
Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
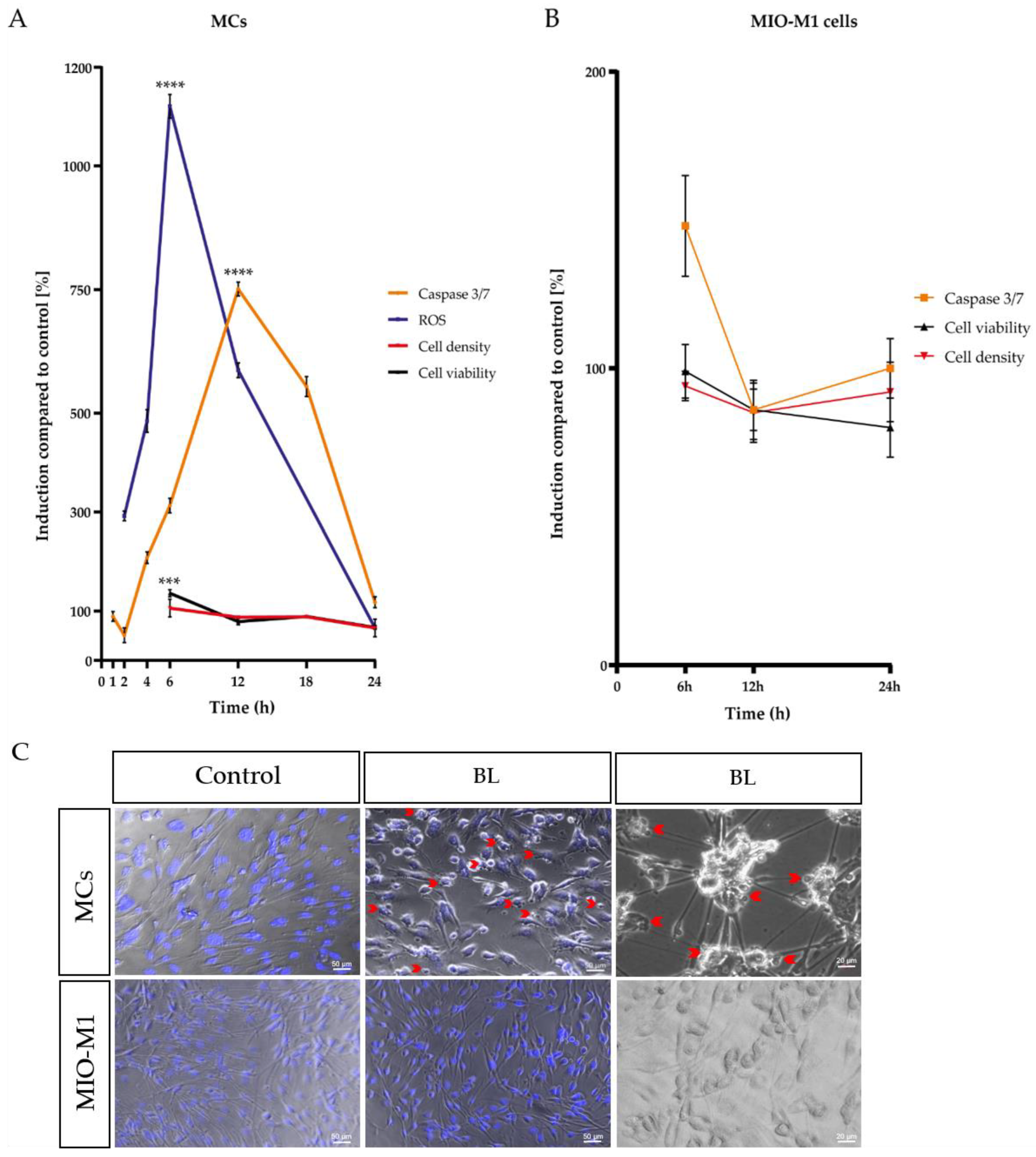
2.1. Single Blue Light Exposure Significantly Enhanced Oxidative Stress Levels in Müller Cell-Derived Cells and MIO-M1 Cells
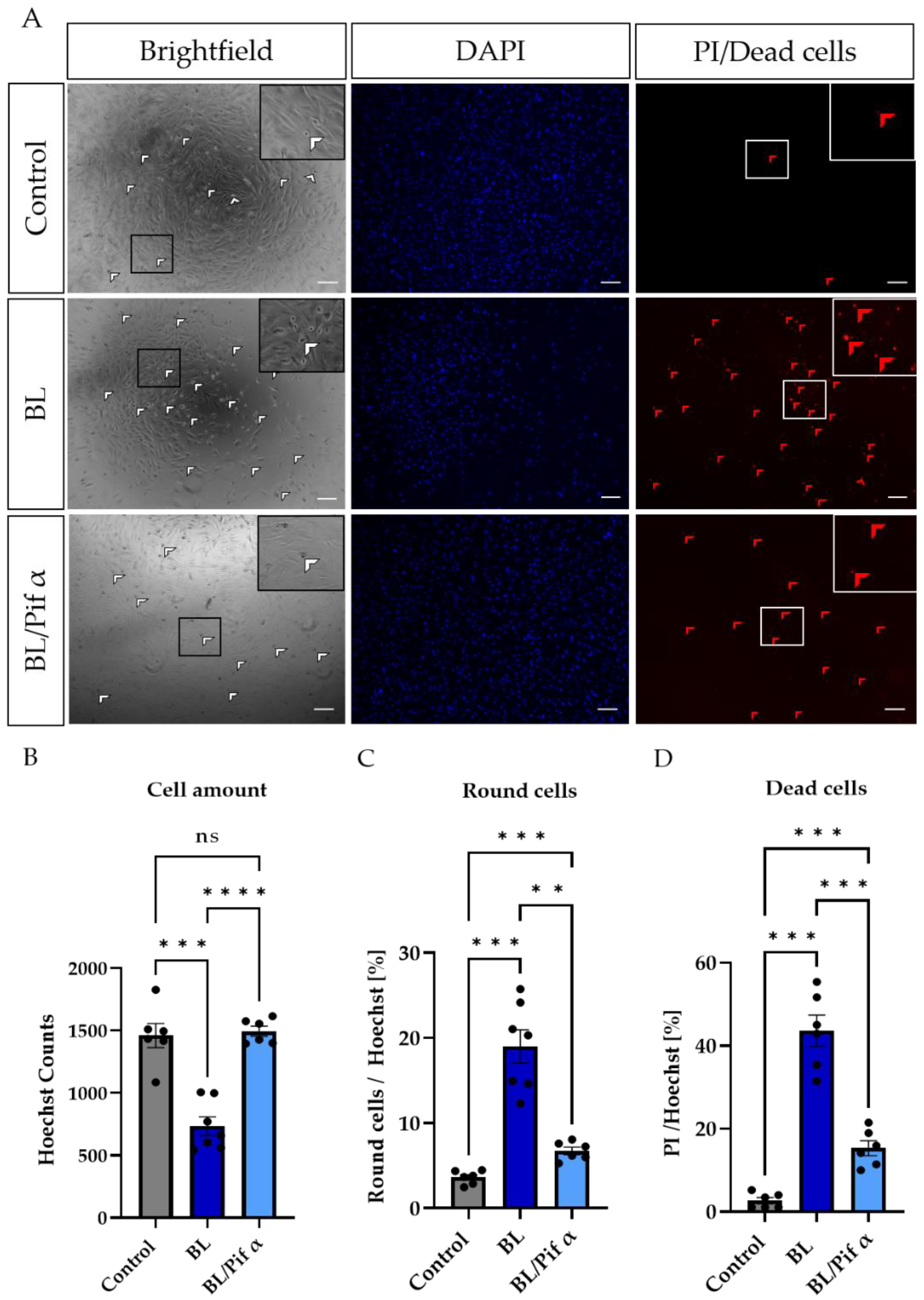
2.2. Blue Light Induced Apoptosis in MCs, but Not in MIO-M1 Cells
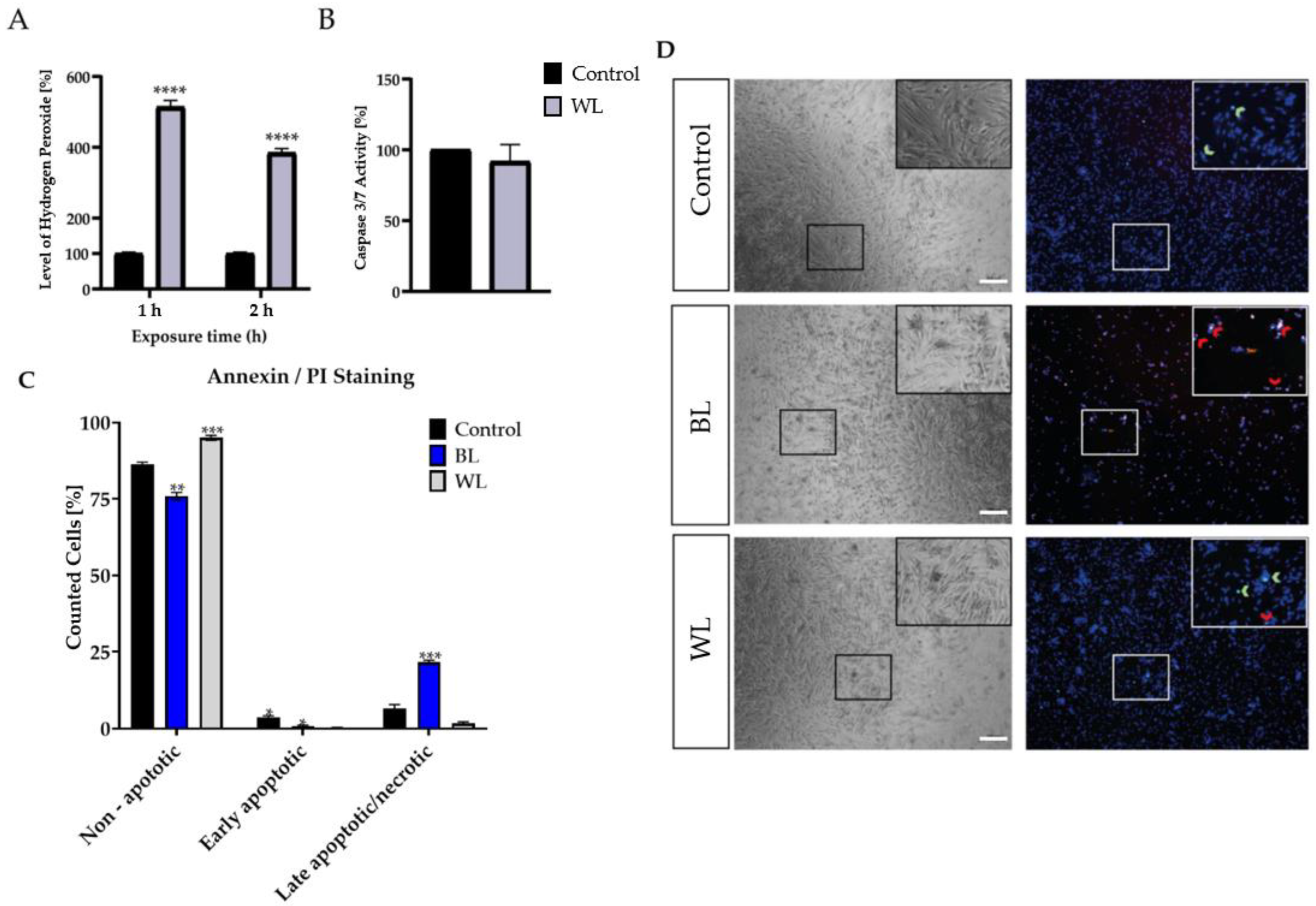
2.3. Particular Blue Light in White Light Artificial Exposure Harms Müller Cell-Derived Cells
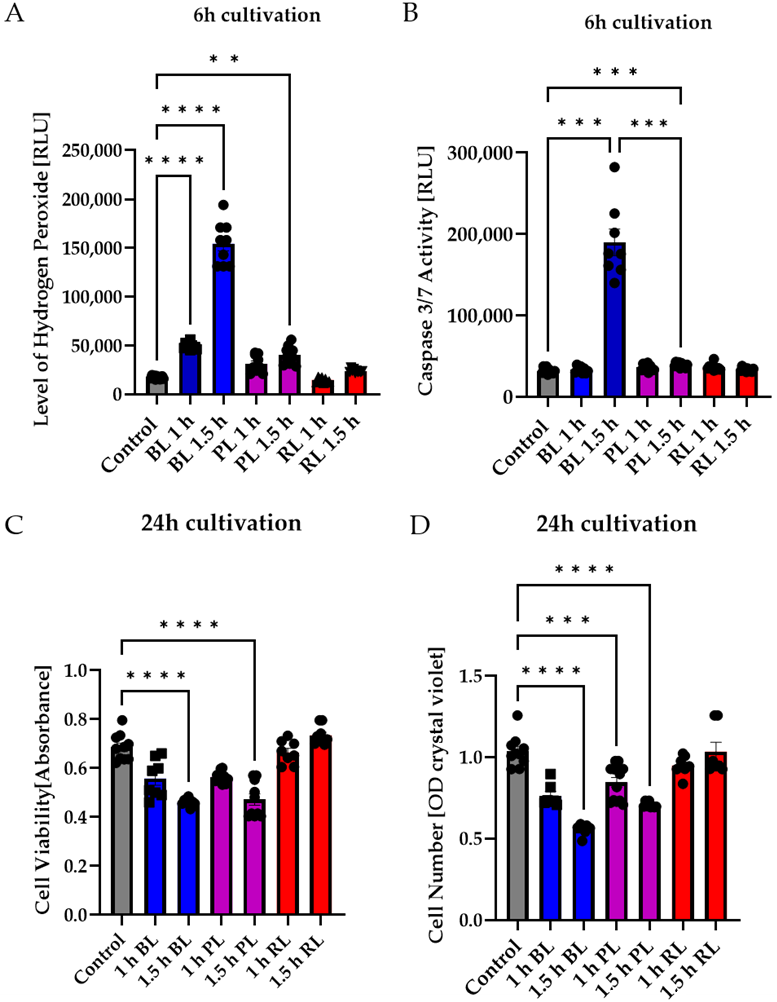
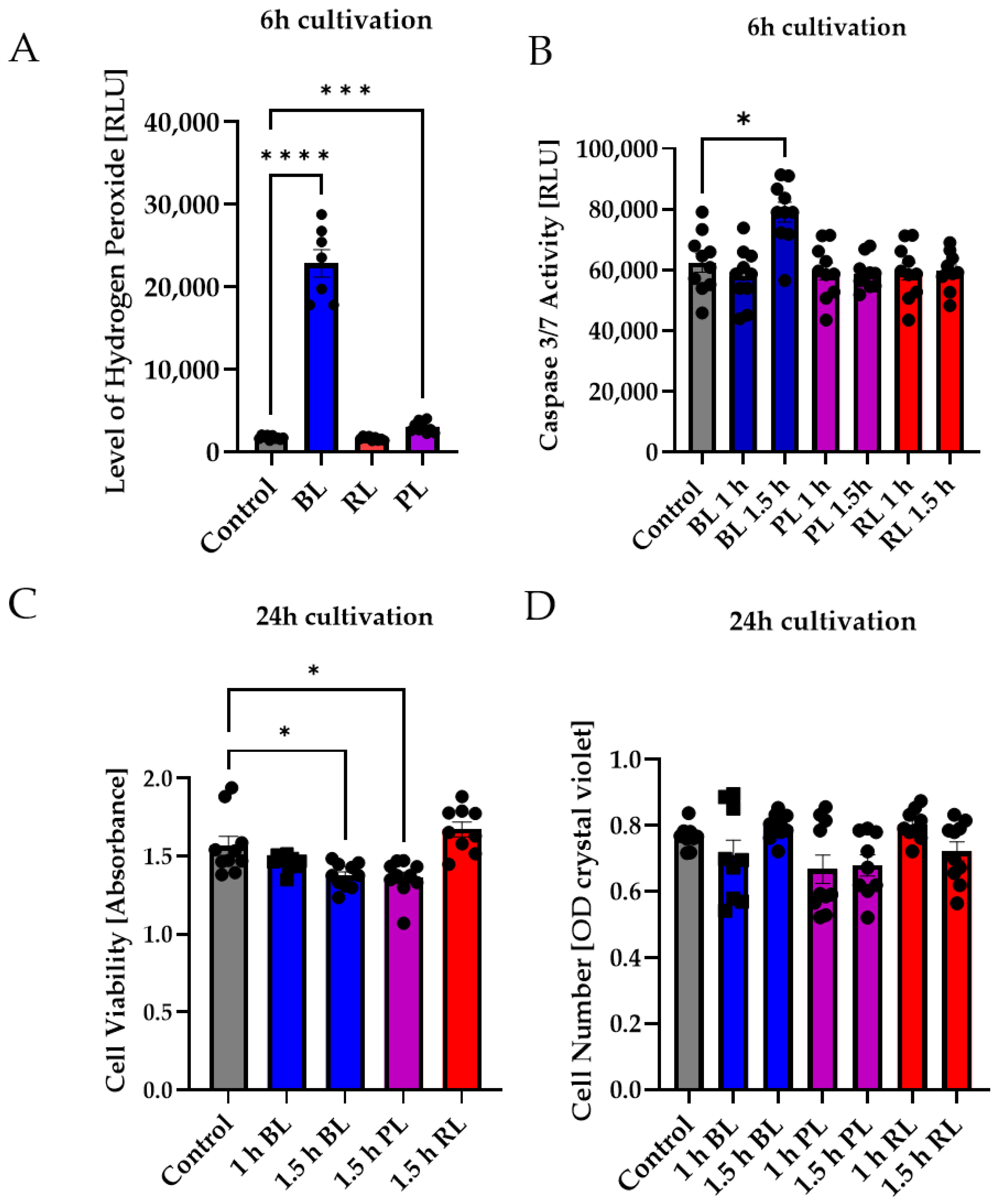
2.4. MIO-M1 Cells Demonstrate Robustness to Blue, Purple, and Red Light Exposure
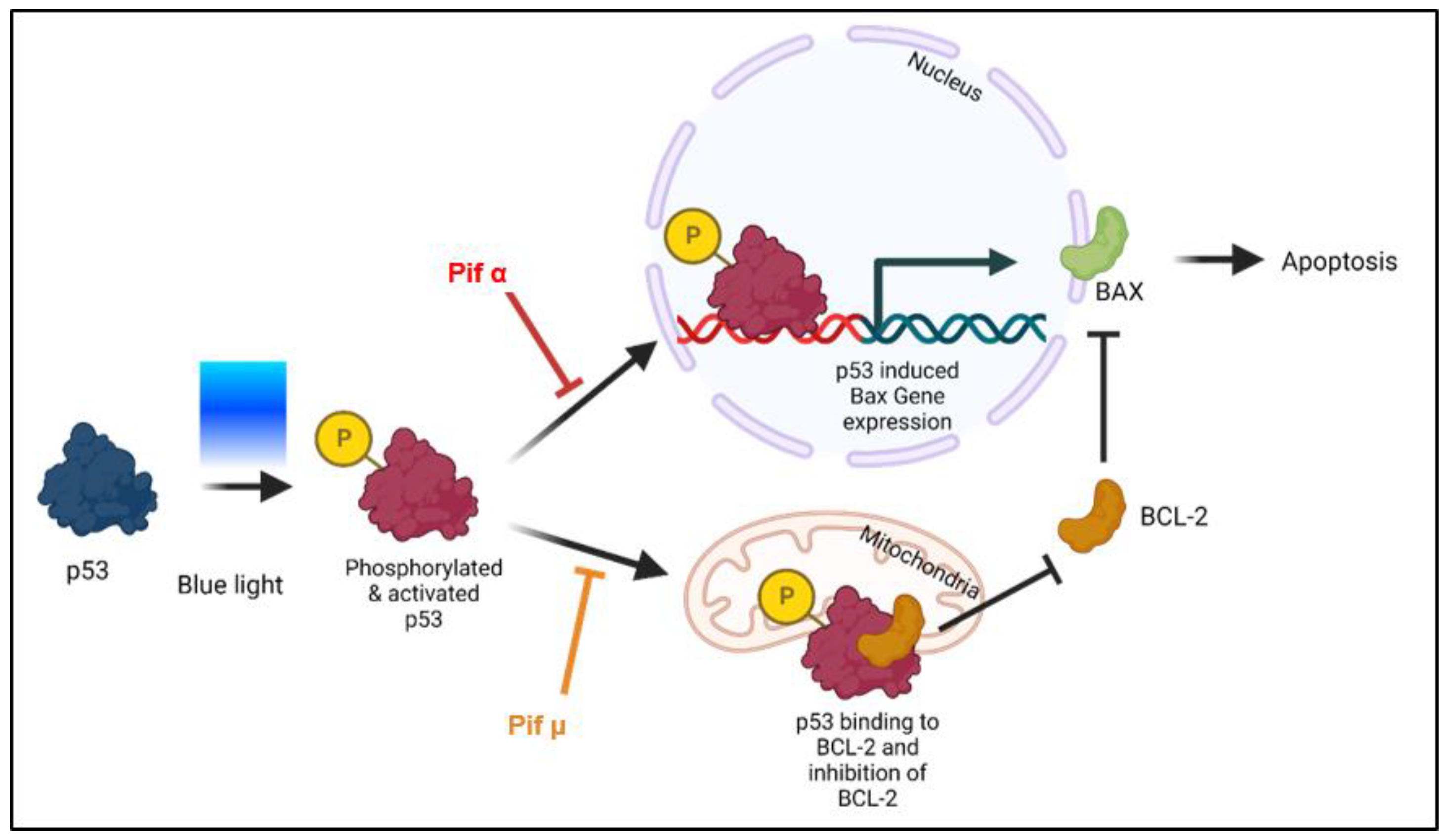
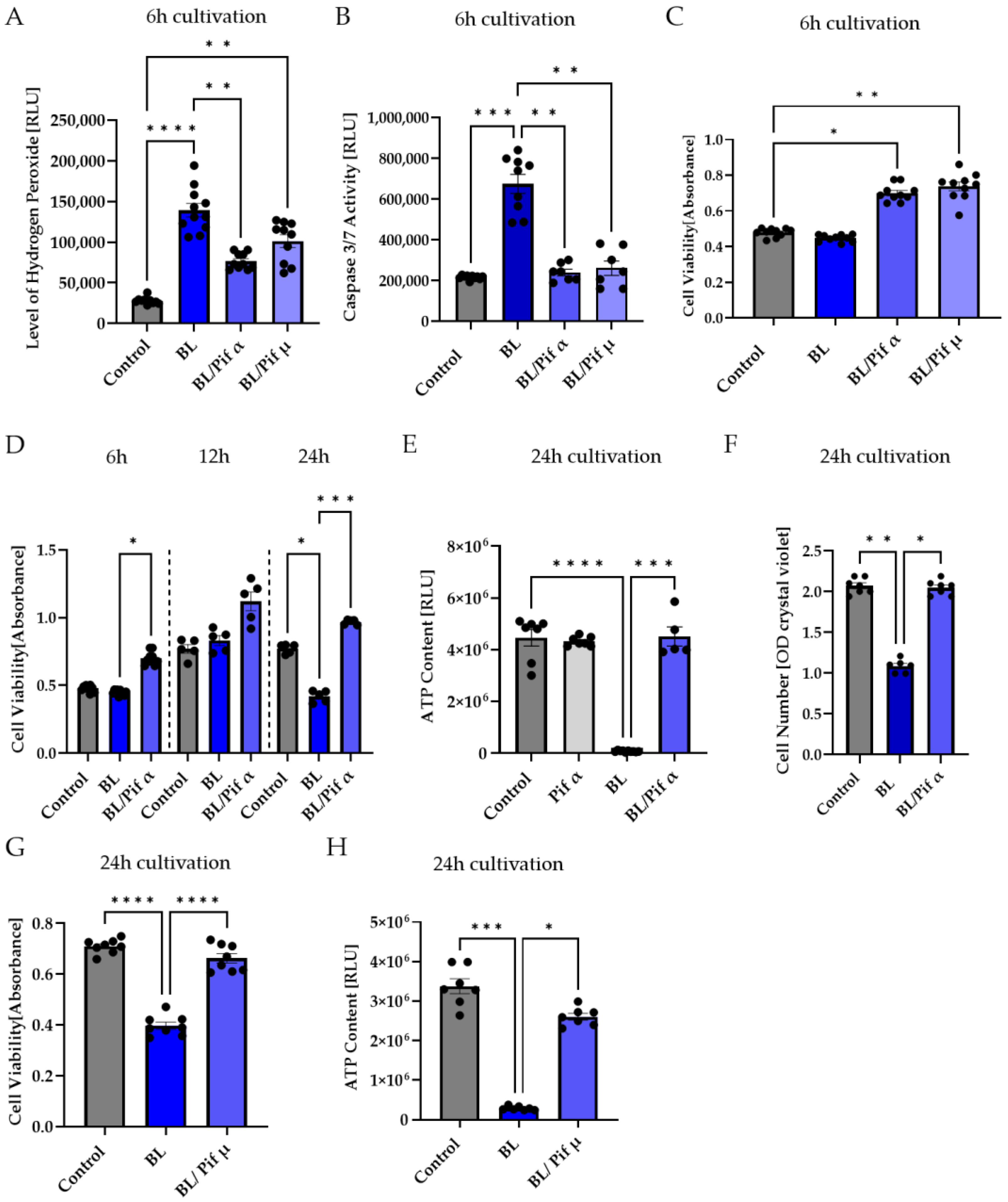
2.5. Blue Light-Induced Apoptosis in Müller Cell-Derived Cells Can Be Prevented by Inhibition of p53
3. Discussion
4. Materials and Methods
4.1. Primary Cells and Cell Lines
4.1.1. Isolation of Müller Cell-Derived Cultures (MC)
4.1.2. Cultivation of MIO-M1 Cell Line
4.2. Light Sources and Exposure Setup
Blue, Red, Purple, and White Light Exposure
4.3. Methods
4.3.1. Cell Viability, Cell Density, ROS ASSAY, Caspase 3/7 Activity, and ATP Assay
4.3.2. Inhibition of p53
4.3.3. Human Apoptosis Array
4.3.4. Annexin V/Propidium Iodide Staining
4.3.5. Propidium Iodide Staining (Dead Staining)
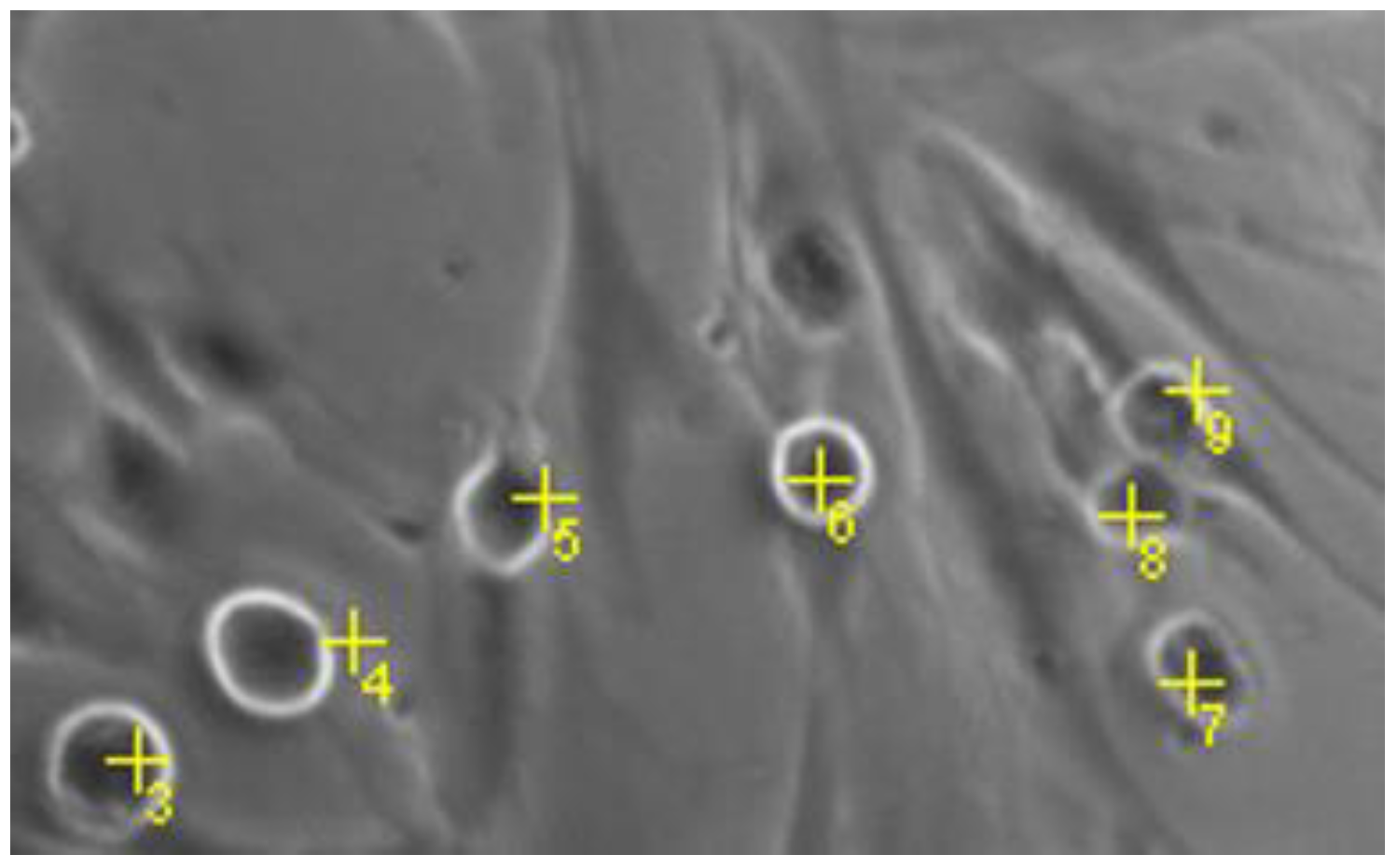
4.3.6. Quantification of Round MCs
4.3.7. Statistical Analysis
4.3.8. Image Generation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Villette, T. Blue Light Hazard: New Knowledge, New Approaches to Maintaining Ocular Health; Essilor International: New York, NY, USA, 2013. [Google Scholar]
- Nakamura, S.; Chichibu, S.F. Introduction to Nitride Semiconductor Blue Lasers and Light Emitting Diodes; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- O’Hagan, J.B.; Khazova, M.; A Price, L.L. Low-energy light bulbs, computers, tablets and the blue light hazard. Eye 2016, 30, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Tsai, C.; Chien, C.; Hsiao, J.C.; Huang, S.; Lee, C.; Lin, H.; Wen, Y.; Tseng, K. The Influence of Low-powered Family LED Lighting on Eyes in Mice Experimental Model. Life Sci. J. 2012, 9, 477–482. [Google Scholar]
- Pardhan, S.; Sapkota, R.P. Eye complications of exposure to blue-violet light. Points Vue Int. Rev. Ophthalmic Opt. 2016. Available online: www.pointsdevue.com (accessed on 12 October 2022).
- Dunaief, J.L.; Dentchev, T.; Ying, G.S.; Milam, A.H. The role of apoptosis in age-related macular degeneration. Arch. Ophthalmol. 2002, 120, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.L.; Mainster, M.A. Circadian photoreception: Ageing and the eye’s important role in systemic health. Ophthalmology 2008, 11, 1439–1444. [Google Scholar] [CrossRef]
- Behar-Cohen, F.; Martinsons, C.; Viénot, F.; Zissis, G.; Barlier-Salsi, A.; Cesarini, J.P.; Enouf, O.; Garcia, M.; Picaud, S.; Attia, D. Light-emitting diodes (LED) for domestic lighting: Any risks for the eye? Prog. Retin. Eye Res. 2011, 30, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Czepita, D.; Mojsa, A.; Ustianowska, M.; Czepita, M.; Lachowicz, E. Reading, writing, working on a computer or watching television, and myopia. Klin Oczna 2010, 112, 293–295. [Google Scholar]
- Lou, L.; Arumugam, B.; Hung, L.F.; Beach, K.M.; She, Z.; Smith, E.L.; Ostrin, L.A. Effects of narrowband light rearing on activity and the pupil in infant rhesus monkeys. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1338. [Google Scholar]
- Jaadane, I.; Villalpando Rodriguez, G.; Boulenguez, P. Retinal phototoxicity and the evaluation of the blue light hazard of a new solid-state lighting technology. Sci. Rep. 2020, 10, 6733. [Google Scholar] [CrossRef]
- Moon, J.; Yun, J.; Yoon, Y.D.; Park, S.I.; Seo, Y.J.; Park, W.S.; Chu, H.Y.; Park, K.H.; Lee, M.Y.; Lee, C.W.; et al. Blue Light Effect on Retinal Pigment Epithelial Cells by Display Devices. Integr. Biol. 2017, 9, 436–443. [Google Scholar] [CrossRef]
- Hunter, J.J.; Morgan, M.J.; Merigan, W.H.; Sliney, D.H.; Sparrow, J.R.; Williams, D.R. The susceptibility of the retina to photochemical damage from visible light. Prog. Retin. Eye Res. 2012, 31, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Margrain, T.H.; Boulton, M.; Marshall, J.; Sliney, D.H. Do blue light filters confer protection against age-related macular degeneration? Prog. Retin. Eye Res. 2004, 23, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Oveson, B.C.; Jo, Y.J.; Lauer, T.W.; Usui, S.; Komeima, K.; Xie, B.; Campochiaro, P.A. Increased expression of glutathione peroxidase 4 strongly protects retina from oxidative damage. Antioxid. Redox Signal. 2009, 11, 715–724. [Google Scholar] [CrossRef]
- Algvere, P.V.; Marshall, J.; Seregard, S. Age-related maculopathy and the impact of blue light hazard. Acta Ophthalmol. Scand. 2006, 84, 4–15. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 signaling is impaired in the aging RPE given an oxidative insult. Exp. Eye Res. 2014, 119, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Nelson, K.C.; Wu, M.; Sternberg, P., Jr.; Jones, D.P. Oxidative damage and protection of the RPE. Prog. Retin. Eye Res. 2000, 19, 205–221. [Google Scholar] [CrossRef]
- Arnault, E.; Barrau, C.; Nanteau, C.; Gondouin, P.; Bigot, K.; Viénot, F.; Gutman, E.; Fontaine, V.; Villette, T.; Cohen-Tannoudji, D.; et al. Phototoxic action spectrum on a retinal pigment epithelium model of age-related macular degeneration exposed to sunlight normalized conditions. PLoS ONE 2013, 8, e71398. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Aspects Med. 2012, 33, 399–417. [Google Scholar] [CrossRef]
- Masuda, T.; Boulton, S.M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid. Med. Cell. Longev. 2017, 33, 399–417. [Google Scholar] [CrossRef]
- Sasaki, M.; Yuki, K.; Kurihara, T.; Miyake, S.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K.; Ozawa, Y. Biological role of lutein in the light-induced retinal degeneration. J. Nutr. Biochem. 2012, 23, 423–429. [Google Scholar] [CrossRef]
- Ozawa, Y. Oxidative stress in the light-exposed retina and its implication in age-related macular degeneration. Redox Biol. 2020, 37, 101779. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sawada, O.; Kohno, H.; Le, Y.Z.; Subauste, C.; Maeda, T.; Maeda, A. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 2013, 288, 7506–7518. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Ozawa, Y.; Osada, H.; Lin, J.B.; Toda, E.; Watanabe, M.; Yuki, K.; Kubota, S.; Apte, R.S.; Tsubota, K. Neuroprotective role of retinal SIRT3 against acute photo-stress. NPJ Aging Mech. Dis. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spector, A. Oxidative stress-induced cataract: Mechanism of action. FASEB J. 1995, 9, 1173–1182. [Google Scholar] [CrossRef]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 3, 11282–11287. [Google Scholar] [CrossRef]
- Gritz, D.C.; Montes, C.; Atalla, L.R.; Wu, G.S.; Sevanian, A.; Rao, N.A. Histochemical localization of superoxide production in experimental autoimmune uveitis. Curr. Eye Res. 1991, 10, 927–931. [Google Scholar] [CrossRef]
- Niesman, M.R.; Johnson, K.A.; Penn, J.S. Therapeutic effect of liposomal superoxide dismutase in an animal model of retinopathy of prematurity. Neurochem. Res. 1997, 22, 597–605. [Google Scholar] [CrossRef]
- Alio, J.L.; Ayala, M.J.; Mulet, M.E.; Artola, A.; Ruiz, J.M.; Bellot, J. Antioxidant therapy in the treatment of experimental acute corneal inflammation. Ophthalmic Res. 1995, 27, 136–143. [Google Scholar] [CrossRef]
- Ham, W.T.; Mueller, H.A.; Ruffolo, J.J.; Millen, J.E.; Cleary, S.F.; Guerry, R.K.; Guerry, D., 3rd. Basic mechanisms underlying the production of photochemical lesions in the mammalian retina. Curr. Eye Res. 1984, 3, 165–174. [Google Scholar] [CrossRef]
- Iandiev, I.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Grimm, C.; Remé, C.E.; Reichenbach, A.; Pannicke, T.; Bringmann, A. Müller Cell Response to Blue Light Injury of the Rat Retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3559–3567. [Google Scholar] [CrossRef]
- Cuenca, N.; Fernandez-Sanchez, L.; Campello, L.; Maneu, V.; De la Villa, P.; Lax, P.; Pinilla, I. Cellular responses following retinal injuries and therapeutic approaches for neurodegenerative diseases. Prog. Retin. Eye Res. 2014, 43, 17–75. [Google Scholar] [CrossRef] [PubMed]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A Journey Into the Retina: Müller Glia Commanding Survival and Death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Bora, N.S.; Matta, B.; Lyzogubov, V.V.; Bora, P.S. Relationship between the complement system, risk factors and prediction models in age-related macular degeneration. Mol. Immunol. 2015, 63, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.M.; McLeod, D.S.; Bhutto, I.A.; Villalonga, M.B.; Seddon, J.M.; Lutty, G.A. Idiopathic preretinal glia in aging and age-related macular degeneration. Exp Eye Res. 2016, 150, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Franze, K.; Grosche, J.; Skatchkov, S.N.; Schinkinger, S.; Foja, C.; Schild, D.; Uckermann, O.; Travis, K.; Reichenbach, A.; Guck, J. Muller cells are living optical fibers in the vertebrate retina. Proc. Natl. Acad. Sci. USA 2007, 104, 8287–8292. [Google Scholar] [CrossRef]
- Ardeljan, D.; Chan, C.C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Retin. Eye Res. 2013, 37, 68–89. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.H.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Sun, Q.; Kim, H.E.; Cho, H.; Shi, S.; Kim, B.; Kim, O. Red light-emitting diode irradiation regulates oxidative stress and inflammation through SPHK1/NF-κB activation in human keratinocytes. J. Photochem. Photobiol. B Biol. 2018, 186, 31–40. [Google Scholar] [CrossRef]
- Zhang, J.; Yue, X.; Luo, H.; Jiang, W.; Mei, Y.; Ai, L.; Gao, G.; Wu, Y.; Yang, H.; An, J.; et al. Illumination with 630 nm Red Light Reduces Oxidative Stress and Restores Memory by Photo-Activating Catalase and Formaldehyde Dehydrogenase in SAMP8 Mice. Antioxid. Redox Signal. 2019, 30, 1432–1449. [Google Scholar] [CrossRef]
- Núñez-Álvarez, C.; Suárez-Barrio, C.; del Olmo Aguado, S.; Osborne, N.N. Blue light negatively affects the survival of ARPE19 cells through an action on their mitochondria and blunted by red light. Acta Ophthalmol. Scand. Found. 2018, 97, e103–e115. [Google Scholar] [CrossRef]
- Godley, B.F.; Shamsi, F.A.; Liang, F.Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Buehl, A.M.; Doepper, H.; Grauthoff, S.; Kiebler, T.; Peters, L.; Hurst, J.; Kuehn, S.; Bartz-Schmidt, K.U.; Dick, H.B.; Joachim, S.C.; et al. Oxidative stress-induced retinal damage is prevented by mild hypothermia in an ex vivo model of cultivated porcine retinas. Clin. Exp. Ophthalmol. 2020, 48, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, F.; Cervellati, C.; Romani, A.; Cremonini, E.; Sticozzi, C.; Belmonte, G.; Pessina, F.; Valacchi, G. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic. Res. 2014, 48, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, S.; Hurst, J.; Rensinghoff, F.; Tsai, T.; Grauthoff, S.; Satgunarajah, Y.; Dick, H.B.; Schnichels, S.; Joachim, S.C. Degenerative effects of cobalt-chloride treatment on neurons and microglia in a porcine retina organ culture model. Exp. Eye Res. 2017, 155, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Sarna, T. Properties and function of the ocular melanin—A photobiophysical view. J. Photochem. Photobiol. 1992, 12, 215–258. [Google Scholar] [CrossRef]
- Boulton, M.; Różanowska, M.; Różanowski, B. Retinal photodamage. J. Photochem. Photobiol. 2001, 64, 144–161. [Google Scholar] [CrossRef]
- Hollborn, M.; Ulbricht, E.; Rillich, K.; Dukic-Stefanovic, S.; Wurm, A.; Wagner, L.; Reichenbach, A.; Wiedemann, P.; Limb, G.A.; Bringmann, A.; et al. The human Müller cell line MIO-M1 expresses opsins. Mol. Vis. 2011, 17, 2738–2750. [Google Scholar]
- Rios, M.N.; Marchese, N.A.; Guido, M.E. Expression of Non-visual Opsins Opn3 and Opn5 in the Developing Inner Retinal Cells of Birds. Light-Responses in Müller Glial Cells. Front. Cell. Neurosci. 2019, 13, 376. [Google Scholar] [CrossRef]
- Kuse, Y.; Ogawa, K.; Tsuruma, K.; Shimazawa, M.; Hara, H. Damage of photoreceptor-derived cells in culture induced by light emitting diode-derived blue light. Sci. Rep. 2014, 4, 5223. [Google Scholar] [CrossRef]
- Mellerio, J. Light effects on the retina. Princ. Pract. Ophthalmol. Basic Sci. 1994, 116, 1326–1345. [Google Scholar]
- García-Silva, M.T.; Ribes, A.; Campos, Y.; Garavaglia, B.; Arenas, J. Syndrome of encephalopathy, petechiae, and ethylmalonic aciduria. Pediatr. Neurol. 1997, 17, 165–170. [Google Scholar] [CrossRef]
- Hockberger, P.E.; Skimina, T.A.; Centonze, V.E.; Lavin, C.; Chu, S.; Dadras, S.; Reddy, J.K.; White, J.G. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc. Natl. Acad. Sci. USA 1999, 96, 6255–6260. [Google Scholar] [CrossRef] [PubMed]
- Helm, K.; Beyreis, M.; Mayr, C.; Ritter, M.; Jakab, M.; Kiesslich, T.; Plaetzer, K. Cell Death Discrimination and Screening Method by Simple and Cost-Effective Viability Analysis. Cell. Physiol. Biochem. 2017, 41, 1011–1019. [Google Scholar] [CrossRef]
- Peters, S.; Griebsch, M.; Klemm, M.; Haueisen, J.; Hammer, M. Hydrogen peroxide modulates energy metabolism and oxidative stress in cultures of permanent human Müller cells MIO-M1. J. Biophotonics 2017, 9, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Ozaki, T.; Nakanishi, M.; Kikuchi, H.; Yoshida, K.; Horie, H.; Kuwano, H.; Nakagawara, A. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes Cells 2007, 4, 461–471. [Google Scholar] [CrossRef]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef]
- Simabuco, F.M.; Morale, M.G.; Pavan, I.C.; Morelli, A.P.; Silva, F.R.; Tamura, R.E. p53 and metabolism: From mechanism to therapeutics. Oncotarget 2018, 9, 23780–23823. [Google Scholar] [CrossRef]
- Toft-Kehler, A.K.; Gurubaran, I.S.; Desler, C.; Rasmussen, L.J.; Skytt, D.M.; Kolko, M. Oxidative Stress-Induced Dysfunction of Müller Cells During Starvation. Invest. Ophthalmol. Vis. Sci. 2016, 6, 2721–2728. [Google Scholar] [CrossRef]
- Pereiro, X.; Ruzafa, N.; Acera, A.; Urcola, A.; Vecino, E. Optimization of a Method to Isolate and Culture Adult Porcine, Rats and Mice Müller Glia in Order to Study Retinal Diseases. Front. Cell. Neurosci. 2020, 14, 7. [Google Scholar] [CrossRef]
- Limb, G.A.; Salt, T.E.; Munro, P.M.; Moss, S.E.; Khaw, P.T. In vitro characterization of a spontaneously immortalized human Müller cell line (MIO-M1). Investig. Ophthalmol. Vis. Sci. 2002, 43, 864–869. [Google Scholar]
- Schnichels, S.; Hagemann, U.; Januschowski, K.; Hofmann, J.; Bartz-Schmidt, K.U.; Szurman, P.; Spitzer, M.S.; Aisenbrey, S. Comparative toxicity and proliferation testing of aflibercept, bevacizumab and ranibizumab on different ocular cells. Br. J. Ophthalmol. 2013, 97, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Schnichels, S.; Schultheiß, M.; Hofmann, J.; Szurman, P.; Bartz-Schmidt, K.U.; Spitzer, M.S. Trichostatin A induces cell death at the concentration recommended to differentiate the RGC-5 cell line. Neurochem. Int. 2012, 6, 581–591. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fietz, A.; Hurst, J.; Schnichels, S. Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells. Int. J. Mol. Sci. 2022, 23, 14540. https://doi.org/10.3390/ijms232314540
Fietz A, Hurst J, Schnichels S. Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells. International Journal of Molecular Sciences. 2022; 23(23):14540. https://doi.org/10.3390/ijms232314540
Chicago/Turabian StyleFietz, Agnes, José Hurst, and Sven Schnichels. 2022. "Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells" International Journal of Molecular Sciences 23, no. 23: 14540. https://doi.org/10.3390/ijms232314540
APA StyleFietz, A., Hurst, J., & Schnichels, S. (2022). Out of the Shadow: Blue Light Exposure Induces Apoptosis in Müller Cells. International Journal of Molecular Sciences, 23(23), 14540. https://doi.org/10.3390/ijms232314540