
Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces cerevisiae
 , , , and
, , , and 
Abstract
1. Introduction
2. Results and Discussion
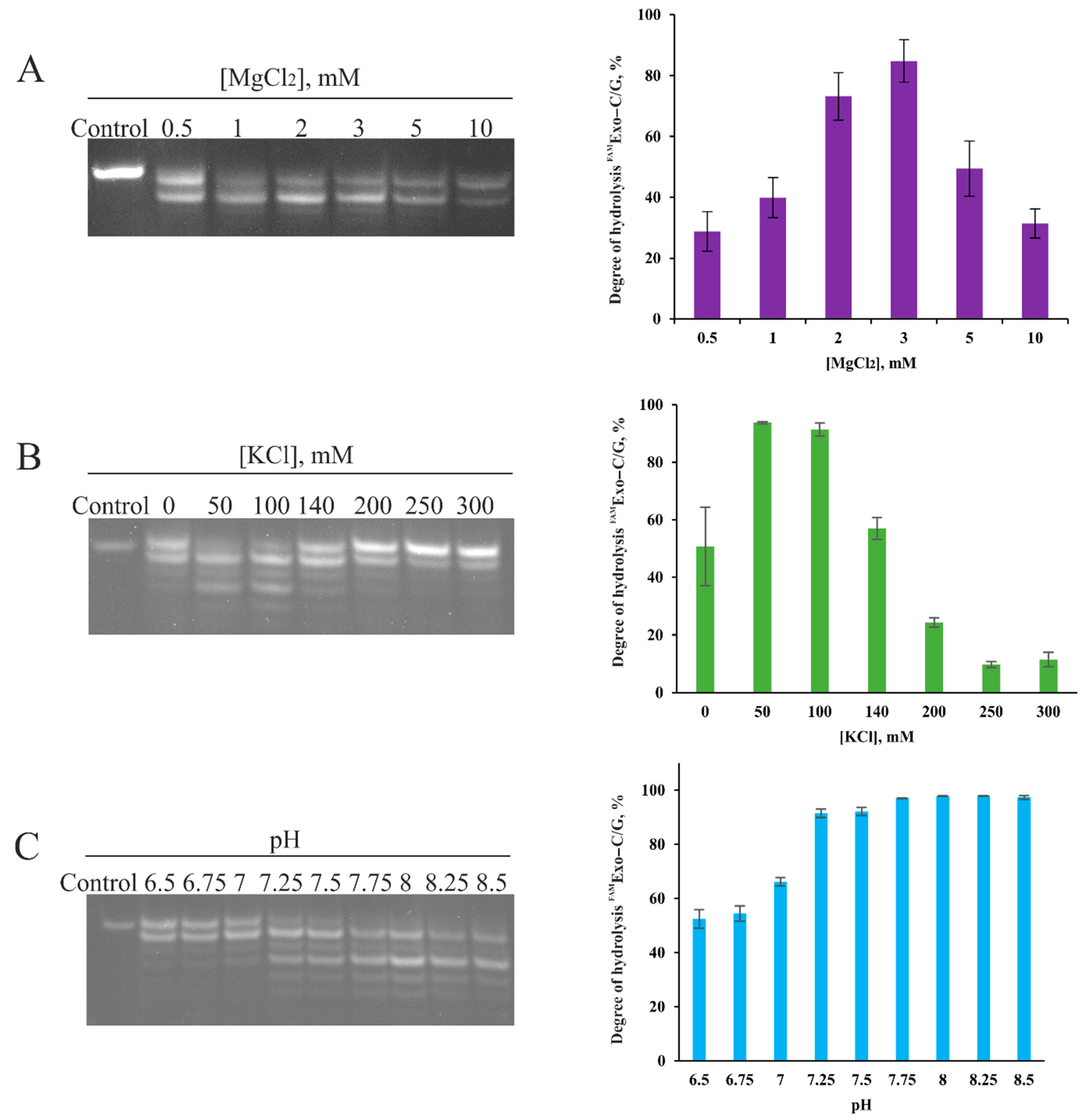
2.1. Influence of Reaction Conditions on 3′–5′–Exonuclease Activity of Apn2
2.2. Accumulation Kinetics of Products of the 3′–5′–Exonuclease Reaction
2.3. Determining the Inhibitory Action of 2′–Deoxynucleoside Monophosphates on the 3′–5′–Exonuclease Activity of Apn2
2.4. Examination of Conformational Changes of Apn2 during the Enzymatic Cycle
2.5. Analysis of Conformational Changes in the DNA Substrate during the Enzymatic Cycle
3. Materials and Methods
3.1. DNA Substrates
3.2. The Enzyme
3.3. Electrophoresis in a Denaturing Polyacrylamide Gel (PAAG)
3.4. The Stopped–Flow Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Friedberg, E.C.; Roger, A.S.; Wolfram, S.; Graham, C.W.; Tom, E.; Richard, D.W. DNA Repair and Mutagenesis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006; ISBN 9781555813192. [Google Scholar]
- McCullough, A.K.; Dodson, M.L.; Lloyd, R.S. Initiation of base excision repair: Glycosylase mechanisms and structures. Annu. Rev. Biochem. 1999, 68, 255–285. [Google Scholar] [CrossRef] [PubMed]
- Memisoglu, A.; Samson, L. Base excision repair in yeast and mammals. Mutat. Res. 2000, 451, 39–51. [Google Scholar] [CrossRef]
- Wallace, S.S. Base excision repair: A critical player in many games. DNA Repair 2014, 19, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef]
- Gros, L.; Saparbaev, M.K.; Laval, J. Enzymology of the repair of free radicals–induced DNA damage. Oncogene 2002, 21, 8905–8925. [Google Scholar] [CrossRef]
- Wilson III, D.M.; Barsky, D. The major human abasic endonuclease: Formation, consequences and repair of abasic lesions in DNA. Mutat. Res. 2001, 485, 283–307. [Google Scholar] [CrossRef]
- Demple, B.; Sung, J.-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef]
- Unk, I.; Haracska, L.; Johnson, R.E.; Prakash, S.; Prakash, L. Apurinic endonuclease activity of yeast Apn2 protein. J. Biol. Chem. 2000, 275, 22427–22434. [Google Scholar] [CrossRef]
- Ide, H.; Tedzuka, K.; Shimzu, H.; Kimura, Y.; Purmal, A.A.; Wallace, S.S.; Kow, Y.W. α–Deoxyadenosine, a Major Anoxic Radiolysis Product of Adenine in DNA, Is a Substrate for Escherichia coli Endonuclease IV. Biochemistry 1994, 33, 7842–7847. [Google Scholar] [CrossRef]
- Ischenko, A.A.; Saparbaev, M.K. Alternative nucleotide incision repair pathway for oxidative DNA damage. Nature 2002, 415, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Guliaev, A.B.; Hang, B.; Singer, B. Structural insights by molecular dynamics simulations into specificity of the major human AP endonuclease toward the benzene–derived DNA adduct, pBQ–C. Nucleic Acids Res. 2004, 32, 2844–2852. [Google Scholar] [CrossRef] [PubMed]
- Daviet, S.; Couve–Privat, S.; Gros, L.; Shinozuka, K.; Ide, H.; Saparbaev, M.; Ishchenko, A.A. Major oxidative products of cytosine are substrates for the nucleotide incision repair pathway. DNA Repair 2007, 6, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Prorok, P.; Saint–Pierre, C.; Gasparutto, D.; Fedorova, O.S.; Ishchenko, A.A.; Leh, H.; Buckle, M.; Tudek, B.; Saparbaev, M. Highly mutagenic exocyclic DNA adducts are substrates for the human nucleotide incision repair pathway. PLoS ONE 2012, 7, e51776. [Google Scholar] [CrossRef] [PubMed]
- Christov, P.P.; Banerjee, S.; Stone, M.P.; Rizzo, C.J. Selective Incision of the alpha–N–Methyl–Formamidopyrimidine Anomer by Escherichia coli Endonuclease IV. J. Nucleic Acids 2010, 2010, 850234. [Google Scholar] [CrossRef]
- Vrouwe, M.G.; Pines, A.; Overmeer, R.M.; Hanada, K.; Mullenders, L.H. UV–induced photolesions elicit ATR–kinase–dependent signaling in non–cycling cells through nucleotide excision repair–dependent and –independent pathways. J. Cell Sci. 2011, 124, 435–446. [Google Scholar] [CrossRef]
- Gros, L.; Ishchenko, A.A.; Ide, H.; Elder, R.H.; Saparbaev, M.K. The major human AP endonuclease (Ape1) is involved in the nucleotide incision repair pathway. Nucleic Acids Res. 2004, 32, 73–81. [Google Scholar] [CrossRef]
- Prorok, P.; Alili, D.; Saint–Pierre, C.; Gasparutto, D.; Zharkov, D.O.; Ishchenko, A.A.; Tudek, B.; Saparbaev, M.K. Uracil in duplex DNA is a substrate for the nucleotide incision repair pathway in human cells. Proc. Natl. Acad. Sci. USA 2013, 110, E3695–E3703. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Takeshita, M.; Grollman, A.P.; Demple, B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 1995, 270, 16002–16007. [Google Scholar] [CrossRef]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′–5′ Exonuclease Activity of Human AP–Endonuclease APE1. Molecules 2018, 23, 2101. [Google Scholar] [CrossRef] [PubMed]
- Senchurova, S.I.; Kuznetsova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. The kinetic mechanism of 3′ –5′ exonucleolytic activity of AP endonuclease Nfo from E. coli. Cells 2022, 11, 2998. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.; Kim, W.C.; Mantha, A.K.; Kim, S.E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of Apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c–myc mRNA. Nucleic Acids Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Gavrilova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of APE1 Endoribonuclease Activity. Mol. Biol. 2021, 55, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Gorrell, A.; Rader, S.D.; Lee, C.H. Endoribonuclease activity of human apurinic/apyrimidinic endonuclease 1 revealed by a real–time fluorometric assay. Anal. Biochem. 2010, 398, 69–75. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Novopashina, D.S.; Fedorova, O.S.; Kuznetsov, N.A. Effect of the Substrate Structure and Metal Ions on the Hydrolysis of Undamaged RNA by Human AP Endonuclease APE1. Acta Nat. 2020, 12, 74–85. [Google Scholar] [CrossRef]
- Davletgildeeva, A.T.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Activity of Human Apurinic/Apyrimidinic Endonuclease APE1 Toward Damaged DNA and Native RNA With Non–canonical Structures. Front. Cell Dev. Biol. 2020, 8, 590848. [Google Scholar] [CrossRef]
- Dyrkheeva, N.S.; Lebedeva, N.A.; Lavrik, O.I. AP endonuclease 1 as a key enzyme in repair of apurinic/apyrimidinic sites. Biochemistry 2016, 81, 951–967. [Google Scholar] [CrossRef]
- Hadi, M.Z.; Ginalski, K.; Nguyen, L.H.; Wilson, D.M. Determinants in nuclease specificity of Ape1 and Ape2, human homologues of Escherichia coli exonuclease III. J. Mol. Biol. 2002, 316, 853–866. [Google Scholar] [CrossRef]
- Lin, Y.; Raj, J.; Li, J.; Ha, A.; Hossain, M.A.; Richardson, C.; Mukherjee, P.; Yan, S. APE1 senses DNA single–strand breaks for repair and signaling. Nucleic Acids Res. 2020, 48, 1925–1940. [Google Scholar] [CrossRef]
- Willis, J.; Patel, Y.; Lentz, B.L.; Yan, S. APE2 is required for ATR–Chk1 checkpoint activation in response to oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 10592–10597. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bai, L.; Cupello, S.; Hossain, M.A.; Deem, B.; McLeod, M.; Raj, J.; Yan, S. APE2 promotes DNA damage response pathway from a single–strand break. Nucleic Acids Res. 2018, 46, 2479–2494. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Lin, Y.; Yan, S. Single–strand break end resection in genome integrity: Mechanism and regulation by APE2. Int. J. Mol. Sci. 2018, 19, 2389. [Google Scholar] [CrossRef]
- Popoff, S.C.; Spira, A.I.; Johnson, A.W.; Demple, B. Yeast structural gene (APN1) for the major apurinic endonuclease: Homology to Escherichia coli endonuclease IV. Proc. Natl. Acad. Sci. USA 1990, 87, 4193–4197. [Google Scholar] [CrossRef]
- Johnson, R.E.; Torres–Ramos, C.A.; Izumi, T.; Mitra, S.; Prakash, S.; Prakash, L. Identification of APN2, the Saccharomyces cerevisiae homolog of the major human AP endonuclease HAP1, and its role in the repair of abasic sites. Genes Dev. 1998, 12, 3137–3143. [Google Scholar] [CrossRef] [PubMed]
- Unk, I.; Haracska, L.; Prakash, S.; Prakash, L. 3′–Phosphodiesterase and 3′→5′ Exonuclease Activities of Yeast Apn2 Protein and Requirement of These Activities for Repair of Oxidative DNA Damage. Mol Cell Biol. 2001, 21, 1656–1661. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, D.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Search for Modified DNA Sites with the Human Methyl–CpG–Binding Enzyme MBD4. Acta Nat. 2017, 9, 88–98. [Google Scholar] [CrossRef]
- Kuznetsova, A.A.; Iakovlev, D.A.; Misovets, I.V.; Ishchenko, A.A.; Saparbaev, M.K.; Kuznetsov, N.A.; Fedorova, O.S. Pre–steady–state kinetic analysis of damage recognition by human single–strand selective monofunctional uracil–DNA glycosylase SMUG1. Mol. Biosyst. 2017, 13, 2638–2649. [Google Scholar] [CrossRef]
- Kladova, O.A.; Kuznetsova, A.A.; Fedorova, O.S.; Kuznetsov, N.A. Mutational and Kinetic Analysis of Lesion Recognition by Escherichia coli Endonuclease VIII. Genes 2017, 8, 140. [Google Scholar] [CrossRef]
- Kladova, O.A.; Krasnoperov, L.N.; Kuznetsov, N.A.; Fedorova, O.S. Kinetics and thermodynamics of DNA processing by wild type DNA–glycosylase endo III and its catalytically inactive mutant forms. Genes 2018, 9, 190. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Substrate | Ks, μM−1 | kcat, ×103 s−1 |
|---|---|---|
| FAMExo–C/G | 0.40 ± 0.20 | 2.7 ± 1.2 |
| FAMExo–G/C | 3.0 ± 0.3 | |
| FAMExo–T/A | 3.3 ± 1.2 | |
| FAMExo–A/T | 7.5 ± 0.7 | |
| FAMExo–C/A | 13.3 ± 3.3 | |
| FAMExo–C/T | 12.0 ± 0.2 | |
| FAMExo–C/C | 16.7 ± 5.0 | |
| FAMExo–CCC/GGG | 1.0 ± 1.2 | 2.4 ± 0.4 |
| FAMExo–GGG/CCC | 0.3 ± 0.4 | 3.4 ± 1.6 |
| FAMExo–TTT/AAA | 0.7 ± 0.4 | 2.2 ± 0.1 |
| FAMExo–AAA/TTT | 7.2 ± 4.1 | 62 ± 25 |
| Name | Sequence |
|---|---|
| FAMExo–X/Y, where X=G/A/T/C, Y=G/A/T/C | 5′–FAM–CAGCTCTGTACGTGAGX–3′ 3′–GTCGAGACATGCACTCYTCACCACTGTG–5′ |
| TrpExo–G/C | 5′–CAGCTCTGTACGTGAGG–3′ 3′–GTCGAGACATGCACTCCTCACCACTGTG–5′ |
| FAMExo–CCC/GGG | 5′–FAM–CAGCTCTGTACGTGCCC–3′ 3′–GTCGAGACATGCACGGGTCACCACTGTG–5′ |
| FAMExo–GGG/CCC | 5′–FAM–CAGCTCTGTACGTGGGG–3′ 3′–GTCGAGACATGCACCCCTCACCACTGTG–5′ |
| FAMExo–TTT/AAA | 5′–FAM–CAGCTCTGTACGTGTTT–3′ 3′–GTCGAGACATGCACAAATCACCACTGTG–5′ |
| FAMExo–AAA/TTT | 5′–FAM–CAGCTCTGTACGTGAAA–3′ 3′–GTCGAGACATGCACTTTTCACCACTGTG–5′ |
| Exo–aPu1 | 5′–CAGCTCTGTACGTGAGaPu–3′ 3′–GTCGAGACATGCACTC T TCACCACTGTG–5′ |
| Exo–aPu2 | 5′–CAGCTCTGTACGTGAaPuT–3′ 3′–GTCGAGACATGCACT T ATCACCACTGTG–5′ |
| Exo–aPuopposite | 5′–CAGCTCTGTACGTGAG T–3′ 3′–GTCGAGACATGCACTCaPuTCACCACTGTG–5′ |
| Exo–1CPy | 5′–CAGCTCTGTACGTGAGCpy–3′ 3′–GTCGAGACATGCACTCGTCACCACTGTG–5′ |
| Exo–2CPy | 5′–CAGCTCTGTACGTGACpyT–3′ 3′–GTCGAGACATGCACT G ATCACCACTGTG–5′ |
| Exo–oppositeCPy | 5′–CAGCTCTGTACGTGAGG–3′ 3′–GTCGAGACATGCACTCCpyTCACCACTGTG–5′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuznetsova, A.A.; Gavrilova, A.A.; Ishchenko, A.A.; Saparbaev, M.; Fedorova, O.S.; Kuznetsov, N.A. Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces cerevisiae. Int. J. Mol. Sci. 2022, 23, 14404. https://doi.org/10.3390/ijms232214404
Kuznetsova AA, Gavrilova AA, Ishchenko AA, Saparbaev M, Fedorova OS, Kuznetsov NA. Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces cerevisiae. International Journal of Molecular Sciences. 2022; 23(22):14404. https://doi.org/10.3390/ijms232214404
Chicago/Turabian StyleKuznetsova, Aleksandra A., Anastasia A. Gavrilova, Alexander A. Ishchenko, Murat Saparbaev, Olga S. Fedorova, and Nikita A. Kuznetsov. 2022. "Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces cerevisiae" International Journal of Molecular Sciences 23, no. 22: 14404. https://doi.org/10.3390/ijms232214404
APA StyleKuznetsova, A. A., Gavrilova, A. A., Ishchenko, A. A., Saparbaev, M., Fedorova, O. S., & Kuznetsov, N. A. (2022). Kinetic Features of 3′–5′–Exonuclease Activity of Apurinic/Apyrimidinic Endonuclease Apn2 from Saccharomyces cerevisiae. International Journal of Molecular Sciences, 23(22), 14404. https://doi.org/10.3390/ijms232214404