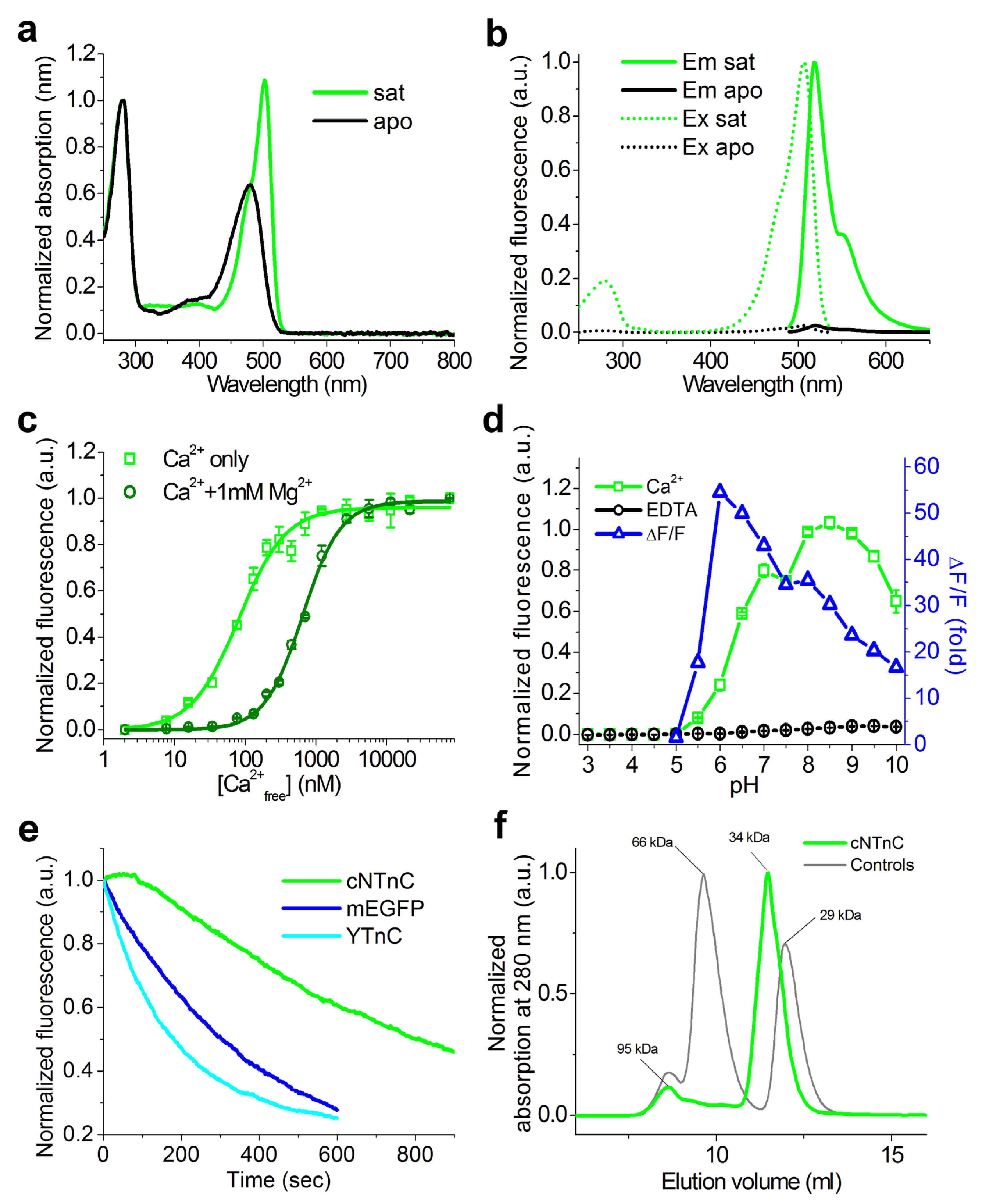
2.5. Properties of cNTnC and fYTnC2 GECIs In Vitro
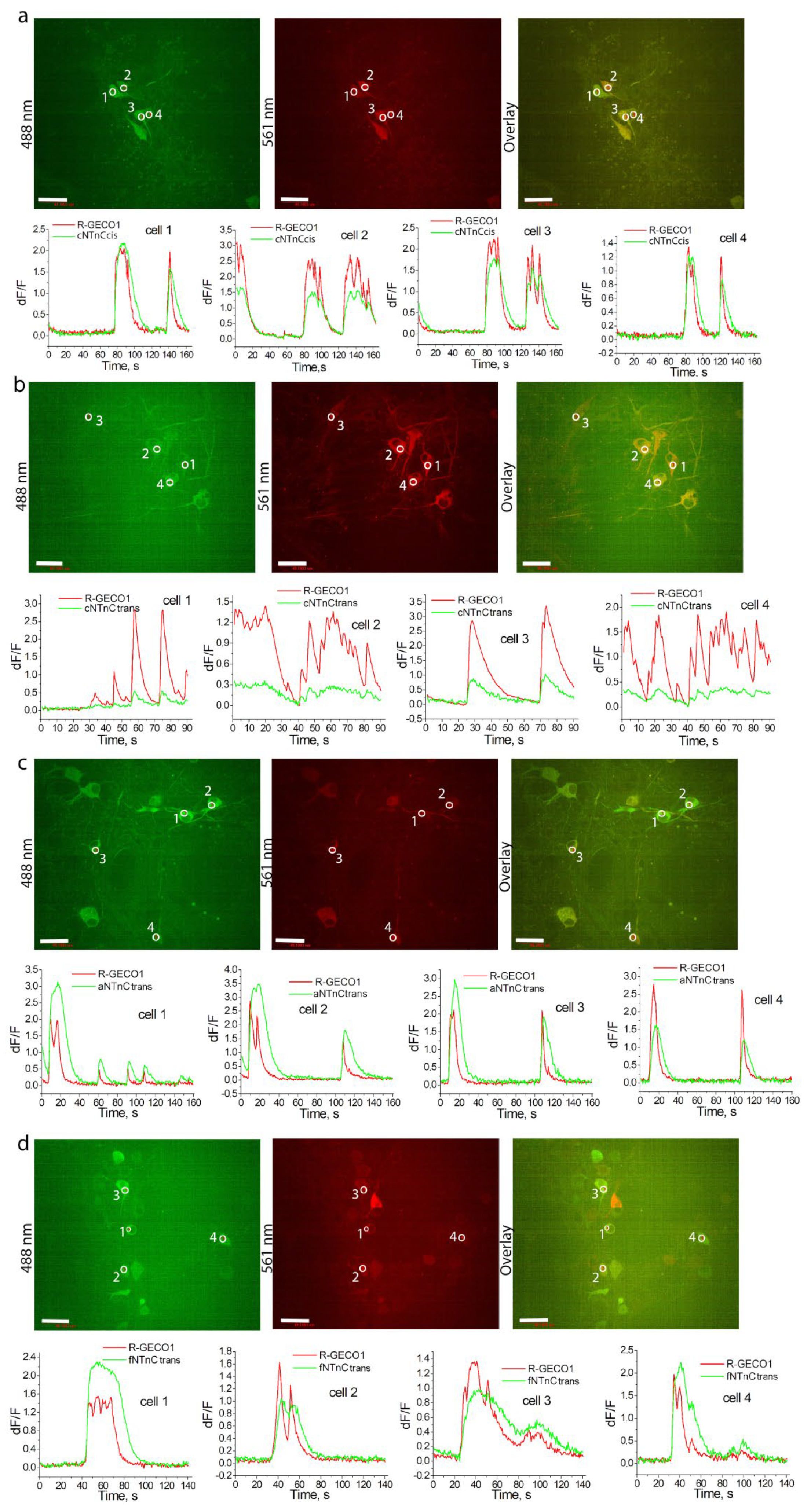
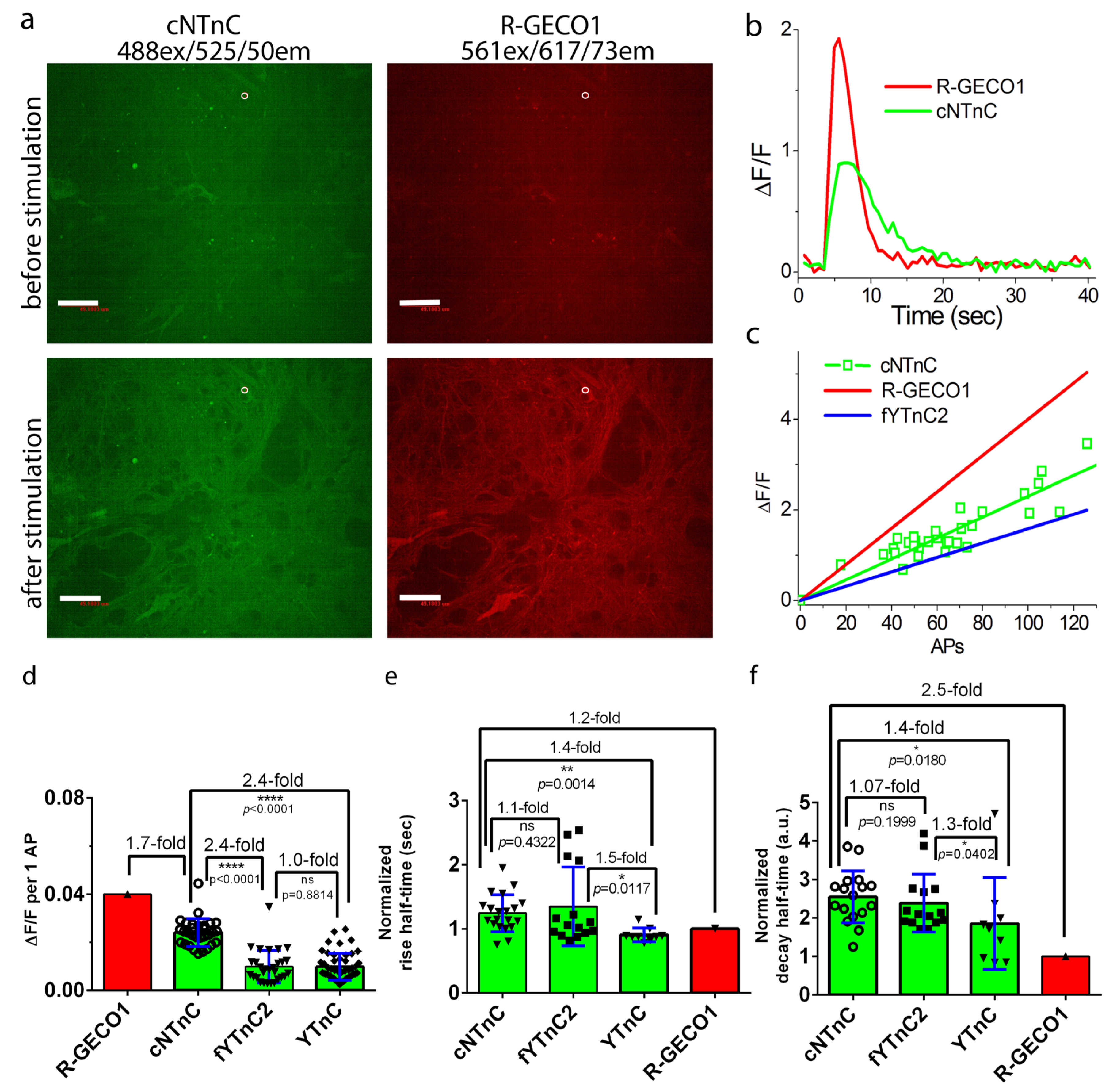
2.6. Validation of cNTnC and fYTnC2 Indicators in Neuronal Cultures
3. Materials and Methods
3.1. Protein Crystallization
3.2. Data Collection, Processing, Structure Solution, and Refinement
3.3. Structure Analysis and Validation
3.4. Cloning of Bacterial Vectors, Mutagenesis, and Library Screening
3.5. Protein Purification and Characterization
3.6. Mammalian Plasmid Construction
3.7. Mammalian Live-Cell Imaging
3.8. Imaging in Primary Mouse Neuronal Cultures
3.9. Statistics
3.10. Ethical Approval and Animal Care
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| RFP | Red fluorescent protein |
| LSSRFP | Red fluorescent protein with large Stokes shift |
| FP | Fluorescent protein |
| PBS | Phosphate-buffered saline |
| QY | Quantum yield |
| SD | Standard deviation |
References
- Piatkevich, K.D.; Murdock, M.H.; Subach, F.V. Advances in Engineering and Application of Optogenetic Indicators for Neuroscience. Appl. Sci. 2019, 9, 562. [Google Scholar] [CrossRef]
- Thestrup, T.; Litzlbauer, J.; Bartholomäus, I.; Mues, M.; Russo, L.; Dana, H.; Kovalchuk, Y.; Liang, Y.; Kalamakis, G.; Laukat, Y.; et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat. Methods 2014, 11, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Goltstein, P.M.; Portugues, R.; Griesbeck, O. Putting a finishing touch on GECIs. Front. Mol. Neurosci. 2014, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Tallini, Y.N.; Ohkura, M.; Choi, B.-R.; Ji, G.; Imoto, K.; Doran, R.; Lee, J.; Plan, P.; Wilson, J.; Xin, H.-B.; et al. Imaging cellular signals in the heart in vivo: Cardiac expression of the high-signal Ca2+ indicator GCaMP2. Proc. Natl. Acad. Sci. USA 2006, 103, 4753–4758. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Hires, S.A.; Mao, T.; Huber, D.; Chiappe, M.E.; Chalasani, S.H.; Petreanu, L.; Akerboom, J.; McKinney, S.A.; Schreiter, E.; et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 2009, 6, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Resendez, S.L.; Jennings, J.H.; Ung, R.L.; Namboodiri, V.M.K.; Zhou, C.Z.; Otis, J.M.; Nomura, H.; McHenry, J.A.; Kosyk, O.; Stuber, G.D. Visualization of cortical, subcortical and deep brain neural circuit dynamics during naturalistic mammalian behavior with head-mounted microscopes and chronically implanted lenses. Nat. Protoc. 2016, 11, 566–597. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9, 1504. [Google Scholar] [CrossRef]
- Steinmetz, N.A.; Buetfering, C.; Lecoq, J.; Lee, C.R.; Peters, A.J.; Jacobs, E.A.K.; Coen, P.; Ollerenshaw, D.R.; Valley, M.T.; de Vries, S.E.J.; et al. Aberrant Cortical Activity in Multiple GCaMP6-Expressing Transgenic Mouse Lines. eNeuro 2017, 4, 1–15. [Google Scholar] [CrossRef]
- Barykina, N.; Subach, O.M.; Doronin, D.A.; Sotskov, V.; Roshchina, M.A.; Kunitsyna, T.A.; Malyshev, A.; Smirnov, I.V.; Azieva, A.; Sokolov, I.S.; et al. A new design for a green calcium indicator with a smaller size and a reduced number of calcium-binding sites. Sci. Rep. 2016, 6, 34447. [Google Scholar] [CrossRef]
- Barykina, N.V.; Doronin, D.A.; Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Ivleva, O.A.; Gruzdeva, A.M.; Kunitsyna, T.A.; Ivashkina, O.I.; Lazutkin, A.A.; et al. NTnC-like genetically encoded calcium indicator with a positive and enhanced response and fast kinetics. Sci. Rep. 2018, 8, 15233. [Google Scholar] [CrossRef]
- Zhao, Y.; Araki, S.; Wu, J.; Teramoto, T.; Chang, Y.F.; Nakano, M.; Abdelfattah, A.S.; Fujiwara, M.; Ishihara, T.; Nagai, T.; et al. An expanded palette of genetically encoded Ca2+ indicators. Science 2011, 333, 1888–1891. [Google Scholar] [CrossRef]
- Subach, O.M.; Barykina, N.V.; Chefanova, E.S.; Vlaskina, A.V.; Sotskov, V.P.; Ivashkina, O.I.; Anokhin, K.V.; Subach, F.V. FRCaMP, A Red Fluorescent Genetically Encoded Calcium Indicator Based on Calmodulin from Schizosaccharomyces Pombe Fungus. Int. J. Mol. Sci. 2020, 22, 111. [Google Scholar] [CrossRef]
- Subach, O.M.; Sotskov, V.P.; Plusnin, V.V.; Gruzdeva, A.M.; Barykina, N.V.; Ivashkina, O.I.; Anokhin, K.V.; Nikolaeva, A.Y.; Korzhenevskiy, D.A.; Vlaskina, A.V.; et al. Novel Genetically Encoded Bright Positive Calcium Indicator NCaMP7 Based on the mNeonGreen Fluorescent Protein. Int. J. Mol. Sci. 2020, 21, 1644. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Grabe, M.; Oster, G. Regulation of Organelle Acidity. J. Gen. Physiol. 2001, 117, 329–344. [Google Scholar] [CrossRef]
- Brocard, J.; Rajdev, S.; Reynolds, I.J. Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron 1993, 11, 751–757. [Google Scholar] [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Shaner, N.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Svetogorov, R.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA Diffraction Beamline for Studying Crystalline Samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Evans, P. Scaling and assessment of data quality. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Collaborative Computational Project. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Struct. Biol. Crystallogr. 2004, D60, 2126–2132. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Subach, O.M.; Vlaskina, A.V.; Agapova, Y.K.; Dorovatovskii, P.V.; Nikolaeva, A.Y.; Ivashkina, O.I.; Popov, V.O.; Piatkevich, K.D.; Khrenova, M.G.; Smirnova, T.A.; et al. LSSmScarlet, dCyRFP2s, dCyOFP2s and CRISPRed2s, Genetically Encoded Red Fluorescent Proteins with a Large Stokes Shift. Int. J. Mol. Sci. 2021, 22, 12887. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Indicator | Kd, nM | dF/F | Kd, hM (Mg2+) | dF/F (Mg2+) | kon obs at 300 nM Ca2+, s−1 a | t1/2off, s | koff, s−1 | dF/F Relative to RGECO1 in Neuronal Culture |
|---|---|---|---|---|---|---|---|---|
| R-GECO1 | 482 b | 15 b | 1138 ± 43 c | 21.0 ± 0.2 c | ND | ND | 0.752 b | 1.0 |
| GCaMP6s | 144 ± 3 d | 43 d | 227.3 ± 0.2 d | 46 d | 0.49 ± 0.05 d | 1.01 c | 0.69 ± 0.01 d | 3.2 ± 2 e |
| NTnC2 | 1408 ± 51 | 90 | 1290 ± 40 | 48 | 0.06 | 3.84 | 0.194 ± 0.001 | 0.95 ± 0.31 |
| iNTnC2 | 49 ± 1 | 30 | 30 ± 1 | 10 | ND | 14.2 | 0.044 ± 0.002 | 0.12 ± 0.06 |
| Indicator | Purified Proteins | HeLa Cells | Neuronal Culture | ||
|---|---|---|---|---|---|
| ΔF/F | Kd, nM [Hill] a | ΔF/F | ΔF/F, Versus R-GECO1 | t1/2off, Versus R-GECO1 | |
| fNTnCtrans | 20 | 502 [1.9] | 9.3 ± 4.9 | 0.5 ± 0.1 | 3.3 ± 1.5 |
| fNTnCcis | 30 | 288 [2.2] | 1.7 ± 0.5 | ND | ND |
| cNTnCtrans | 64 | 571 [1.8] | 11.4 ± 3.2 | 0.2 ± 0.1 | 1.9 ± 0.3 |
| cNTnCcis | 28 | 556 [1.72] | 6.9 ± 0.6 | 0.8 ± 0.2 | 2.4 ± 0.5 |
| aNTnCtrans | 33 | 558 [1.7] | 4.4 ±2.2 | 0.8 ± 0.3 | 4.5 ± 1.6 |
| aNTnCcis | 137 | 721 [2.4] | 0.8 ± 0.2 | ND | ND |
| Indicator | Purified Proteins | HeLa Cells | |
|---|---|---|---|
| ΔF/F | Kd, nM a | ΔF/F | |
| fYTnC2 | 17 | 709 | 4.0 ± 1.5 |
| cYTnC2 | 4 | 564 | 2.3 ± 0.2 |
| aYTnC2 | 10 | 620 | 0.8 ± 0.1 |
| Properties | Proteins | |||||
|---|---|---|---|---|---|---|
| cNTnC | YTnC a | |||||
| Apo | Sat | Apo | Sat | |||
| Absorption maximum (nm) | 480 | 503 | 413 | 495 (405) | ||
| Excitation maximum (nm) | 506 | 506 | 412, 501 | 502 (413) | ||
| Emission maximum (nm) | 520 | 518 | 514 | 516 (516) | ||
| Quantum yield b | 0.021 ± 0.004 | 0.53 ± 0.03 | 0.012 | 0.19 (0.03) | ||
| ε (mM−1 cm−1) c | 53.8 ± 4.8 | 82.3 ± 3.0 | 28 ± 2 | 29 ± 3 (20 ± 2) | ||
| Brightness vs. EGFP (%) d | 3.4 | 130 | 1 | 17 (2) | ||
| ΔF/F | Purified protein | 0 mM Mg2+ | 38.3 ± 7.2 | 10.6 ± 0.4 | ||
| 1 mM Mg2+ | 19.4 ± 0.8 | 2.9 ± 0.2 | ||||
| HeLa cells | 4.9 ± 1.4 | 2.0 ± 0.4 | ||||
| pKa | 7.36 ± 0.24 | 6.24 ± 0.01 7.80 ± 0.04 | 5.2 ± 0.1, 8.2 ± 0.1 | 6.3 ± 0.1 | ||
| Kd (nM) e | 0 mM Mg2+ | 81 ± 6 [n = 1.3 ± 0.1] | 223 ± 10 [n = 1.4 ± 0.1] | |||
| 1 mM Mg2+ | 651 ± 22 [n = 1.6 ± 0.1] | 410 ± 19 [n = 1.7 ± 0.2] | ||||
| Maturation half-time (min) f | 76 | 16 | ||||
| Photobleaching half-time (s) g | 803 ± 90 | 193 ± 36 * | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subach, O.M.; Vlaskina, A.V.; Agapova, Y.K.; Korzhenevskiy, D.A.; Nikolaeva, A.Y.; Varizhuk, A.M.; Subach, M.F.; Patrushev, M.V.; Piatkevich, K.D.; Boyko, K.M.; et al. cNTnC and fYTnC2, Genetically Encoded Green Calcium Indicators Based on Troponin C from Fast Animals. Int. J. Mol. Sci. 2022, 23, 14614. https://doi.org/10.3390/ijms232314614
Subach OM, Vlaskina AV, Agapova YK, Korzhenevskiy DA, Nikolaeva AY, Varizhuk AM, Subach MF, Patrushev MV, Piatkevich KD, Boyko KM, et al. cNTnC and fYTnC2, Genetically Encoded Green Calcium Indicators Based on Troponin C from Fast Animals. International Journal of Molecular Sciences. 2022; 23(23):14614. https://doi.org/10.3390/ijms232314614
Chicago/Turabian StyleSubach, Oksana M., Anna V. Vlaskina, Yuliya K. Agapova, Dmitriy A. Korzhenevskiy, Alena Y. Nikolaeva, Anna M. Varizhuk, Maksim F. Subach, Maxim V. Patrushev, Kiryl D. Piatkevich, Konstantin M. Boyko, and et al. 2022. "cNTnC and fYTnC2, Genetically Encoded Green Calcium Indicators Based on Troponin C from Fast Animals" International Journal of Molecular Sciences 23, no. 23: 14614. https://doi.org/10.3390/ijms232314614
APA StyleSubach, O. M., Vlaskina, A. V., Agapova, Y. K., Korzhenevskiy, D. A., Nikolaeva, A. Y., Varizhuk, A. M., Subach, M. F., Patrushev, M. V., Piatkevich, K. D., Boyko, K. M., & Subach, F. V. (2022). cNTnC and fYTnC2, Genetically Encoded Green Calcium Indicators Based on Troponin C from Fast Animals. International Journal of Molecular Sciences, 23(23), 14614. https://doi.org/10.3390/ijms232314614