
Fully Symmetric Cyclodextrin Polycarboxylates: How to Determine Reliable Protonation Constants from NMR Titration Data



Abstract
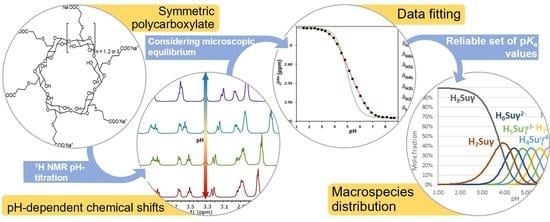
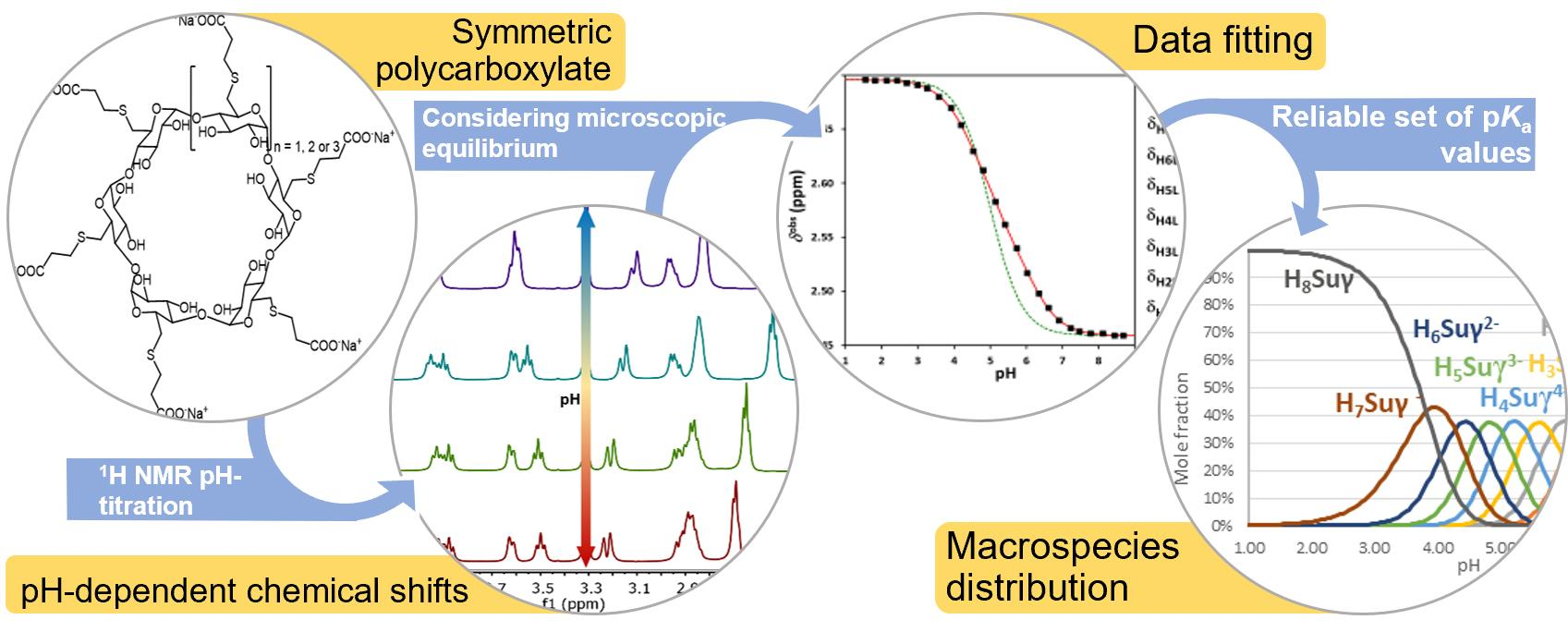
1. Introduction
2. Results
2.1. Acid-Base Characterization of Sualphadex
2.2. The General Macroscopic Evaluation Model
2.3. The Equidistant Macroscopic Evaluation Model
2.4. The Q-Fitting Model
2.5. The Microscopic Site-Binding Model
3. Discussion
3.1. Protonation of Subetadex and Sugammadex
3.2. Basicity Comparison with Related Compounds
4. Materials and Methods
4.1. Materials
4.1.1. Synthesis of Sugammadex-Analogues (General Procedure)
- Su-αCD: 1H NMR (400 MHz, 300 K, D2O) δ(ppm) 5.10 (d, J = 2.8 Hz, 6H, H1), 4.13 (bt, J = 8.5 Hz, 6H, H5), 3.99 (t, J = 9.2 Hz, 6H, H3), 3.67 (dd, J = 9.8, 2.8 Hz, 6H, H2), 3.58 (t, J = 8.8 Hz, 6H, H4), 3.29 (m, 6H, H6a), 2.98 (m, 18H, H6b, H7), 2.72 (m, 12H, H8).
- 13C NMR (100 MHz, 300 K, D2O) δ(ppm) 101.2 (C1), 84.6 (C4), 73.1 (C3), 71.8 (C2), 71.2 (C5), 35.4 (C8), 33.8 (C6), 28.3 (C7). The 13C chemical shifts were read from the DEPT-edited HSQC spectrum.
- Su-βCD: 1H NMR (600 MHz, 300 K, D2O) δ(ppm) 5.18 (d, J = 3.5 Hz, 7H, H1), 4.14–3.85 (m, 14H, H3, H5), 3.77–3.51 (m, 14H, H2, H4), 3.33–3.10 (m, 7H, H6a), 3.12–2.75 (m, 18H, H6b, H7), 2.61 (t, J = 7.3 Hz, 14H, H8).
- 13C NMR (150 MHz, 300 K, D2O) δ(ppm) 178.5 (C9), 100.7 (C1), 83.1 (C4), 72.8 (C2), 71.9 (C3), 71.2 (C5), 36.6 (C8), 33.4 (C6), 28.8 (C7).
- Su-γCD: 1H NMR (600 MHz, 300 K, D2O) δ(ppm) 5.22 (d, J = 3.8 Hz, 8H, H1), 4.010–4.02 (m, 8H, H5), 3.97 (d, J = 9.4 Hz, 8H, H3), 3.70 (dd, J = 9.8, 3.8 Hz, 8H, H2), 3.61 (t, J = 9.4 Hz, 8H, H4), 3.31–3.19 (m, 8H, H6a), 3.01 (dd, J = 13.5, 8.5 Hz, 8H, H6b), 2.97–2.87 (m, 16H, H7), 2.67 (t, J = 7.2 Hz, 16H, H8).
- 13C NMR (150 MHz, 298 K, D2O) δ(ppm) 178.4 (C9), 100.8 (C1), 82.7 (C4), 72.7 (C2), 72.2 (C3), 71.2 (C5), 36.1 (C8), 33.4 (C6), 28.5 (C7).
4.1.2. Synthesis of Mono-Sugammadex
- Mono-Su-γCD: 1H NMR (600 MHz, D2O, 300 K) δ(ppm) 5.22–5.04 (m, 8d, J = 3.8 Hz, 8H, H1, H1′), 4.08–3.99 (m, 1H, H5′), 3.99–3.80 (m, 29H, H3, H3′, H5, H5′, H6a,b), 3.72–3.63 (m, 8H, H2, H2′), 3.59 (m, 8H, H4, H4′), 3.14 (dd, J = 13.9, 2.5 Hz, 1H, H6′a), 2.93 (t, J = 7.1 Hz, 4H, H7*), 2.91–2.85 (m, 1H, H6′b), 2.83 (t, J = 7.4 Hz, 2H, H7′), 2.60 (t, J = 7.1 Hz, 4H, H8*), 2.48 (t, J = 7.4 Hz, 2H, H8′).
- 13C NMR (151 MHz, D2O) δ(ppm) 180.6 (C9′, C9*), 101.7–101.6 (C1), 101.4 (C1), 83.48 (C4′), 80.6–80.3 (C4), 72.9–72.7 (C3, C3′), 72.3 (C4), 71.7 (C5), 71.1 (C5′), 60.3–60.1 (C6), 37.48 (C8′), 36.76 (C8*), 34.58 (C7*), 32.85 (C6′), 29.01 (C7′).
4.1.3. Chemicals
4.2. Methods
4.2.1. ESI-MS Measurements
4.2.2. pH Measurements
4.2.3. NMR Experiments
4.2.4. Evaluation of Titration Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loftsson, T.; Jarho, P.; Másson, M.; Järvinen, T. Cyclodextrins in drug delivery. Expert Opin. Drug Deliv. 2005, 2, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Challa, R.; Ahuja, A.; Ali, J.; Khar, R.K. Cyclodextrins in drug delivery: An updated review. AAPS PharmSciTech 2005, 6, E329–E357. [Google Scholar] [CrossRef] [PubMed]
- Matencio, A.; Navarro-Orcajada, S.; García-Carmona, F.; López-Nicolás, J.M. Applications of cyclodextrins in food science. A review. Trends Food Sci. Technol. 2020, 104, 132–143. [Google Scholar] [CrossRef]
- Fenyvesi, É.; Vikmon, M.; Szente, L. Cyclodextrins in Food Technology and Human Nutrition: Benefits and Limitations. Crit. Rev. Food Sci. Nutr. 2016, 56, 1981–2004. [Google Scholar] [CrossRef]
- Szente, L.; Szemán, J. Cyclodextrins in analytical chemistry: Host-guest type molecular recognition. Anal. Chem. 2013, 85, 8024–8030. [Google Scholar] [CrossRef]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of Cyclodextrins and Drug/Cyclodextrin Complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef] [PubMed]
- Pinho, E.; Grootveld, M.; Soares, G.; Henriques, M. Cyclodextrins as encapsulation agents for plant bioactive compounds. Carbohydr. Polym. 2014, 101, 121–135. [Google Scholar] [CrossRef]
- Capelezzo, A.P.; Mohr, L.C.; Dalcanton, F.; de Mello, J.M.M.; Fiori, M.A. β-Cyclodextrins as Encapsulating Agents of Essential Oils. In Cyclodextrin—A Versatile Ingredient; InTech: London, UK, 2018; ISBN 978-1-78923-069-7. [Google Scholar]
- Schneiderman, E.; Stalcup, A.M. Cyclodextrins: A versatile tool in separation science. J. Chromatogr. B Biomed. Sci. Appl. 2000, 745, 83–102. [Google Scholar] [CrossRef]
- Garibyan, A.; Delyagina, E.; Agafonov, M.; Khodov, I.; Terekhova, I. Effect of pH, temperature and native cyclodextrins on aqueous solubility of baricitinib. J. Mol. Liq. 2022, 360, 119548. [Google Scholar] [CrossRef]
- Várnai, B.; Grabarics, M.; Szakács, Z.; Pagel, K.; Malanga, M.; Sohajda, T.; Béni, S. Structural characterization of fondaparinux interaction with per-6-amino-beta-cyclodextrin: An NMR and MS study. J. Pharm. Biomed. Anal. 2021, 197, 113947. [Google Scholar] [CrossRef]
- Singh, D.; Sivashanmugam, T.; Kumar, H.; Nag, K.; Parthasarathy, S.; Shetti, A. Sugammadex: A revolutionary drug in neuromuscular pharmacology. Anesth. Essays Res. 2013, 7, 302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hill, D.R.; Rees, D. Use of Cortisol-Sequestering Agents for the Treatment of Hypercortisolaemia Related Disorders. WO Patent 02/36105 A2, 10 May 2002. [Google Scholar]
- Kennedy, D.J.; Mayer, B.P.; Valdez, C.A. Modified Cyclodextrins for the Selective Sequestration of Fentanyl Related Compounds and Uses Thereof. U.S. Patent 10,442,871 B2, 15 October 2019. [Google Scholar]
- Darwish, K.A.; Mrestani, Y.; Neubert, R.H.H. Study of Interactions Between Sugammadex and Penicillins Using Affinity Capillary Electrophoresis. Chromatographia 2013, 76, 1767–1771. [Google Scholar] [CrossRef]
- Ujj, D.; Kalydi, E.; Malanga, M.; Varga, E.; Sohajda, T.; Béni, S.; Benkovics, G. Sugammadex analogue cyclodextrins as chiral selectors for enantioseparation of cathinone derivatives by capillary electrophoresis. J. Chromatogr. A 2022, 1683, 463506. [Google Scholar] [CrossRef]
- Adam, J.M.; Bennett, D.J.; Bom, A.; Clark, J.K.; Feilden, H.; Hutchinson, E.J.; Palin, R.; Prosser, A.; Rees, D.C.; Rosair, G.M.; et al. Cyclodextrin-Derived Host Molecules as Reversal Agents for the Neuromuscular Blocker Rocuronium Bromide: Synthesis and Structure−Activity Relationships. J. Med. Chem. 2002, 45, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Cameron, K.S.; Fielding, L. NMR diffusion coefficient study of steroid-cyclodextrin inclusion complexes. Magn. Reson. Chem. 2002, 40, S106–S109. [Google Scholar] [CrossRef]
- Möller, N.; Hellwig, T.; Stricker, L.; Engel, S.; Fallnich, C.; Ravoo, B.J. Near-infrared photoswitching of cyclodextrin–guest complexes using lanthanide-doped LiYF 4 upconversion nanoparticles. Chem. Commun. 2017, 53, 240–243. [Google Scholar] [CrossRef]
- Wenz, G.; Strassnig, C.; Thiele, C.; Engelke, A.; Morgenstern, B.; Hegetschweiler, K. Recognition of Ionic Guests by Ionic β-Cyclodextrin Derivatives. Chem. A Eur. J. 2008, 14, 7202–7211. [Google Scholar] [CrossRef]
- Agnes, M.; Thanassoulas, A.; Stavropoulos, P.; Nounesis, G.; Miliotis, G.; Miriagou, V.; Athanasiou, E.; Benkovics, G.; Malanga, M.; Yannakopoulou, K. Designed positively charged cyclodextrin hosts with enhanced binding of penicillins as carriers for the delivery of antibiotics: The case of oxacillin. Int. J. Pharm. 2017, 531, 480–491. [Google Scholar] [CrossRef]
- Benkovics, G.; Fejős, I.; Darcsi, A.; Varga, E.; Malanga, M.; Fenyvesi, É.; Sohajda, T.; Szente, L.; Béni, S.; Szemán, J. Single-isomer carboxymethyl-γ-cyclodextrin as chiral resolving agent for capillary electrophoresis. J. Chromatogr. A 2016, 1467, 445–453. [Google Scholar] [CrossRef]
- Řezanka, P.; Navrátilová, K.; Řezanka, M.; Král, V.; Sýkora, D. Application of cyclodextrins in chiral capillary electrophoresis. Electrophoresis 2014, 35, 2701–2721. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Chiral Selectors in Capillary Electrophoresis: Trends During 2017–2018. Molecules 2019, 24, 1135. [Google Scholar] [CrossRef] [PubMed]
- Fejős, I.; Kalydi, E.; Malanga, M.; Benkovics, G.; Béni, S. Single isomer cyclodextrins as chiral selectors in capillary electrophoresis. J. Chromatogr. A 2020, 1627, 461375. [Google Scholar] [CrossRef] [PubMed]
- Rocco, A.; Maruška, A.; Fanali, S. Cyclodextrins as a chiral mobile phase additive in nano-liquid chromatography: Comparison of reversed-phase silica monolithic and particulate capillary columns. Anal. Bioanal. Chem. 2012, 402, 2935–2943. [Google Scholar] [CrossRef] [PubMed]
- Fejős, I.; Kalydi, E.; Kukk, E.L.; Seggio, M.; Malanga, M.; Béni, S. Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis. Molecules 2021, 26, 5271. [Google Scholar] [CrossRef] [PubMed]
- Servais, A.-C.; Rousseau, A.; Fillet, M.; Lomsadze, K.; Salgado, A.; Crommen, J.; Chankvetadze, B. Separation of propranolol enantiomers by CE using sulfated β-CD derivatives in aqueous and non-aqueous electrolytes: Comparative CE and NMR study. Electrophoresis 2010, 31, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Salgado, A.; Chankvetadze, B. Applications of nuclear magnetic resonance spectroscopy for the understanding of enantiomer separation mechanisms in capillary electrophoresis. J. Chromatogr. A 2016, 1467, 95–144. [Google Scholar] [CrossRef] [PubMed]
- Várnai, B.; Malanga, M.; Sohajda, T.; Béni, S. Molecular interactions in remdesivir-cyclodextrin systems. J. Pharm. Biomed. Anal. 2022, 209, 114482. [Google Scholar] [CrossRef]
- Várnai, B.; Zsila, F.; Szakács, Z.; Garádi, Z.; Malanga, M.; Béni, S. Sulfobutylation of Beta-Cyclodextrin Enhances the Complex Formation with Mitragynine: An NMR and Chiroptical Study. Int. J. Mol. Sci. 2022, 23, 3844. [Google Scholar] [CrossRef] [PubMed]
- Mirzahosseini, A.; Orgován, G.; Tóth, G.; Hosztafi, S.; Noszál, B. The complete microspeciation of ovothiol A disulfide: A hexabasic symmetric biomolecule. J. Pharm. Biomed. Anal. 2015, 107, 209–216. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, H.; Yang, J.; Liang, L. Determination of complex 12-grade phytic acid dissociation constants. Bulg. Chem. Commun. 2015, 47, 22–29. [Google Scholar]
- Reijenga, J.; van Hoof, A.; van Loon, A.; Teunissen, B. Development of Methods for the Determination of pK a Values. Anal. Chem. Insights 2013, 8, ACI. S12304. [Google Scholar] [CrossRef] [PubMed]
- Cakara, D.; Kleimann, J.; Borkovec, M. Microscopic Protonation Equilibria of Poly(amidoamine) Dendrimers from Macroscopic Titrations. Macromolecules 2003, 36, 4201–4207. [Google Scholar] [CrossRef]
- van Duijvenbode, R.C.; Rajanayagam, A.; Koper, G.J.M.; Baars, M.W.P.L.; de Waal, B.F.M.; Meijer, E.W.; Borkovec, M. Synthesis and Protonation Behavior of Carboxylate-Functionalized Poly(propyleneimine) Dendrimers. Macromolecules 2000, 33, 46–52. [Google Scholar] [CrossRef]
- Szakács, Z.; Kraszni, M.; Noszál, B. Determination of microscopic acid-base parameters from NMR-pH titrations. Anal. Bioanal. Chem. 2004, 378, 1428–1448. [Google Scholar] [CrossRef]
- Hägele, G.; Szakács, Z.; Ollig, J.; Hermens, S.; Pfaff, C. NMR-controlled titrations: Characterizing aminophosphonates and related structures. Heteroat. Chem. 2000, 11, 562–582. [Google Scholar] [CrossRef]
- Frassineti, C.; Ghelli, S.; Gans, P.; Sabatini, A.; Moruzzi, M.S.; Vacca, A. Nuclear Magnetic Resonance as a Tool for Determining Protonation Constants of Natural Polyprotic Bases in Solution. Anal. Biochem. 1995, 231, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A.; Ghelli, S.; Frassineti, C.; Alderighi, L.; Gans, P.; Sabatini, A. Determination of protonation constants of some fluorinated polyamines by means of 13C NMR data processed by the new computer program HypNMR2000. Protonation sequence in polyamines. Anal. Bioanal. Chem. 2003, 376, 1041–1052. [Google Scholar] [CrossRef]
- Kyvala, M.; Lukes, I. OPIUM computer program. Available online: https://web.natur.cuni.cz/~kyvala/opium.html (accessed on 5 May 2021).
- Mazák, K.; Noszál, B. Advances in microspeciation of drugs and biomolecules: Species-specific concentrations, acid-base properties and related parameters. J. Pharm. Biomed. Anal. 2016, 130, 390–403. [Google Scholar] [CrossRef]
- Szakács, Z.; Noszál, B. Protonation microequilibrium treatment of polybasic compounds with any possible symmetry. J. Math. Chem. 1999, 26, 139–155. [Google Scholar] [CrossRef]
- Ullmann, G.M. Relations between Protonation Constants and Titration Curves in Polyprotic Acids: A Critical View. J. Phys. Chem. B 2003, 107, 1263–1271. [Google Scholar] [CrossRef]
- Noszál, B.; Szakács, Z. Microscopic Protonation Equilibria of Oxidized Glutathione. J. Phys. Chem. B 2003, 107, 5074–5080. [Google Scholar] [CrossRef]
- Szakács, Z.; Béni, S.; Noszál, B. Resolution of carboxylate protonation microequilibria of NTA, EDTA and related complexones. Talanta 2008, 74, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Borkovec, M.; Koper, G.J.M. A Cluster Expansion Method for the Complete Resolution of Microscopic Ionization Equilibria from NMR Titrations. Anal. Chem. 2000, 72, 3272–3279. [Google Scholar] [CrossRef] [PubMed]
- Borkovec, M.; Brynda, M.; Koper, G.J.M.; Spiess, B. Resolution of microscopic protonation mechanisms in polyprotic molecules. Chimia 2002, 56, 695–701. [Google Scholar] [CrossRef]
- Borkovec, M.; Koper, G.J.M.; Spiess, B. The intrinsic view of ionization equilibria of polyprotic molecules. New J. Chem. 2014, 38, 5679–5685. [Google Scholar] [CrossRef]
- Al-Soufi, W.; Cabrer, P.R.; Jover, A.; Budal, R.M.; Tato, J.V. Determination of second-order association constants by global analysis of 1H and 13C NMR chemical shifts. Steroids 2003, 68, 43–53. [Google Scholar] [CrossRef]
- Noszal, B. Group constant: A measure of submolecular basicity. J. Phys. Chem. 1986, 90, 4104–4110. [Google Scholar] [CrossRef]
- Hawkins, C.J.; Perrin, D.D. Polynuclear Complex Formation. II. Copper(II) with Cystine and Related Ligands. Inorg. Chem. 1963, 2, 843–849. [Google Scholar] [CrossRef]
- Xu, L.; Kamon, Y.; Hashidzume, A. Synthesis of a New Polyanion Possessing Dense 1,2,3-Triazole Backbone. Polymers 2021, 13, 1614. [Google Scholar] [CrossRef]
- Li, X.-B.; Li, Z.-J.; Gao, Y.-J.; Meng, Q.-Y.; Yu, S.; Weiss, R.G.; Tung, C.-H.; Wu, L.-Z. Mechanistic Insights into the Interface-Directed Transformation of Thiols into Disulfides and Molecular Hydrogen by Visible-Light Irradiation of Quantum Dots. Angew. Chemie Int. Ed. 2014, 53, 2085–2089. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water Suppression That Works. Excitation Sculpting Using Arbitrary Wave-Forms and Pulsed-Field Gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Hu, W.; Xie, J.; Chau, H.W.; Si, B.C. Evaluation of parameter uncertainties in nonlinear regression using Microsoft Excel Spreadsheet. Environ. Syst. Res. 2015, 4, 4. [Google Scholar] [CrossRef]
- Madurga, S.; Nedyalkova, M.; Mas, F.; Garcés, J.L. Ionization and Conformational Equilibria of Citric Acid: Delocalized Proton Binding in Solution. J. Phys. Chem. A 2017, 121, 5894–5906. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Suα-CD | Suβ-CD | Suγ-CD | |||||
|---|---|---|---|---|---|---|---|
| ED or Q | SB Micro | ED or Q | SB Micro | Lit. [20] | ED or Q | SB Micro | |
| log K1 | 6.43 (0.07) | 6.28 (0.02) | 6.60 (0.03) | 6.54 (0.02) | 6.49 (1) | 6.68 (0.08) | 6.64 (0.02) |
| log K2 | 5.79 (0.09) | 5.78 (0.02) | 6.18 (0.04) | 6.06 (0.02) | 5.69 (1) | 6.17 (0.12) | 6.18 (0.02) |
| log K3 | 5.26 (0.11) | 5.31 (0.02) | 5.27 (0.08) | 5.64 (0.02) | 5.25 (1) | 5.88 (0.16) | 5.81 (0.02) |
| log K4 | 4.70 (0.14) | 4.81 (0.02) | 5.53? (0.06) | 5.18 (0.01) | 4.75 (1) | 5.28 (0.22) | 5.43 (0.01) |
| log K5 | 4.53? (0.11) | 4.35 (0.02) | 4.54 (0.06) | 4.73 (0.02) | 4.31 (1) | 5.14 (0.22) | 5.03 (0.01) |
| log K6 | 3.77 (0.07) | 3.84 (0.02) | 4.46? (0.05) | 4.31 (0.02) | 3.78 (1) | 4.59 (0.17) | 4.65 (0.02) |
| log K7 | - | - | 3.69 (0.03) | 3.82 (0.02) | <3 | 4.34? (0.12) | 4.28 (0.02) |
| log K8 | - | - | - | - | - | 3.78 (0.07) | 3.83 (0.02) |
| 5.65 (0.07) | 5.51 (0.02) | 5.75 (0.03) | 5.70 (0.02) | - | 5.77 (0.08) | 5.73 (0.02) | |
| 5.39 (0.09) | 5.38 (0.02) | 5.71 (0.04) | 5.58 (0.02) | - | 5.63 (0.12) | 5.64 (0.02) | |
| 5.13 (0.12) | 5.18 (0.02) | 5.04 (0.08) | 5.42 (0.02) | - | 5.58 (0.16) | 5.51 (0.02) | |
| 4.83 (0.14) | 4.94 (0.02) | 5.53? (0.08) | 5.18 (0.01) | - | 5.18 (0.22) | 5.33 (0.01) | |
| 4.93? (0.11) | 4.75 (0.02) | 4.76 (0.06) | 4.95 (0.02) | - | 5.24 (0.22) | 5.13 (0.01) | |
| 4.55 (0.07) | 4.62 (0.02) | 4.94? (0.05) | 4.78 (0.02) | - | 4.89 (0.17) | 4.95 (0.02) | |
| - | - | 4.53 (0.03) | 4.67 (0.02) | - | 4.89? (0.12) | 4.82 (0.02) | |
| - | - | - | - | - | 4.69 (0.07) | 4.73 (0.02) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalydi, E.; Malanga, M.; Ujj, D.; Benkovics, G.; Szakács, Z.; Béni, S. Fully Symmetric Cyclodextrin Polycarboxylates: How to Determine Reliable Protonation Constants from NMR Titration Data. Int. J. Mol. Sci. 2022, 23, 14448. https://doi.org/10.3390/ijms232214448
Kalydi E, Malanga M, Ujj D, Benkovics G, Szakács Z, Béni S. Fully Symmetric Cyclodextrin Polycarboxylates: How to Determine Reliable Protonation Constants from NMR Titration Data. International Journal of Molecular Sciences. 2022; 23(22):14448. https://doi.org/10.3390/ijms232214448
Chicago/Turabian StyleKalydi, Eszter, Milo Malanga, Dóra Ujj, Gábor Benkovics, Zoltán Szakács, and Szabolcs Béni. 2022. "Fully Symmetric Cyclodextrin Polycarboxylates: How to Determine Reliable Protonation Constants from NMR Titration Data" International Journal of Molecular Sciences 23, no. 22: 14448. https://doi.org/10.3390/ijms232214448
APA StyleKalydi, E., Malanga, M., Ujj, D., Benkovics, G., Szakács, Z., & Béni, S. (2022). Fully Symmetric Cyclodextrin Polycarboxylates: How to Determine Reliable Protonation Constants from NMR Titration Data. International Journal of Molecular Sciences, 23(22), 14448. https://doi.org/10.3390/ijms232214448