

Role of Biomarkers in the Management of Acute Myeloid Leukemia


Abstract

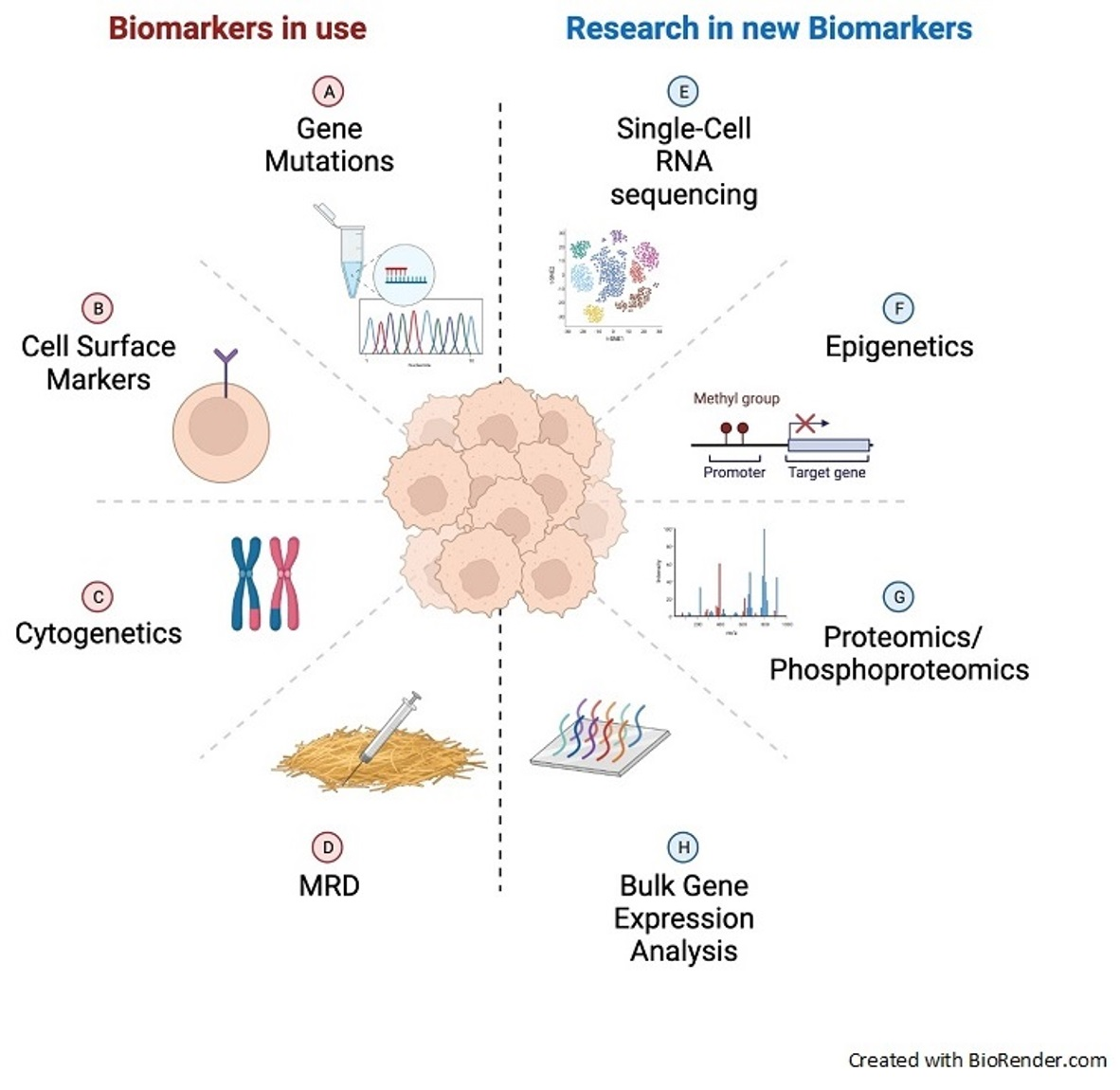
1. Introduction
2. Established Biomarkers in AML
2.1. FLT3
2.2. IDH1/2
2.3. NPM1
2.4. CD33
2.5. TP53
2.6. ASXL1
2.7. RUNX1
2.8. Cytogenetics
3. Emerging Biomarkers in AML
Measurable Residual Disease
4. The Future of Biomarkers in AML
4.1. Gene Expression Analysis
4.2. Biomarkers for Immunotherapy
4.3. Epigenetics: Methylation Patterns
4.4. Ex Vivo Drug Testing
4.5. Other Proposed Biomarkers
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bennett, J.M.; Catovsky, D.; Daniel, M.-T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-operative Group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; Iarc: Lyon, France, 2001; Volume 3. [Google Scholar]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 ELN Recommendations from an International Expert Panel. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. Midostaurin approved for FLT3-mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Tallman, M.S.; Pollyea, D.A. AML (Version 3.2021). Available online: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf (accessed on 8 September 2021).
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef]
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Rollig, C.; Wollmer, E.; Wasch, R.; Bornhauser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia with FLT3-Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.J. Targeting IDH1 and IDH2 Mutations in Acute Myeloid Leukemia: Emerging Options and Pending Questions. Hemasphere 2021, 5, e583. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; DiNardo, C.D. Acute myeloid leukemia with IDH1 and IDH2 mutations: 2021 treatment algorithm. Blood Cancer J. 2021, 11, 107. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Fathi, A.T. Targeting IDH Mutations in AML: Wielding the Double-edged Sword of Differentiation. Curr. Cancer Drug Targets 2020, 20, 490–500. [Google Scholar] [CrossRef] [PubMed]
- McMurry, H.; Fletcher, L.; Traer, E. IDH Inhibitors in AML-Promise and Pitfalls. Curr. Hematol. Malig. Rep. 2021, 16, 207–217. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Nomdedeu, J.; Hoyos, M.; Carricondo, M.; Esteve, J.; Bussaglia, E.; Estivill, C.; Ribera, J.M.; Duarte, R.; Salamero, O.; Gallardo, D.; et al. Adverse impact of IDH1 and IDH2 mutations in primary AML: Experience of the Spanish CETLAM group. Leuk. Res. 2012, 36, 990–997. [Google Scholar] [CrossRef]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Cerchione, C.; Romano, A.; Daver, N.; DiNardo, C.; Jabbour, E.J.; Konopleva, M.; Ravandi-Kashani, F.; Kadia, T.; Martelli, M.P.; Isidori, A.; et al. IDH1/IDH2 Inhibition in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 639387. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; Seet, C.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: A phase 1 study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Luo, L.; Hsu, V.; Gudi, R.; Dorff, S.E.; Przepiorka, D.; Deisseroth, A.; Shen, Y.L.; Sheth, C.M.; Charlab, R.; et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clin. Cancer Res. 2019, 25, 3205–3209. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Brunetti, L.; Martelli, M.P. How I diagnose and treat NPM1-mutated AML. Blood 2021, 137, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabadran, R.; Gionfriddo, I.; Mezzasoma, F.; Milano, F.; Nabet, B.; et al. Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 2018, 34, 499–512.e499. [Google Scholar] [CrossRef]
- Falini, B.; Bolli, N.; Liso, A.; Martelli, M.P.; Mannucci, R.; Pileri, S.; Nicoletti, I. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: Molecular basis and clinical implications. Leukemia 2009, 23, 1731–1743. [Google Scholar] [CrossRef]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terre, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Ostronoff, F.; Othus, M.; Lazenby, M.; Estey, E.; Appelbaum, F.R.; Evans, A.; Godwin, J.; Gilkes, A.; Kopecky, K.J.; Burnett, A.; et al. Prognostic significance of NPM1 mutations in the absence of FLT3-internal tandem duplication in older patients with acute myeloid leukemia: A SWOG and UK National Cancer Research Institute/Medical Research Council report. J. Clin. Oncol. 2015, 33, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Prata, P.H.; Bally, C.; Prebet, T.; Recher, C.; Venton, G.; Thomas, X.; Raffoux, E.; Pigneux, A.; Cluzeau, T.; Desoutter, J.; et al. NPM1 mutation is not associated with prolonged complete remission in acute myeloid leukemia patients treated with hypomethylating agents. Haematologica 2018, 103, e455–e457. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Loghavi, S.; Kadia, T.M.; Daver, N.; Borthakur, G.; Pemmaraju, N.; Naqvi, K.; Alvarado, Y.; Yilmaz, M.; Short, N.; et al. Outcomes of older patients with NPM1-mutated AML: Current treatments and the promise of venetoclax-based regimens. Blood Adv. 2020, 4, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Ravandi, F.; DiNardo, C.D.; Jabbour, E.; Kantarjian, H.M.; Andreeff, M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia 2021, 35, 2482–2495. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.; Yacko, L.R.; Glode, A.E. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pract. Oncol. 2019, 10, 68–82. [Google Scholar]
- Linenberger, M.L. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: Progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia 2005, 19, 176–182. [Google Scholar] [CrossRef]
- Walter, R.B.; Appelbaum, F.R.; Estey, E.H.; Bernstein, I.D. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood 2012, 119, 6198–6208. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef]
- McGavin, J.K.; Spencer, C.M. Gemtuzumab ozogamicin. Drugs 2001, 61, 1317–1322, discussion 1323–1314. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar] [PubMed]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Lowenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Lowenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B.; Beck, J.; Graux, C.; van Putten, W.; Schouten, H.C.; Verdonck, L.F.; Ferrant, A.; Sonneveld, P.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: Results of a multicenter phase 3 study. Blood 2010, 115, 2586–2591. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Wei, A.H. How I treat acute myeloid leukemia in the era of new drugs. Blood 2020, 135, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Ladha, A.; Hui, G.; Cheung, E.; Berube, C.; Coutre, S.E.; Gotlib, J.; Liedtke, M.; Zhang, T.Y.; Muffly, L.; Mannis, G.N. Routine use of gemtuzumab ozogamicin in 7 + 3-based inductions for all ‘non-adverse’ risk AML. Leuk. Lymphoma 2021, 62, 1510–1513. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Grob, T.; Al Hinai, A.S.A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.; Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk Kooy, M.; et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022, 139, 2347–2354. [Google Scholar] [CrossRef] [PubMed]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 mutations in newly diagnosed acute myeloid leukemia: Clinicomolecular characteristics, response to therapy, and outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, O.K.; Siddon, A.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Dal Cin, P.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 mutation defines a unique subgroup within complex karyotype de novo and therapy-related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Hasserjian, R.P. Revealing the dark secrets of TP53-mutated AML. Blood 2022, 140, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, R.R.; Kovtonyuk, L.V.; Klemm, N.; Fullin, J.; Stolz, S.M.; Mueller, J.; Caiado, F.; Kurppa, K.J.; Ebert, B.L.; Manz, M.G.; et al. TP53 mutations confer resistance to hypomethylating agents and BCL-2 inhibition in myeloid neoplasms. Blood Adv. 2022, 6, 3201–3206. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Chilkulwar, A.; Saliba, R.M.; Chen, J.; Rondon, G.; Patel, K.P.; Khogeer, H.; Shah, A.R.; Randolph, B.V.; Perez, J.M.R.; et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood 2018, 131, 2989–2992. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Vyas, P.; Kambhampati, S.; Malki, M.M.A.; Larson, R.A.; Asch, A.S.; Mannis, G.N.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in frontline TP53m AML patients: Phase 1b results. J. Clin. Oncol. 2022, 40, 7020. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Maslah, N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin, P.; Naoui, N.; Schlageter, M.H.; Bally, C.; et al. Synergistic effects of PRIMA-1(Met) (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica 2020, 105, 1539–1551. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahme, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Beve, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myelodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Brecqueville, M.; Devillier, R.; Murati, A.; Mozziconacci, M.J.; Birnbaum, D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J. Hematol. Oncol. 2012, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Gelsi-Boyer, V.; Trouplin, V.; Adelaide, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.L.; Randazzo, F.; Humphries, R.K.; Brock, H.W. Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 2006, 369, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Asada, S.; Kitamura, T. Aberrant histone modifications induced by mutant ASXL1 in myeloid neoplasms. Int. J. Hematol. 2019, 110, 179–186. [Google Scholar] [CrossRef]
- Chou, W.C.; Huang, H.H.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010, 116, 4086–4094. [Google Scholar] [CrossRef]
- Pratcorona, M.; Abbas, S.; Sanders, M.A.; Koenders, J.E.; Kavelaars, F.G.; Erpelinck-Verschueren, C.A.; Zeilemakers, A.; Lowenberg, B.; Valk, P.J. Acquired mutations in ASXL1 in acute myeloid leukemia: Prevalence and prognostic value. Haematologica 2012, 97, 388–392. [Google Scholar] [CrossRef]
- Fan, Y.; Liao, L.; Liu, Y.; Wu, Z.; Wang, C.; Jiang, Z.; Wang, S.; Liu, Y. Risk factors affect accurate prognosis in ASXL1-mutated acute myeloid leukemia. Cancer Cell Int. 2021, 21, 526. [Google Scholar] [CrossRef]
- Hurtado, R.; Guirales, F.; Tirado, C.A. ASXL1 Gene in AML. J. Assoc. Genet. Technol. 2021, 47, 60–68. [Google Scholar]
- Yang, H.; Kurtenbach, S.; Guo, Y.; Lohse, I.; Durante, M.A.; Li, J.; Li, Z.; Al-Ali, H.; Li, L.; Chen, Z.; et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood 2018, 131, 328–341. [Google Scholar] [CrossRef]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 2019, 76, 2511–2523. [Google Scholar] [CrossRef]
- Mill, C.P.; Fiskus, W.; DiNardo, C.D.; Qian, Y.; Raina, K.; Rajapakshe, K.; Perera, D.; Coarfa, C.; Kadia, T.M.; Khoury, J.D.; et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood 2019, 134, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Huo, L.; Lan, F.; Wu, J.; Huang, G. The Clinical, Molecular, and Mechanistic Basis of RUNX1 Mutations Identified in Hematological Malignancies. Mol. Cells 2020, 43, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Teleanu, V.; Papaemmanuil, E.; Weber, D.; Paschka, P.; Hahn, J.; Wallrabenstein, T.; Kolbinger, B.; Kohne, C.H.; Horst, H.A.; et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 2016, 30, 2282. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Rock, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 mutations in acute myeloid leukemia: Results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 2011, 29, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Jalili, M.; Yaghmaie, M.; Ahmadvand, M.; Alimoghaddam, K.; Mousavi, S.A.; Vaezi, M.; Ghavamzadeh, A. Prognostic Value of RUNX1 Mutations in AML: A Meta-Analysis. Asian Pac. J. Cancer Prev. 2018, 19, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Mill, C.P.; Fiskus, W.; DiNardo, C.D.; Birdwell, C.; Davis, J.A.; Kadia, T.M.; Takahashi, K.; Short, N.; Daver, N.; Ohanian, M.; et al. Effective therapy for AML with RUNX1 mutation by cotreatment with inhibitors of protein translation and BCL2. Blood 2022, 139, 907–921. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Rollig, C.; Montesinos, P.; Martinez-Cuadron, D.; Barragan, E.; Garcia, R.; Botella, C.; Martinez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal Abnormalities and Prognosis in NPM1-Mutated Acute Myeloid Leukemia: A Pooled Analysis of Individual Patient Data from Nine International Cohorts. J. Clin. Oncol. 2019, 37, 2632–2642. [Google Scholar] [CrossRef]
- Lugthart, S.; Groschel, S.; Beverloo, H.B.; Kayser, S.; Valk, P.J.; van Zelderen-Bhola, S.L.; Jan Ossenkoppele, G.; Vellenga, E.; van den Berg-de Ruiter, E.; Schanz, U.; et al. Clinical, molecular, and prognostic significance of WHO type inv(3)(q21q26.2)/t(3;3)(q21;q26.2) and various other 3q abnormalities in acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 3890–3898. [Google Scholar] [CrossRef]
- Kayser, S.; Hills, R.K.; Langova, R.; Kramer, M.; Guijarro, F.; Sustkova, Z.; Estey, E.H.; Shaw, C.M.; Racil, Z.; Mayer, J.; et al. Characteristics and outcome of patients with acute myeloid leukaemia and t(8;16)(p11;p13): Results from an International Collaborative Study. Br. J. Haematol. 2021, 192, 832–842. [Google Scholar] [CrossRef]
- Chilton, L.; Hills, R.K.; Harrison, C.J.; Burnett, A.K.; Grimwade, D.; Moorman, A.V. Hyperdiploidy with 49-65 chromosomes represents a heterogeneous cytogenetic subgroup of acute myeloid leukemia with differential outcome. Leukemia 2014, 28, 321–328. [Google Scholar] [CrossRef]
- Jahn, N.; Terzer, T.; Sträng, E.; Dolnik, A.; Cocciardi, S.; Panina, E.; Corbacioglu, A.; Herzig, J.; Weber, D.; Schrade, A.; et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv. 2020, 4, 6342–6352. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Maurillo, L.; Del Principe, M.I.; Del Poeta, G.; Sconocchia, G.; Lo-Coco, F.; Arcese, W.; Amadori, S.; Venditti, A. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood 2012, 119, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Freeman, S.D.; Hills, R.K.; Virgo, P.; Khan, N.; Couzens, S.; Dillon, R.; Gilkes, A.; Upton, L.; Nielsen, O.J.; Cavenagh, J.D.; et al. Measurable Residual Disease at Induction Redefines Partial Response in Acute Myeloid Leukemia and Stratifies Outcomes in Patients at Standard Risk without NPM1 Mutations. J. Clin. Oncol. 2018, 36, 1486–1497. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Zhou, S.; Fu, C.; Berry, D.A.; Walter, R.B.; Freeman, S.D.; Hourigan, C.S.; Huang, X.; Nogueras Gonzalez, G.; Hwang, H.; et al. Association of Measurable Residual Disease with Survival Outcomes in Patients with Acute Myeloid Leukemia. JAMA Oncol. 2020, 6, 1890. [Google Scholar] [CrossRef]
- Shah, M.V.; Jorgensen, J.L.; Saliba, R.M.; Wang, S.A.; Alousi, A.M.; Andersson, B.S.; Bashir, Q.; Ciurea, S.O.; Kebriaei, P.; Marin, D.; et al. Early Post-Transplant Minimal Residual Disease Assessment Improves Risk Stratification in Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2018, 24, 1514–1520. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in acute myeloid leukemia: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef]
- Curran, E.; Stock, W. Taking a “BiTE out of ALL”: Blinatumomab approval for MRD-positive ALL. Blood 2019, 133, 1715–1719. [Google Scholar] [CrossRef]
- Walker, C.J.; Mrózek, K.; Ozer, H.G.; Nicolet, D.; Kohlschmidt, J.; Papaioannou, D.; Genutis, L.K.; Bill, M.; Powell, B.L.; Uy, G.L.; et al. Gene expression signature predicts relapse in adult patients with cytogenetically normal acute myeloid leukemia. Blood Adv. 2021, 5, 1474–1482. [Google Scholar] [CrossRef]
- Zhu, Y.-M.; Wang, P.-P.; Huang, J.-Y.; Chen, Y.-S.; Chen, B.; Dai, Y.-J.; Yan, H.; Hu, Y.; Cheng, W.-Y.; Ma, T.-T.; et al. Gene mutational pattern and expression level in 560 acute myeloid leukemia patients and their clinical relevance. J. Transl. Med. 2017, 15. [Google Scholar] [CrossRef]
- Van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e1224. [Google Scholar] [CrossRef]
- Stetson, L.C.; Balasubramanian, D.; Ribeiro, S.P.; Stefan, T.; Gupta, K.; Xu, X.; Fourati, S.; Roe, A.; Jackson, Z.; Schauner, R.; et al. Single cell RNA sequencing of AML initiating cells reveals RNA-based evolution during disease progression. Leukemia 2021, 35, 2799–2812. [Google Scholar] [CrossRef] [PubMed]
- Wiatrowski, K.; Kim, T.H.; Przespolewski, A. Cellular and Molecular Biomarkers Predictive of Response to Immunotherapy in Acute Myeloid Leukemia. Front. Oncol. 2022, 12, 826768. [Google Scholar] [CrossRef] [PubMed]
- Olombel, G.; Guerin, E.; Guy, J.; Perrot, J.Y.; Dumezy, F.; de Labarthe, A.; Bastie, J.N.; Legrand, O.; Raffoux, E.; Plesa, A.; et al. The level of blast CD33 expression positively impacts the effect of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Blood 2016, 127, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.; Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020, 4, 5011–5024. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int. J. Mol. Sci. 2019, 20, 4576. [Google Scholar] [CrossRef]
- Božić, T.; Lin, Q.; Frobel, J.; Wilop, S.; Hoffmann, M.; Müller-Tidow, C.; Brümmendorf, T.H.; Jost, E.; Wagner, W. DNA-methylation in C1R is a prognostic biomarker for acute myeloid leukemia. Clin. Epigenetics 2015, 7, 116. [Google Scholar] [CrossRef]
- Hu, L.; Gao, Y.; Shi, Z.; Liu, Y.; Zhao, J.; Xiao, Z.; Lou, J.; Xu, Q.; Tong, X. DNA methylation-based prognostic biomarkers of acute myeloid leukemia patients. Ann. Transl. Med. 2019, 7, 737. [Google Scholar] [CrossRef]
- Šestáková, Š.; Cerovská, E.; Šálek, C.; Kundrát, D.; Ježíšková, I.; Folta, A.; Mayer, J.; Ráčil, Z.; Cetkovský, P.; Remešová, H. A validation study of potential prognostic DNA methylation biomarkers in patients with acute myeloid leukemia using a custom DNA methylation sequencing panel. Clin. Epigenetics 2022, 14, 22. [Google Scholar] [CrossRef]
- Lin, L.; Tong, Y.; Straube, J.; Zhao, J.; Gao, Y.; Bai, P.; Li, J.; Wang, J.; Wang, H.; Wang, X.; et al. Ex-vivo drug testing predicts chemosensitivity in acute myeloid leukemia. J. Leukoc. Biol. 2020, 107, 859–870. [Google Scholar] [CrossRef]
- Kita, R.; Richardson, M.A.; Anderson, E.K.; Strachan, D.C.; Rashid, R.; Terrell, M.; Marcogliese, A.N.; Santaguida, M.T.; Stevens, A.M. Correlation of ex vivo drug sensitivity with clinical response in pediatric AML. J. Clin. Oncol. 2021, 39, 10032. [Google Scholar] [CrossRef]
- Dowling, P.; Tierney, C.; Dunphy, K.; Miettinen, J.J.; Heckman, C.A.; Bazou, D.; O’Gorman, P. Identification of Protein Biomarker Signatures for Acute Myeloid Leukemia (AML) Using Both Nontargeted and Targeted Approaches. Proteomes 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.W.; Kim, H.; Hur, W.; Jung, J.H.; Jeong, S.J.; Shin, H.; Seo, D.; Jeong, H.; Choi, B.; Hong, S.; et al. A Proteomic Approach to Understand the Clinical Significance of Acute Myeloid Leukemia-Derived Extracellular Vesicles Reflecting Essential Characteristics of Leukemia. Mol. Cell. Proteom. 2021, 20, 100017. [Google Scholar] [CrossRef] [PubMed]
- Gosline, S.J.C.; Tognon, C.; Nestor, M.; Joshi, S.; Modak, R.; Damnernsawad, A.; Posso, C.; Moon, J.; Hansen, J.R.; Hutchinson-Bunch, C.; et al. Proteomic and phosphoproteomic measurements enhance ability to predict ex vivo drug response in AML. Clin. Proteom. 2022, 19, 30. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Biomarker | Definition | Examples of Clinical Applications in AML | Clinical Trials |
|---|---|---|---|
| Measurable residual disease (MRD) | Presence of leukemia cells on the scale of one in 106 to one in 104 cells. Detectable by flow cytometry, qRT-PCR, dPCR, NGS. | Flow cytometry- and NGS-based assays for determining MRD in first complete remission | NCT05339204 NCT01452646 NCT02870777 NCT03769532 |
| Gene expression signatures | Specific genomic alterations and transcriptional profiles identified using RNA sequencing. | NCT00897936 NCT01421862 NCT00897936 NCT01338974 NCT01229956 NCT01057199 | |
| Targets for immunotherapy | Tumor or immune cell surface receptors and cytokine expression in malignant cells. Identified via single-cell RNA sequencing, mass cytometry, single cell cytokine analysis. | Monoclonal antibody-drug conjugates
T-cell directed therapies
Checkpoint inhibitors
| NCT02944162 NCT04884984 NCT05023707 NCT04351022 |
| Epigenetics | Changes upstream of gene expression. For example, dysregulated DNA methylation patterns identified by methods such as bisulfite sequencing. | Hypomethylating agents (azacitidine, decitabine) | NCT00897936 NCT01421862 NCT00897936 NCT01229956 |
| Ex vivo drug testing | Measuring viability of patient samples after treating with various drug combinations in vitro | NCT04267081 NCT03197714 NCT02551718 | |
| Proteomics/ Phosphoproteomics | Identification of specific proteins and phosphorylated proteins using non-targeted (e.g., mass spectrometry) and targeted (e.g., multiplex immunoassays) approaches. | NCT01360125 NCT01338974 NCT01057199 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Small, S.; Oh, T.S.; Platanias, L.C. Role of Biomarkers in the Management of Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 14543. https://doi.org/10.3390/ijms232314543
Small S, Oh TS, Platanias LC. Role of Biomarkers in the Management of Acute Myeloid Leukemia. International Journal of Molecular Sciences. 2022; 23(23):14543. https://doi.org/10.3390/ijms232314543
Chicago/Turabian StyleSmall, Sara, Timothy S. Oh, and Leonidas C. Platanias. 2022. "Role of Biomarkers in the Management of Acute Myeloid Leukemia" International Journal of Molecular Sciences 23, no. 23: 14543. https://doi.org/10.3390/ijms232314543
APA StyleSmall, S., Oh, T. S., & Platanias, L. C. (2022). Role of Biomarkers in the Management of Acute Myeloid Leukemia. International Journal of Molecular Sciences, 23(23), 14543. https://doi.org/10.3390/ijms232314543