
Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
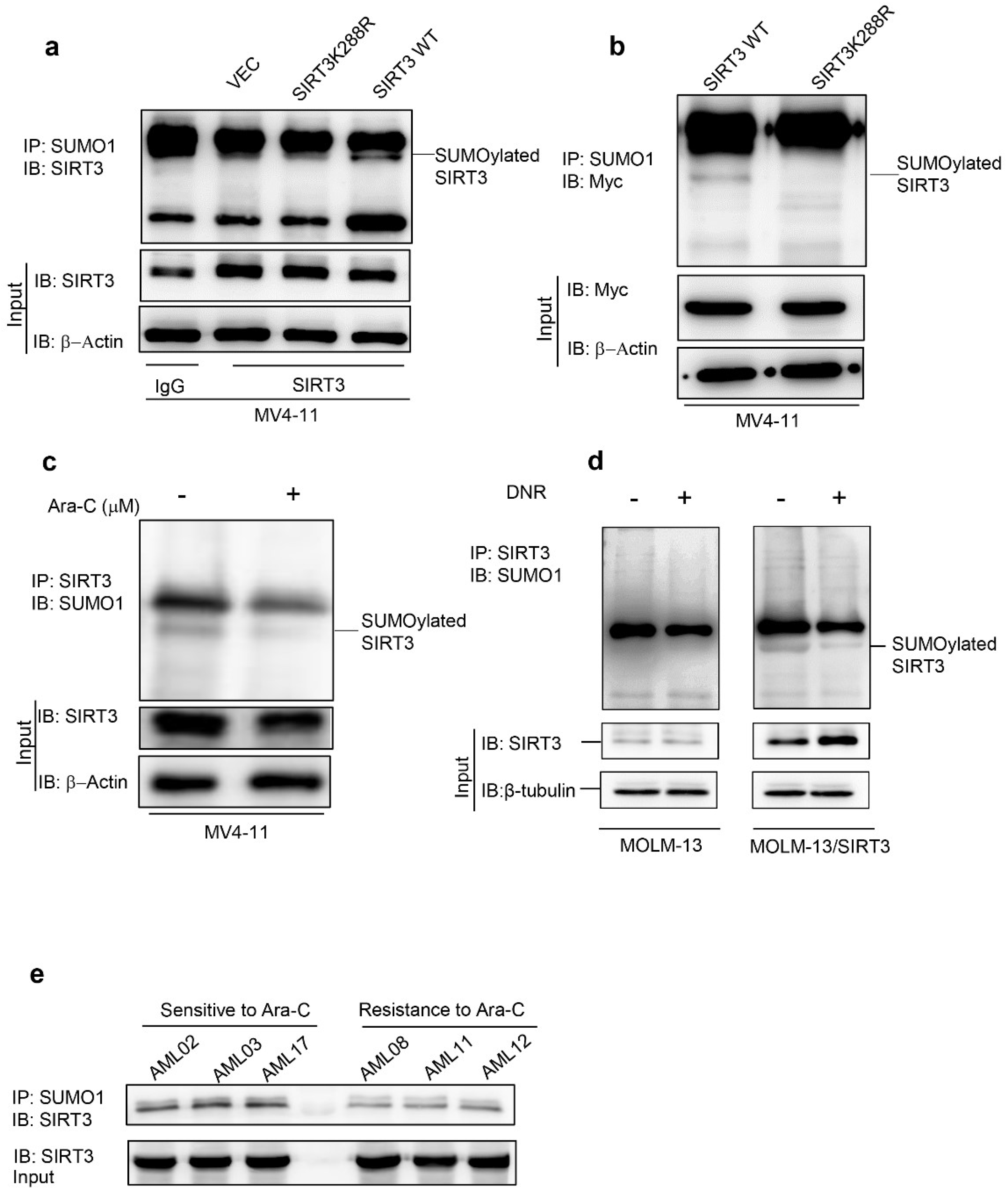
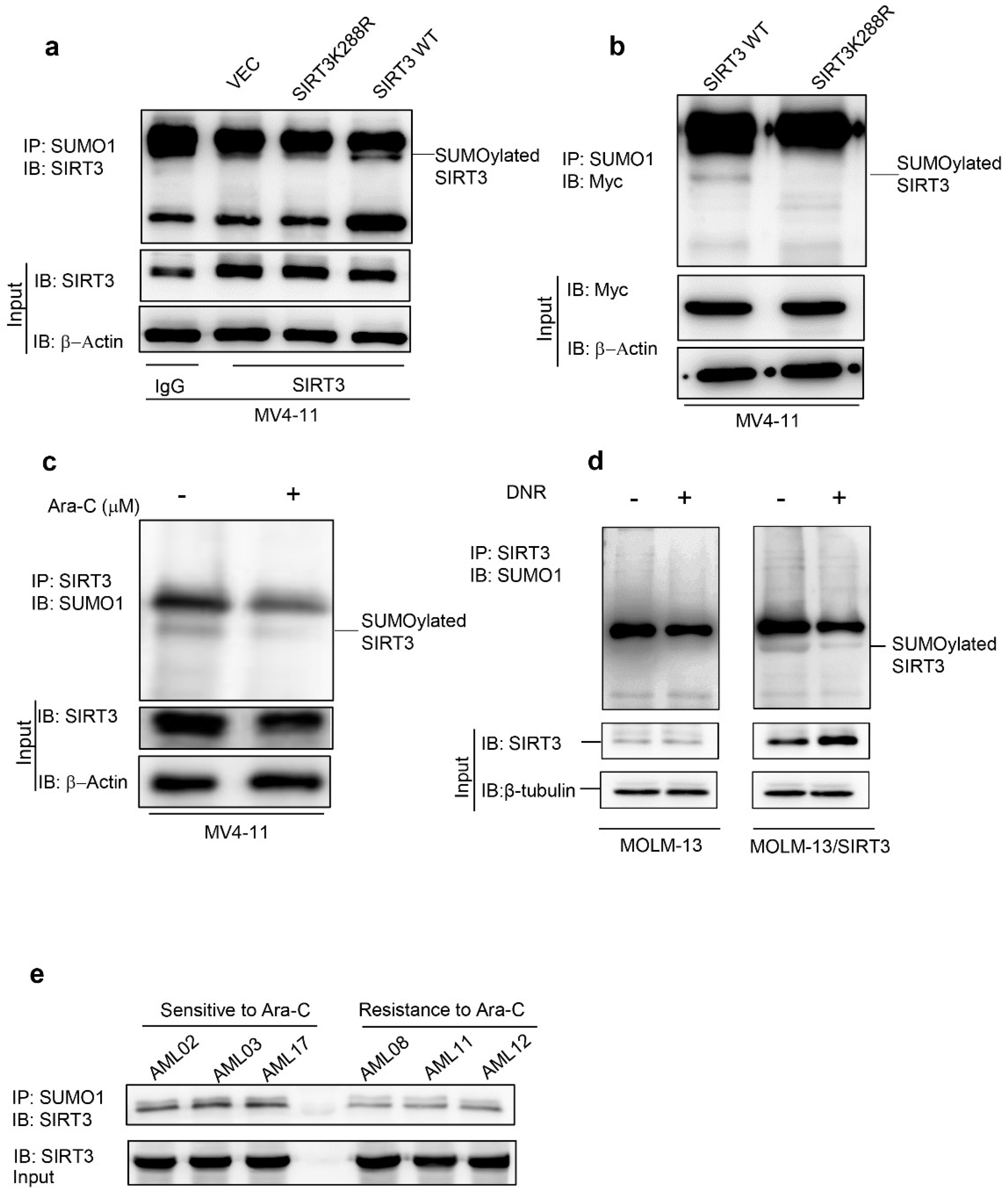
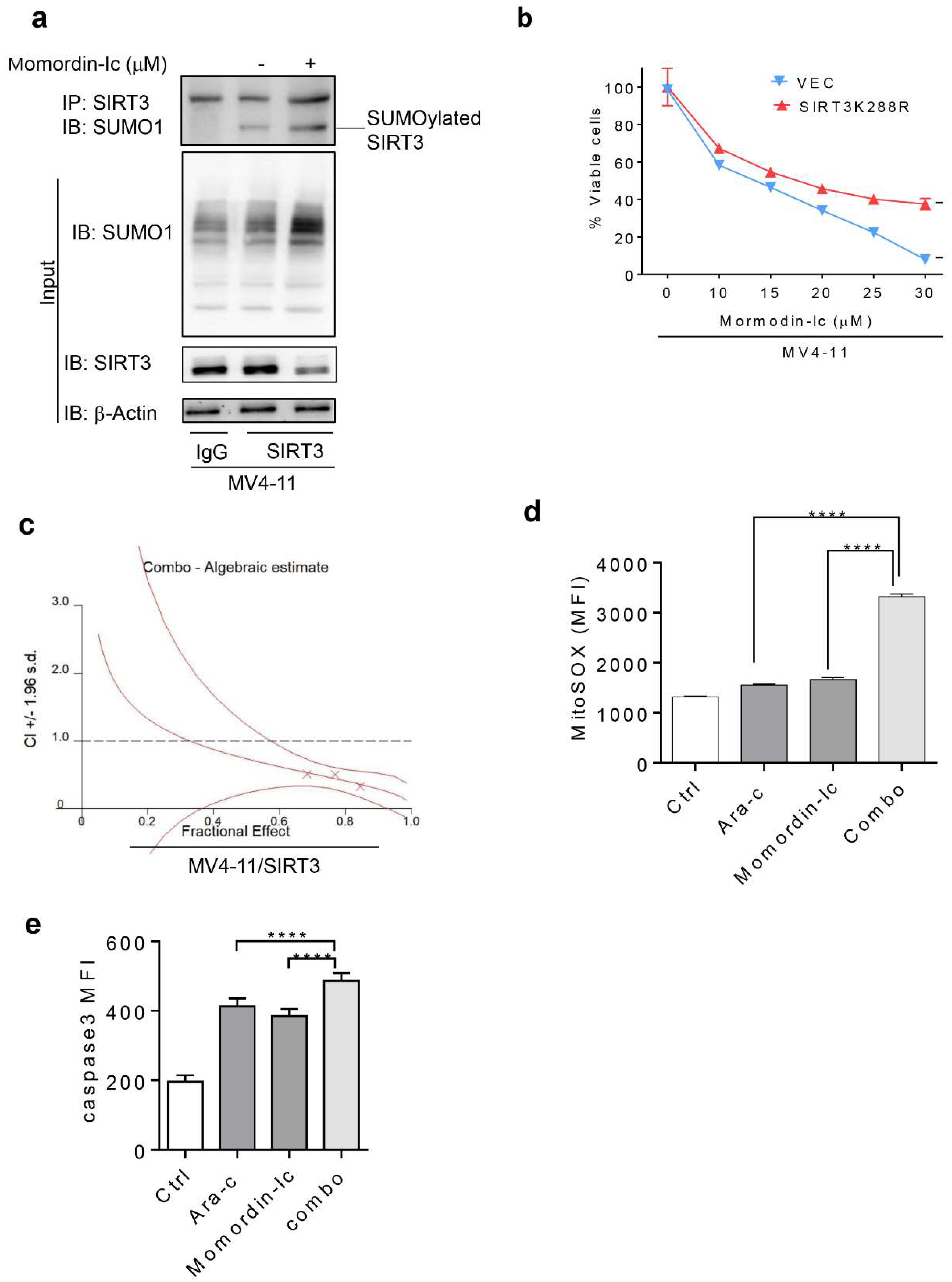
2.1. Chemotherapy Attenuates SIRT3 SUMOylation in AML
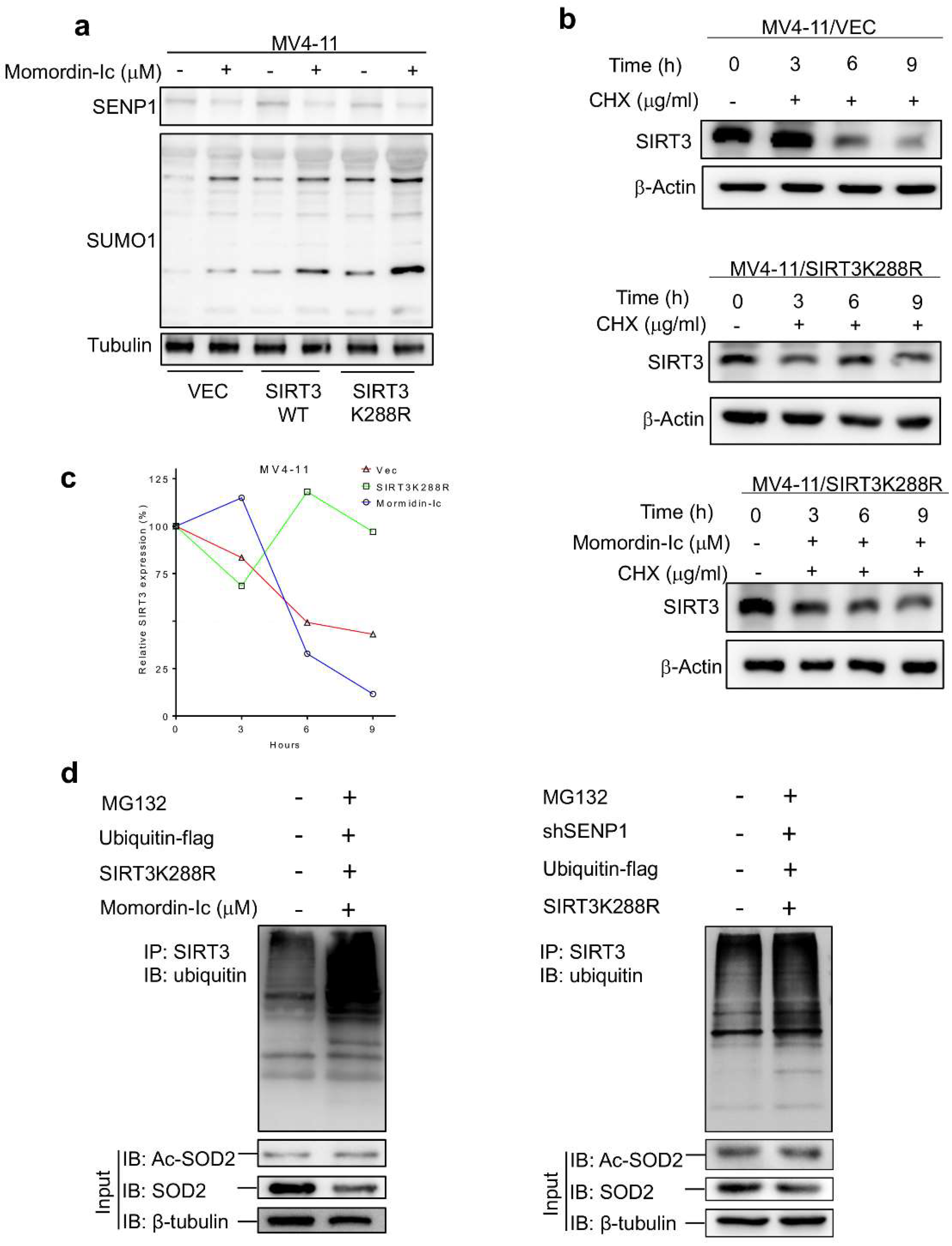
2.2. De-SUMOylation Activates SIRT3 via Inhibition of Its Protein Degradation
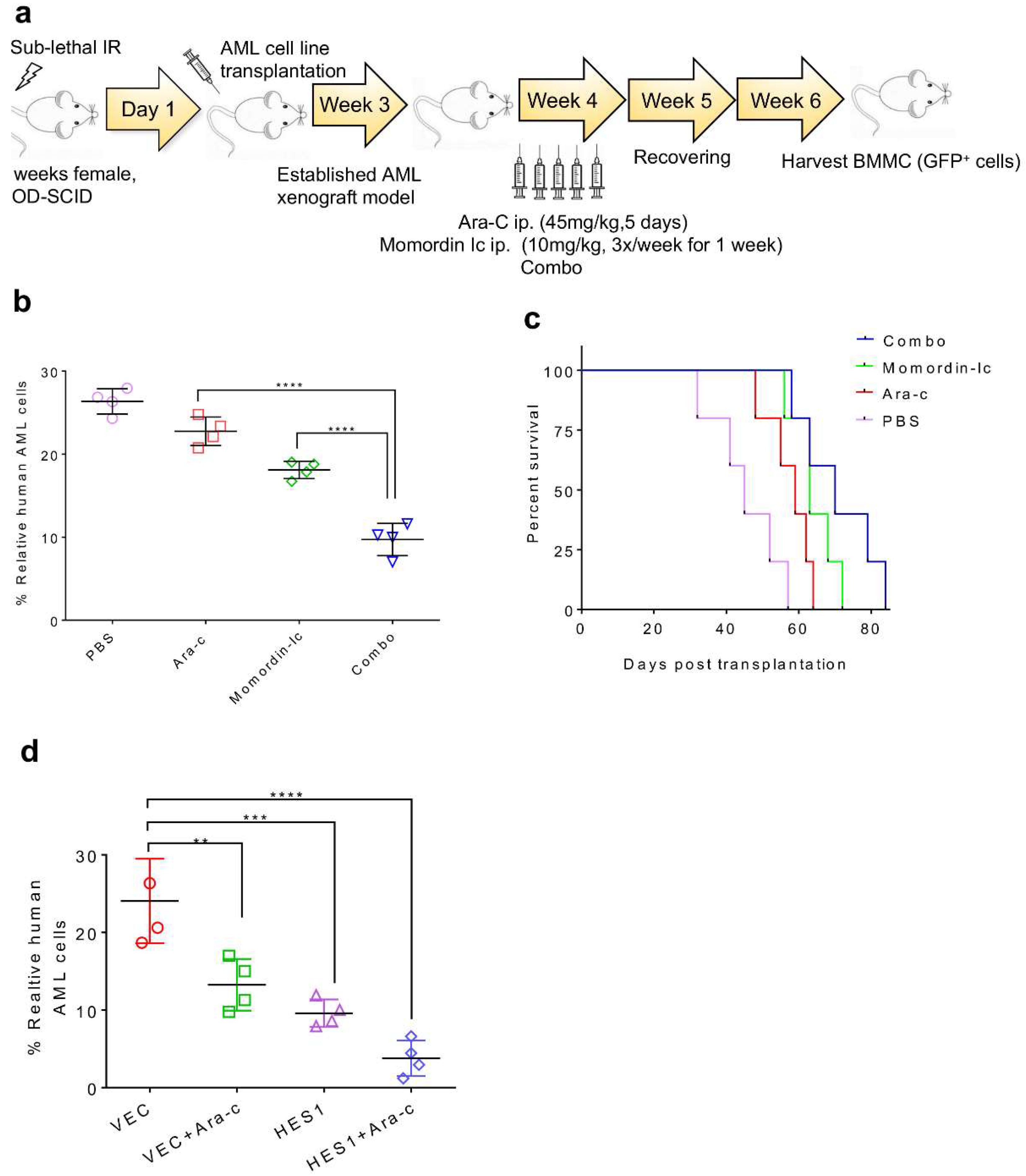
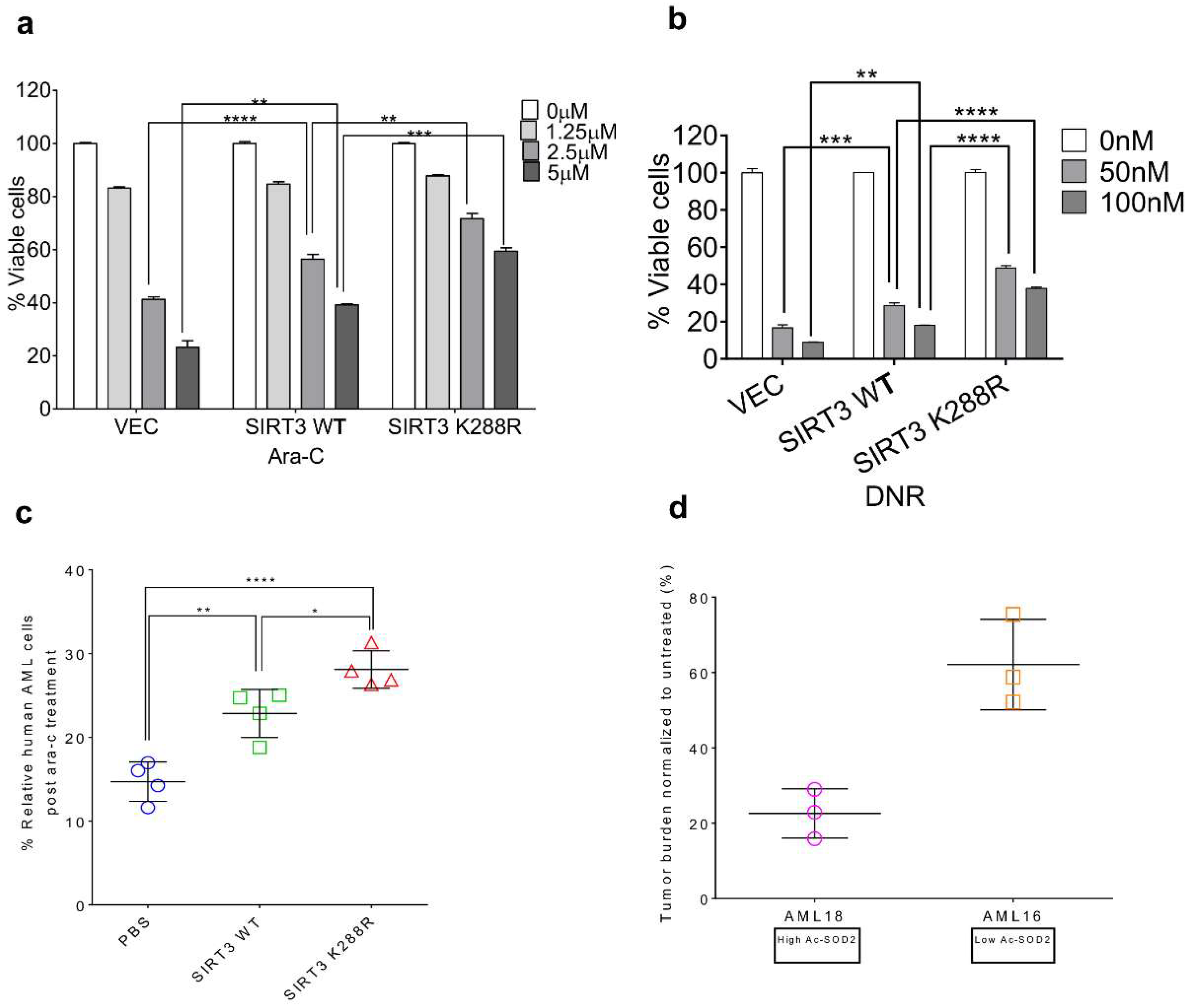
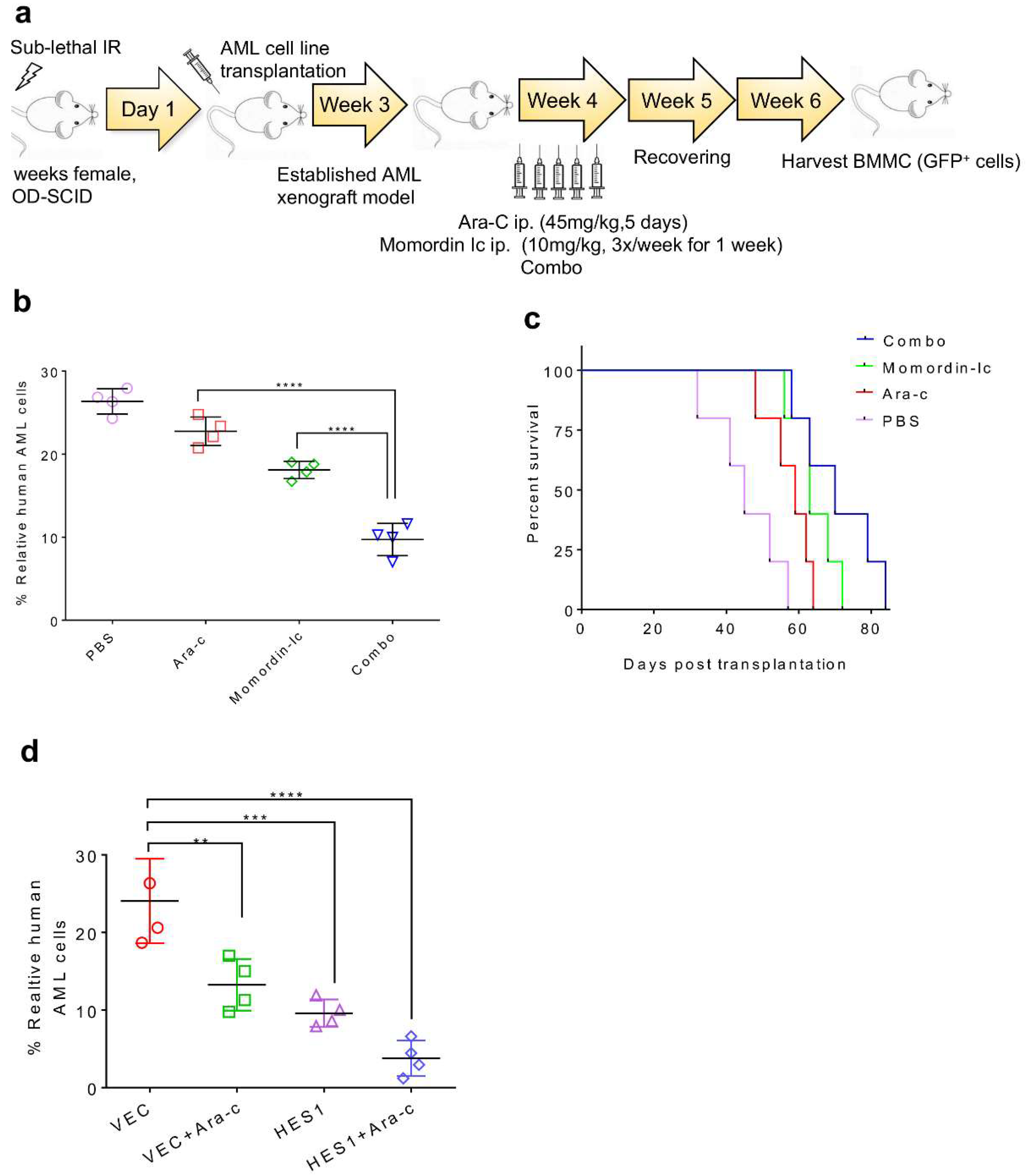
2.3. SIRT3 de-SUMOylation Confers AML Chemoresistance In Vitro and In Vivo
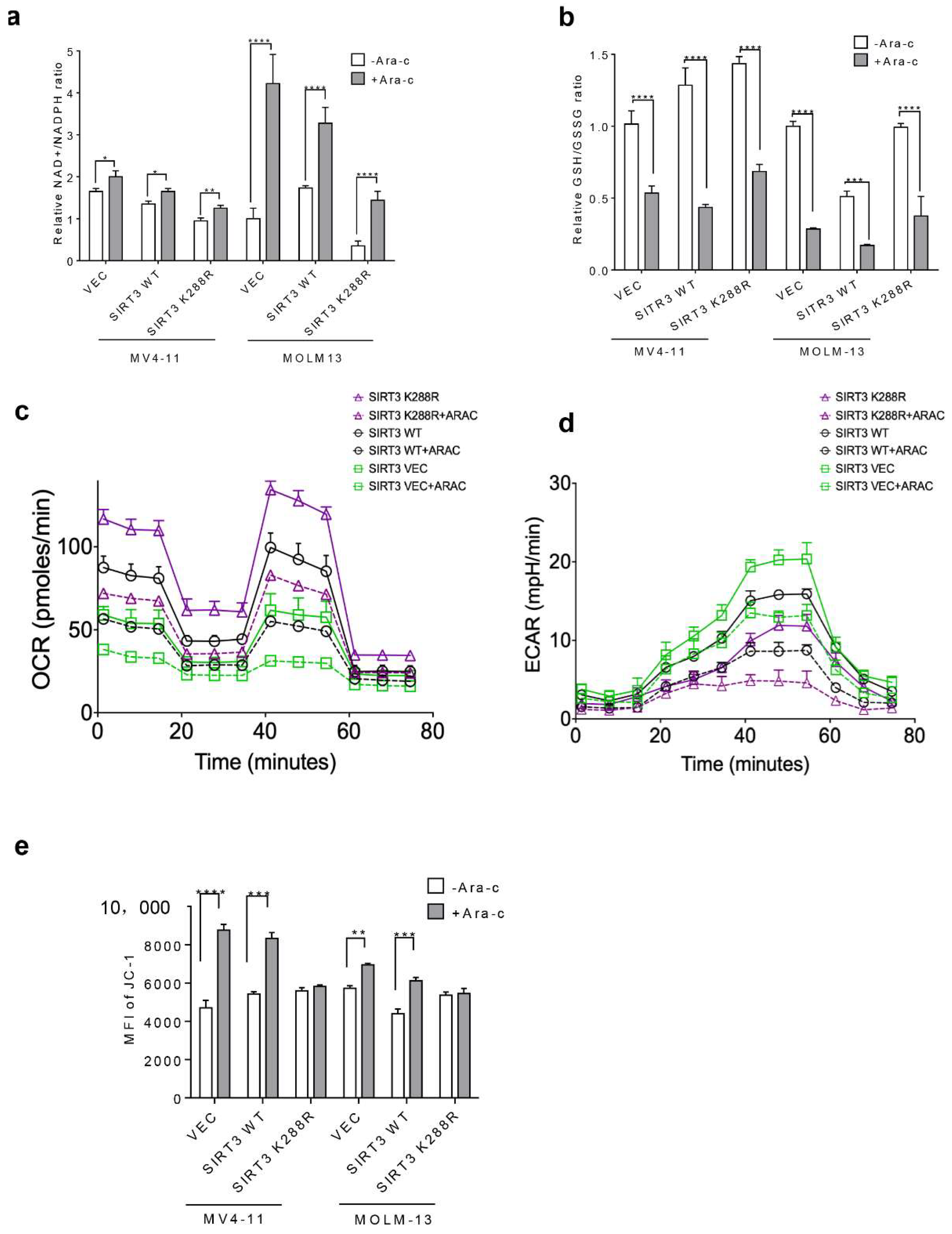
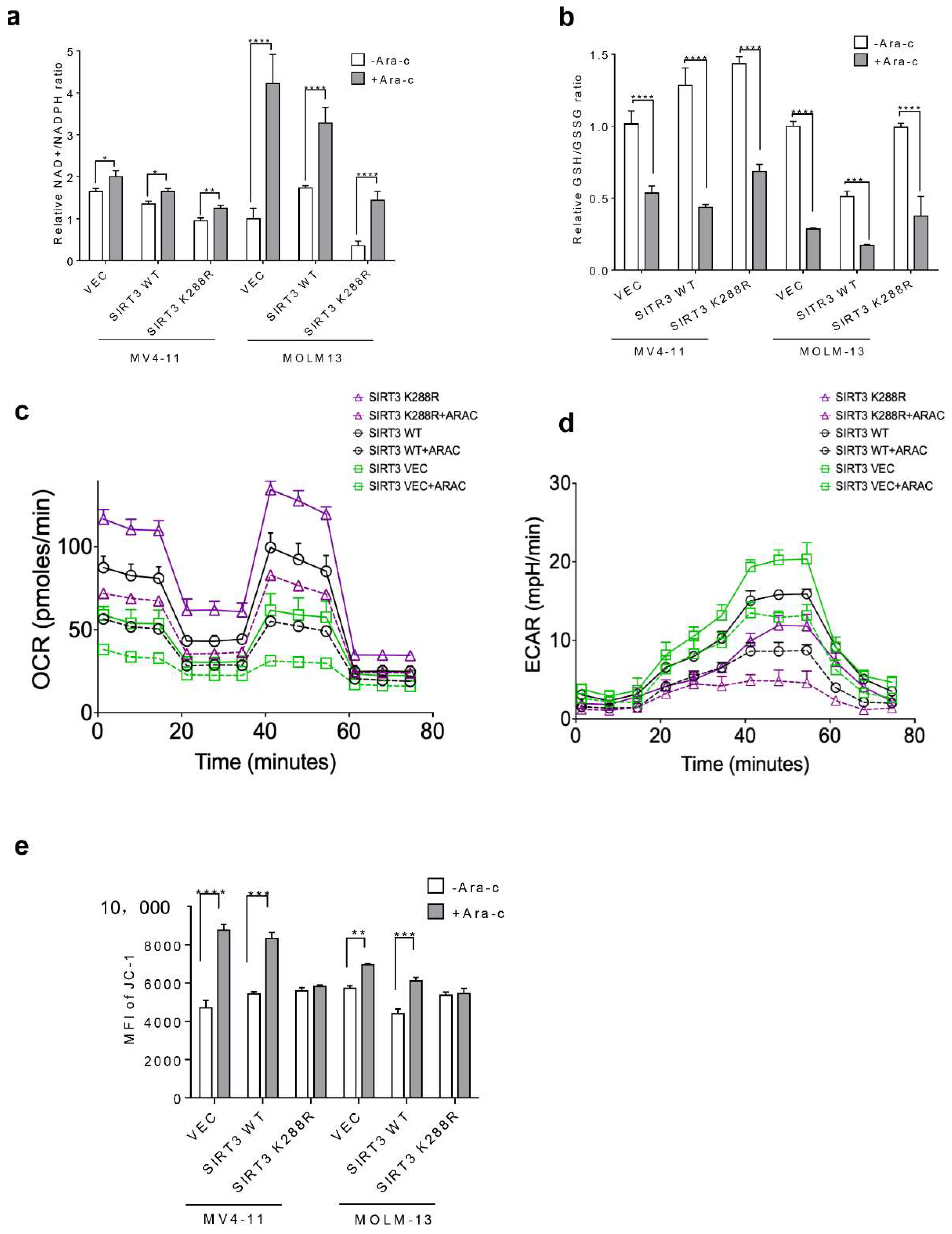
2.4. SIRT3 SUMOylation Regulates Mitochondrial Biogenesis in AML
2.5. SIRT3 de-SUMOylation Confers AML Chemoresistance via Down-Regulating HES1 Dependent FAO
2.6. Inhibition of SIRT3 de-SUMOylation Synergizes with Ara-C in AML In Vitro
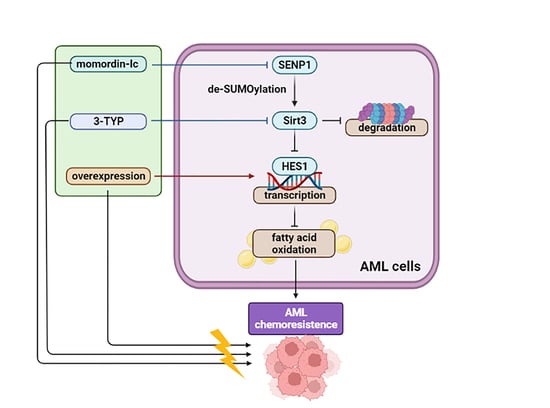
2.7. Combination Therapy Targeting SIRT3 SUMOylation Pathways
3. Discussion
4. Materials and Methods
4.1. Drug Compounds and Antibodies
4.2. Cell Lines, Primary Cells and Culture Conditions
4.3. Lentiviral Plasmids Packaging
4.4. Flow Cytometry Sorting of Stable Transfectants
4.5. Ubiquitination Assay
4.6. Co-Immunoprecipitation and Immunoblotting
4.7. Annexin V/7AAD Apoptosis Detection Assay
4.8. Metabolism Assays
4.9. Extracellular Acidification Rate and Basal Oxygen Consumption Rate
4.10. Transcriptome Sequencing (RNA-Seq) and Data Processing
4.11. Drug Synergy
4.12. Animal Studies
4.13. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD(+) and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The sirtuin family in cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes. Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes. Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661. [Google Scholar] [CrossRef]
- Aventaggiato, M.; Vernucci, E.; Barreca, F.; Russo, M.A.; Tafani, M. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol. Ther. 2021, 221, 107748. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L. The many faces of sirtuins: Sirtuins and the Warburg effect. Nat. Med. 2014, 20, 24–25. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO. Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Paku, M.; Haraguchi, N.; Takeda, M.; Fujino, S.; Ogino, T.; Takahashi, H.; Miyoshi, N.; Uemura, M.; Mizushima, T.; Yamamoto, H.; et al. SIRT3-Mediated SOD2 and PGC-1alpha Contribute to Chemoresistance in Colorectal Cancer Cells. Ann. Surg. Oncol. 2021, 28, 4720–4732. [Google Scholar] [CrossRef]
- Ma, J.; Liu, B.; Yu, D.; Zuo, Y.; Cai, R.; Yang, J.; Cheng, J. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br. J. Haematol. 2019, 187, 49–64. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Gupta, M.; Kim, G.; Rajamohan, S.B.; Isbatan, A.; Gupta, M.P. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 2009, 119, 2758–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Cao, Y.; Zheng, Q.; Tu, J.; Zhou, W.; He, J.; Zhong, J.; Chen, Y.; Wang, J.; Cai, R.; et al. SENP1-Sirt3 Signaling Controls Mitochondrial Protein Acetylation and Metabolism. Mol. Cell 2019, 75, 823–834.e5. [Google Scholar] [CrossRef]
- Ma, Z.; Xu, J.; Wu, L.; Wang, J.; Lin, Q.; Chowdhury, F.A.; Mazumder, M.H.H.; Hu, G.; Li, X.; Du, W. Hes1 deficiency causes hematopoietic stem cell exhaustion. Stem Cells 2020, 38, 756–768. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.; Look, A.T. NOTCH and PI3K-AKT pathways intertwined. Cancer Cell 2007, 12, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Gill, G. SUMO and ubiquitin in the nucleus: Different functions, similar mechanisms? Genes Dev. 2004, 18, 2046–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xie, F.; Zhang, J.; Dijke, P.T.; Zhou, F. SUMO-triggered ubiquitination of NR4A1 controls macrophage cell death. Cell Death Differ. 2017, 24, 1530–1539. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Ma, L.; Weng, W.; Wang, Y.; Liu, H.; Guo, R.; Gao, Y.; Tu, J.; Xu, T.L.; Cheng, J.; et al. DUSP6 SUMOylation protects cells from oxidative damage via direct regulation of Drp1 dephosphorylation. Sci. Adv. 2020, 6, eaaz0361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Zheng, Q.; Wan, G.; Guo, F.; Zeng, X.; Shi, P. Impact of posttranslational modifications in pancreatic carcinogenesis and treatments. Cancer Metastasis Rev. 2021, 40, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Zhou, T.Y.; Wu, Y.G.; Zhang, Y.Z.; Bao, Y.W.; Zhao, Y. SIRT3 retards intervertebral disc degeneration by anti-oxidative stress by activating the SIRT3/FOXO3/SOD2 signaling pathway. Eur. Rev. Med. Pharm. Sci. 2019, 23, 9180–9188. [Google Scholar] [CrossRef]
- Cao, Y.; Li, P.; Wang, H.; Li, L.; Li, Q. SIRT3 promotion reduces resistance to cisplatin in lung cancer by modulating the FOXO3/CDT1 axis. Cancer Med. 2021, 10, 1394–1404. [Google Scholar] [CrossRef]
- Wu, H.; Liu, B.; Chen, Z.; Li, G.; Zhang, Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1alpha/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. 2020, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Loo, S.Y.; Toh, L.P.; Xie, W.H.; Pathak, E.; Tan, W.; Ma, S.; Lee, M.Y.; Shatishwaran, S.; Yeo, J.Z.Z.; Yuan, J.; et al. Fatty acid oxidation is a druggable gateway regulating cellular plasticity for driving metastasis in breast cancer. Sci. Adv. 2021, 7, eabh2443. [Google Scholar] [CrossRef]
- Heiserman, J.P.; Nallanthighal, S.; Gifford, C.C.; Graham, K.; Samarakoon, R.; Gao, C.; Sage, J.J.; Zhang, W.; Higgins, P.J.; Cheon, D.J. Heat Shock Protein 27, a Novel Downstream Target of Collagen Type XI alpha 1, Synergizes with Fatty Acid Oxidation to Confer Cisplatin Resistance in Ovarian Cancer Cells. Cancers 2021, 13, 4855. [Google Scholar] [CrossRef]
- Chen, Z.; Wu, L.; Zhou, J.; Lin, X.; Peng, Y.; Ge, L.; Chiang, C.M.; Huang, H.; Wang, H.; He, W. N6-methyladenosine-induced ERRgamma triggers chemoresistance of cancer cells through upregulation of ABCB1 and metabolic reprogramming. Theranostics 2020, 10, 3382–3396. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Saitoh, K.; Yang, H.; Sekihara, K.; Yamatani, K.; Ruvolo, V.; Taka, H.; Kaga, N.; Kikkawa, M.; Arai, H.; et al. Inhibition of FAO in AML co-cultured with BM adipocytes: Mechanisms of survival and chemosensitization to cytarabine. Sci. Rep. 2018, 8, 16837. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Wang, W.; Zhou, B.; Cui, X.; Yang, H.; Zhang, S. Monensin suppresses cell proliferation and invasion in ovarian cancer by enhancing MEK1 SUMOylation. Exp. Ther. Med. 2021, 22, 1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Jiang, X. SUMOylation of vascular endothelial growth factor receptor 2 inhibits the proliferation, migration, and angiogenesis signaling pathway in non-small cell lung cancer. Anticancer Drugs 2020, 31, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Shen, Y.; Wei, W.; Wang, W.; Jiang, D.; Ren, Y.; Peng, Z.; Fan, Q.; Cheng, J.; Ma, J. Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation. Int. J. Mol. Sci. 2022, 23, 8282. https://doi.org/10.3390/ijms23158282
Zhang Y, Shen Y, Wei W, Wang W, Jiang D, Ren Y, Peng Z, Fan Q, Cheng J, Ma J. Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation. International Journal of Molecular Sciences. 2022; 23(15):8282. https://doi.org/10.3390/ijms23158282
Chicago/Turabian StyleZhang, Yirong, Yajie Shen, Weiqing Wei, Wenhan Wang, Daiji Jiang, Yizhuo Ren, Zijing Peng, Qiuju Fan, Jinke Cheng, and Jiao Ma. 2022. "Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation" International Journal of Molecular Sciences 23, no. 15: 8282. https://doi.org/10.3390/ijms23158282
APA StyleZhang, Y., Shen, Y., Wei, W., Wang, W., Jiang, D., Ren, Y., Peng, Z., Fan, Q., Cheng, J., & Ma, J. (2022). Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation. International Journal of Molecular Sciences, 23(15), 8282. https://doi.org/10.3390/ijms23158282