
Role of Proteostasis Regulation in the Turnover of Stress Granules


Abstract
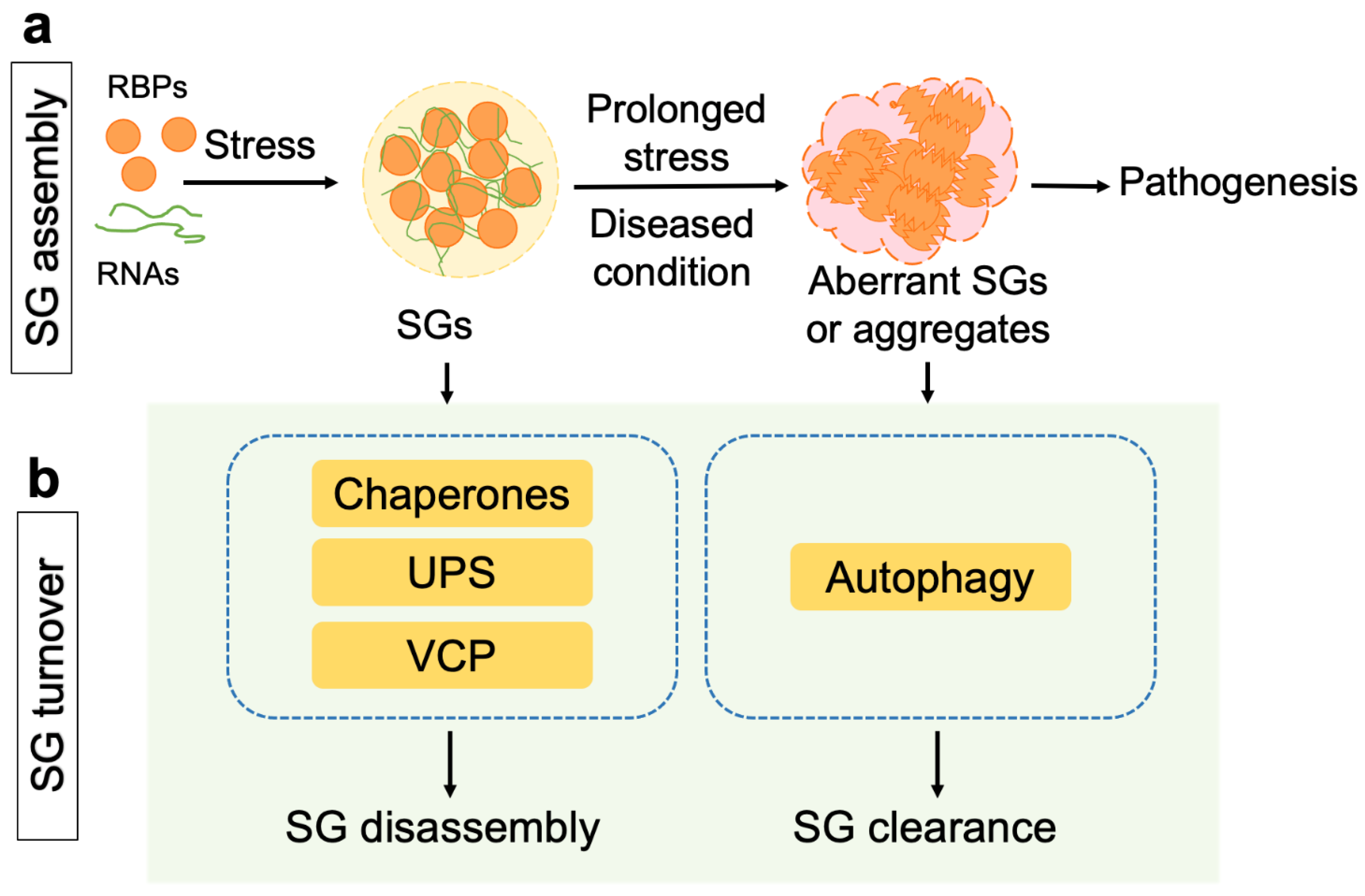
1. Introduction
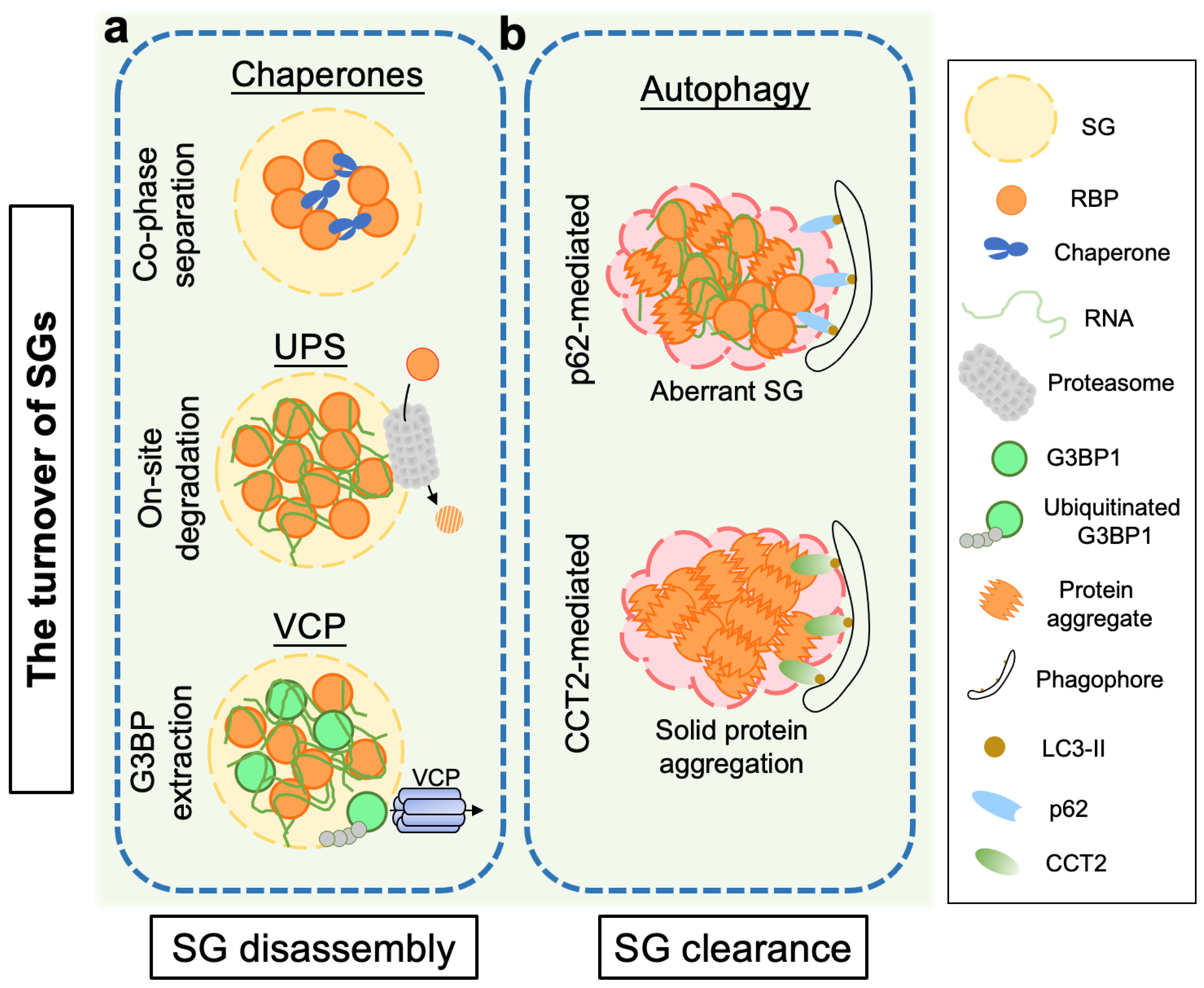
2. Proteostasis Regulation in the Disassembly of Stress Granules
2.1. Molecular Chaperones
2.2. The Ubiquitin-Proteasome System (UPS)
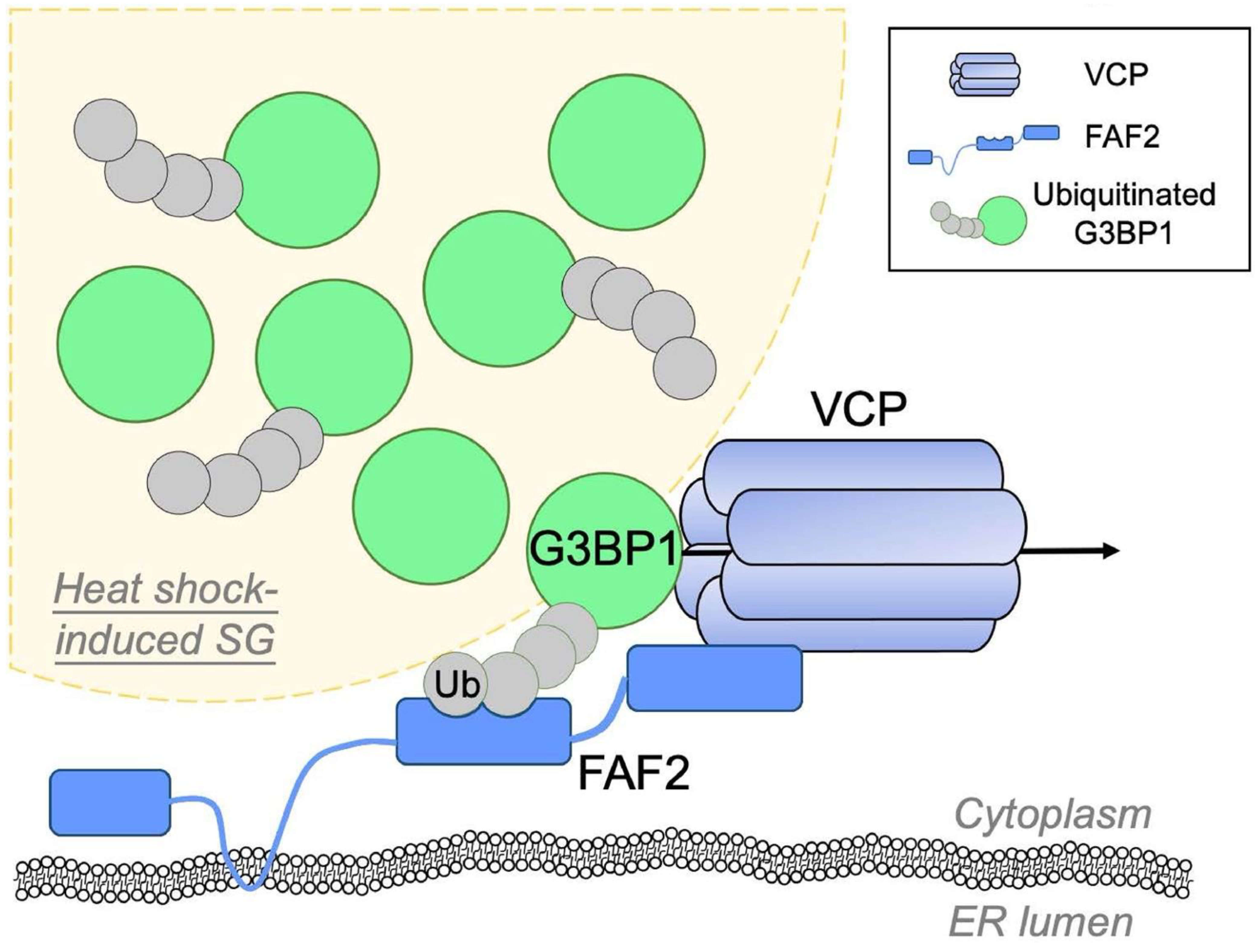
2.3. Valosin-Containing Protein (VCP)/p97
3. Clearance of Stress Granules and Aggregates via the Autophagy Pathway
3.1. Autophagy
3.2. p62/Sequestosome 1 (SQSTM1)
3.3. Chaperonin-Containing TCP-1 Subunit 2 (CCT2)
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Glauninger, H.; Wong Hickernell, C.J.; Bard, J.A.M.; Drummond, D.A. Stressful steps: Progress and challenges in understanding stress-induced mRNA condensation and accumulation in stress granules. Mol. Cell 2022, 82, 2544–2556. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Hofweber, M.; Dormann, D. Friend or foe-Post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem. 2019, 294, 7137–7150. [Google Scholar] [CrossRef]
- Wolozin, B.; Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019, 20, 649–666. [Google Scholar] [CrossRef]
- Krause, L.J.; Herrera, M.G.; Winklhofer, K.F. The Role of Ubiquitin in Regulating Stress Granule Dynamics. Front. Physiol. 2022, 13, 910759. [Google Scholar] [CrossRef]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032813. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, J.; Sun, Q. Aberrant Stress Granule Dynamics and Aggrephagy in ALS Pathogenesis. Cells 2021, 10, 2247. [Google Scholar] [CrossRef]
- Clausen, L.; Abildgaard, A.B.; Gersing, S.K.; Stein, A.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Protein stability and degradation in health and disease. Adv. Protein Chem. Struct. Biol. 2019, 114, 61–83. [Google Scholar]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Cao, X.; Jin, X.; Liu, B. The involvement of stress granules in aging and aging-associated diseases. Aging Cell 2020, 19, e13136. [Google Scholar] [CrossRef]
- Cadena Sandoval, M.; Heberle, A.M.; Rehbein, U.; Barile, C.; Ramos Pittol, J.M.; Thedieck, K. mTORC1 Crosstalk with Stress Granules in Aging and Age-Related Diseases. Front. Aging 2021, 2, 761333. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The Hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Parfitt, D.A.; Li, W.; Gittings, L.M.; Cheetham, M.E. DNAJ Proteins in neurodegeneration: Essential and protective factors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20160534. [Google Scholar] [CrossRef]
- Vendredy, L.; Adriaenssens, E.; Timmerman, V. Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperones 2020, 25, 679–699. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Ganassi, M.; Mateju, D.; Bigi, I.; Mediani, L.; Poser, I.; Lee, H.O.; Seguin, S.J.; Morelli, F.F.; Vinet, J.; Leo, G.; et al. A Surveillance Function of the HSPB8-BAG3-HSP70 Chaperone Complex Ensures Stress Granule Integrity and Dynamism. Mol. Cell 2016, 63, 796–810. [Google Scholar] [CrossRef]
- Mateju, D.; Franzmann, T.M.; Patel, A.; Kopach, A.; Boczek, E.E.; Maharana, S.; Lee, H.O.; Carra, S.; Hyman, A.A.; Alberti, S. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017, 36, 1669–1687. [Google Scholar] [CrossRef]
- Gu, J.; Wang, C.; Hu, R.; Li, Y.; Zhang, S.; Sun, Y.; Wang, Q.; Li, D.; Fang, Y.; Liu, C. Hsp70 chaperones TDP-43 in dynamic, liquid-like phase and prevents it from amyloid aggregation. Cell Res. 2021, 31, 1024–1027. [Google Scholar] [CrossRef]
- Li, Y.; Gu, J.; Wang, C.; Hu, J.; Zhang, S.; Liu, C.; Zhang, S.; Fang, Y.; Li, D. Hsp70 exhibits a liquid-liquid phase separation ability and chaperones condensed FUS against amyloid aggregation. iScience 2022, 25, 104356. [Google Scholar] [CrossRef]
- Gu, J.; Liu, Z.; Zhang, S.; Li, Y.; Xia, W.; Wang, C.; Xiang, H.; Liu, Z.; Tan, L.; Fang, Y.; et al. Hsp40 proteins phase separate to chaperone the assembly and maintenance of membraneless organelles. Proc. Natl. Acad. Sci. USA 2020, 117, 31123–31133. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, S.; Gu, J.; Tong, Y.; Li, Y.; Gui, X.; Long, H.; Wang, C.; Zhao, C.; Lu, J.; et al. Hsp27 chaperones FUS phase separation under the modulation of stress-induced phosphorylation. Nat. Struct. Mol. Biol. 2020, 27, 363–372. [Google Scholar] [CrossRef]
- Mediani, L.; Antoniani, F.; Galli, V.; Vinet, J.; Carra, A.D.; Bigi, I.; Tripathy, V.; Tiago, T.; Cimino, M.; Leo, G.; et al. Hsp90-mediated regulation of DYRK3 couples stress granule disassembly and growth via mTORC1 signaling. EMBO Rep. 2021, 22, e51740. [Google Scholar] [CrossRef]
- Cherkasov, V.; Hofmann, S.; Druffel-Augustin, S.; Mogk, A.; Tyedmers, J.; Stoecklin, G.; Bukau, B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013, 23, 2452–2462. [Google Scholar] [CrossRef]
- Walters, R.W.; Muhlrad, D.; Garcia, J.; Parker, R. Differential effects of Ydj1 and Sis1 on Hsp70-mediated clearance of stress granules in Saccharomyces cerevisiae. RNA 2015, 21, 1660–1671. [Google Scholar] [CrossRef]
- Wang, A.M.; Miyata, Y.; Klinedinst, S.; Peng, H.M.; Chua, J.P.; Komiyama, T.; Li, X.; Morishima, Y.; Merry, D.E.; Pratt, W.B.; et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 2013, 9, 112–118. [Google Scholar] [CrossRef]
- Pinho, B.R.; Almeida, L.M.; Duchen, M.R.; Oliveira, J.M.A. Allosteric activation of Hsp70 reduces mutant huntingtin levels, the clustering of N-terminal fragments, and their nuclear accumulation. Life Sci. 2021, 285, 120009. [Google Scholar] [CrossRef]
- Chiang, A.N.; Liang, M.; Dominguez-Meijide, A.; Masaracchia, C.; Goeckeler-Fried, J.L.; Mazzone, C.S.; Newhouse, D.W.; Kendsersky, N.M.; Yates, M.E.; Manos-Turvey, A.; et al. Synthesis and evaluation of esterified Hsp70 agonists in cellular models of protein aggregation and folding. Bioorg. Med. Chem. 2019, 27, 79–91. [Google Scholar] [CrossRef]
- Carra, S.; Seguin, S.J.; Lambert, H.; Landry, J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J. Biol. Chem. 2008, 283, 1437–1444. [Google Scholar] [CrossRef]
- Moran Luengo, T.; Mayer, M.P.; Rudiger, S.G.D. The Hsp70-Hsp90 Chaperone Cascade in Protein Folding. Trends Cell Biol. 2019, 29, 164–177. [Google Scholar] [CrossRef]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H. Priming the Proteasome to Protect against Proteotoxicity. Trends Mol. Med. 2020, 26, 639–648. [Google Scholar] [CrossRef]
- Mazroui, R.; Di Marco, S.; Kaufman, R.J.; Gallouzi, I.E. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell. 2007, 18, 2603–2618. [Google Scholar] [CrossRef]
- Turakhiya, A.; Meyer, S.R.; Marincola, G.; Bohm, S.; Vanselow, J.T.; Schlosser, A.; Hofmann, K.; Buchberger, A. ZFAND1 Recruits p97 and the 26S Proteasome to Promote the Clearance of Arsenite-Induced Stress Granules. Mol. Cell 2018, 70, 906–919.e7. [Google Scholar] [CrossRef]
- Gwon, Y.; Maxwell, B.A.; Kolaitis, R.-M.; Zhang, P.; Kim, H.J.; Taylor, J.P. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science 2021, 372, eabf6548. [Google Scholar] [CrossRef]
- Tolay, N.; Buchberger, A. Comparative profiling of stress granule clearance reveals differential contributions of the ubiquitin system. Life Sci. Alliance 2021, 4, e202000927. [Google Scholar] [CrossRef]
- Xie, X.; Matsumoto, S.; Endo, A.; Fukushima, T.; Kawahara, H.; Saeki, Y.; Komada, M. Deubiquitylases USP5 and USP13 are recruited to and regulate heat-induced stress granules through their deubiquitylating activities. J. Cell Sci. 2018, 131, jcs210856. [Google Scholar] [CrossRef]
- Sa-Moura, B.; Funakoshi, M.; Tomko, R.J., Jr.; Dohmen, R.J.; Wu, Z.; Peng, J.; Hochstrasser, M. A conserved protein with AN1 zinc finger and ubiquitin-like domains modulates Cdc48 (p97) function in the ubiquitin-proteasome pathway. J. Biol. Chem. 2013, 288, 33682–33696. [Google Scholar] [CrossRef]
- Yasuda, S.; Tsuchiya, H.; Kaiho, A.; Guo, Q.; Ikeuchi, K.; Endo, A.; Arai, N.; Ohtake, F.; Murata, S.; Inada, T.; et al. Stress- and ubiquitylation-dependent phase separation of the proteasome. Nature 2020, 578, 296–300. [Google Scholar] [CrossRef]
- Carrettiero, D.C.; Almeida, M.C.; Longhini, A.P.; Rauch, J.N.; Han, D.; Zhang, X.; Najafi, S.; Gestwicki, J.E.; Kosik, K.S. Stress routes clients to the proteasome via a BAG2 ubiquitin-independent degradation condensate. Nat. Commun. 2022, 13, 3074. [Google Scholar] [CrossRef]
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 2022, 23, 715–731. [Google Scholar] [CrossRef]
- Marmor-Kollet, H.; Siany, A.; Kedersha, N.; Knafo, N.; Rivkin, N.; Danino, Y.M.; Moens, T.G.; Olender, T.; Sheban, D.; Cohen, N.; et al. Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol. Cell 2020, 80, 876–891.e6. [Google Scholar] [CrossRef]
- Keiten-Schmitz, J.; Wagner, K.; Piller, T.; Kaulich, M.; Alberti, S.; Muller, S. The Nuclear SUMO-Targeted Ubiquitin Quality Control Network Regulates the Dynamics of Cytoplasmic Stress Granules. Mol. Cell 2020, 79, 54–67.e7. [Google Scholar] [CrossRef]
- van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Ferrari, V.; Cristofani, R.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Piccolella, M.; Galbiati, M.; et al. Valosin Containing Protein (VCP): A Multistep Regulator of Autophagy. Int. J. Mol. Sci. 2022, 23, 1939. [Google Scholar] [CrossRef]
- Buchan, J.R.; Kolaitis, R.M.; Taylor, J.P.; Parker, R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef]
- Wang, B.; Maxwell, B.A.; Joo, J.H.; Gwon, Y.; Messing, J.; Mishra, A.; Shaw, T.I.; Ward, A.L.; Quan, H.; Sakurada, S.M.; et al. ULK1 and ULK2 Regulate Stress Granule Disassembly Through Phosphorylation and Activation of VCP/p97. Mol. Cell 2019, 74, 742–757.e8. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Ryu, H.H.; Jun, M.H.; Min, K.J.; Jang, D.J.; Lee, Y.S.; Kim, H.K.; Lee, J.A. Autophagy regulates amyotrophic lateral sclerosis-linked fused in sarcoma-positive stress granules in neurons. Neurobiol. Aging 2014, 35, 2822–2831. [Google Scholar] [CrossRef]
- Krisenko, M.O.; Higgins, R.L.; Ghosh, S.; Zhou, Q.; Trybula, J.S.; Wang, W.H.; Geahlen, R.L. Syk Is Recruited to Stress Granules and Promotes Their Clearance through Autophagy. J. Biol. Chem. 2015, 290, 27803–27815. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, C.J.; Flores, J.C.; Valenzuela, J.A.; Rodriguez, G.J.; Zumkehr, J.; Tran, D.N.; Kimonis, V.E.; Kitazawa, M. The Myoblast C2C12 Transfected with Mutant Valosin-Containing Protein Exhibits Delayed Stress Granule Resolution on Oxidative Stress. Am. J. Pathol. 2016, 186, 1623–1634. [Google Scholar] [CrossRef]
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018, 9, 2794. [Google Scholar] [CrossRef]
- Anderson, E.N.; Gochenaur, L.; Singh, A.; Grant, R.; Patel, K.; Watkins, S.; Wu, J.Y.; Pandey, U.B. Traumatic injury induces stress granule formation and enhances motor dysfunctions in ALS/FTD models. Hum. Mol. Genet. 2018, 27, 1366–1381. [Google Scholar] [CrossRef]
- Scrivo, A.; Bourdenx, M.; Pampliega, O.; Cuervo, A.M. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018, 17, 802–815. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, L.; Zhang, N.; Pan, L.; Hadano, S.; Fan, D. Six SQSTM1 mutations in a Chinese amyotrophic lateral sclerosis cohort. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 378–384. [Google Scholar] [CrossRef]
- Le Ber, I.; Camuzat, A.; Guerreiro, R.; Bouya-Ahmed, K.; Bras, J.; Nicolas, G.; Gabelle, A.; Didic, M.; De Septenville, A.; Millecamps, S.; et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar]
- Van der Zee, J.; Van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matej, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef]
- Arai, T.; Nonaka, T.; Hasegawa, M.; Akiyama, H.; Yoshida, M.; Hashizume, Y.; Tsuchiya, K.; Oda, T.; Ikeda, K. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci. Lett. 2003, 342, 41–44. [Google Scholar] [CrossRef]
- Hiji, M.; Takahashi, T.; Fukuba, H.; Yamashita, H.; Kohriyama, T.; Matsumoto, M. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008, 116, 183–191. [Google Scholar] [CrossRef]
- Mizuno, Y.; Amari, M.; Takatama, M.; Aizawa, H.; Mihara, B.; Okamoto, K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2006, 249, 13–18. [Google Scholar] [CrossRef]
- Zhang, P.; Fan, B.; Yang, P.; Temirov, J.; Messing, J.; Kim, H.J.; Taylor, J.P. Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology. eLife 2019, 8, e39578. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Jain, S.; Wheeler, J.R.; Walters, R.W.; Agrawal, A.; Barsic, A.; Parker, R. ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 2016, 164, 487–498. [Google Scholar] [CrossRef]
- Ma, X.; Lu, C.; Chen, Y.; Li, S.; Ma, N.; Tao, X.; Li, Y.; Wang, J.; Zhou, M.; Yan, Y.B.; et al. CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell 2022, 185, 1325–1345.e22. [Google Scholar] [CrossRef]
- Grantham, J. The Molecular Chaperone CCT/TRiC: An Essential Component of Proteostasis and a Potential Modulator of Protein Aggregation. Front. Genet. 2020, 11, 172. [Google Scholar] [CrossRef]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hosp, F.; Frottin, F.; Ge, H.; Mann, M.; Hayer-Hartl, M.; Hartl, F.U. Soluble Oligomers of PolyQ-Expanded Huntingtin Target a Multiplicity of Key Cellular Factors. Mol. Cell 2016, 63, 951–964. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Murakami, T.; Qamar, S.; Lin, J.Q.; Schierle, G.S.; Rees, E.; Miyashita, A.; Costa, A.R.; Dodd, R.B.; Chan, F.T.; Michel, C.H.; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [Google Scholar] [CrossRef]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef]
- Murray, D.T.; Kato, M.; Lin, Y.; Thurber, K.R.; Hung, I.; McKnight, S.L.; Tycko, R. Structure of FUS Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627.e16. [Google Scholar] [CrossRef]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Guillen-Boixet, J.; Kopach, A.; Holehouse, A.S.; Wittmann, S.; Jahnel, M.; Schlussler, R.; Kim, K.; Trussina, I.; Wang, J.; Mateju, D.; et al. RNA-Induced Conformational Switching and Clustering of G3BP Drive Stress Granule Assembly by Condensation. Cell 2020, 181, 346–361.e17. [Google Scholar] [CrossRef]
- Yang, P.; Mathieu, C.; Kolaitis, R.M.; Zhang, P.; Messing, J.; Yurtsever, U.; Yang, Z.; Wu, J.; Li, Y.; Pan, Q.; et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell 2020, 181, 325–345.e28. [Google Scholar] [CrossRef]
- Sanders, D.W.; Kedersha, N.; Lee, D.S.W.; Strom, A.R.; Drake, V.; Riback, J.A.; Bracha, D.; Eeftens, J.M.; Iwanicki, A.; Wang, A.; et al. Competing Protein-RNA Interaction Networks Control Multiphase Intracellular Organization. Cell 2020, 181, 306–324.e28. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Name | Function in SG Disassembly | References |
|---|---|---|
| Hsp70 | Prevent misfolded proteins from accumulation | [17,18] |
| Prevent liquid-to-solid phase transition of FUS and TDP-43 | [19,20] | |
| Hdj1 | Prevent liquid-to-solid phase transition of FUS | [21] |
| HspB8 | Prevent misfolded proteins from accumulation | [17] |
| HspB1 | Inhibit LLPS of FUS and its association with SGs | [22] |
| Hsp90 | Enhance DYRK3 activity that promotes SG disassembly | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, R.; Qian, B.; Li, A.; Fang, Y. Role of Proteostasis Regulation in the Turnover of Stress Granules. Int. J. Mol. Sci. 2022, 23, 14565. https://doi.org/10.3390/ijms232314565
Hu R, Qian B, Li A, Fang Y. Role of Proteostasis Regulation in the Turnover of Stress Granules. International Journal of Molecular Sciences. 2022; 23(23):14565. https://doi.org/10.3390/ijms232314565
Chicago/Turabian StyleHu, Rirong, Beituo Qian, Ang Li, and Yanshan Fang. 2022. "Role of Proteostasis Regulation in the Turnover of Stress Granules" International Journal of Molecular Sciences 23, no. 23: 14565. https://doi.org/10.3390/ijms232314565
APA StyleHu, R., Qian, B., Li, A., & Fang, Y. (2022). Role of Proteostasis Regulation in the Turnover of Stress Granules. International Journal of Molecular Sciences, 23(23), 14565. https://doi.org/10.3390/ijms232314565