
Transgenic Mouse Models to Study the Development and Maintenance of the Adrenal Cortex


Abstract
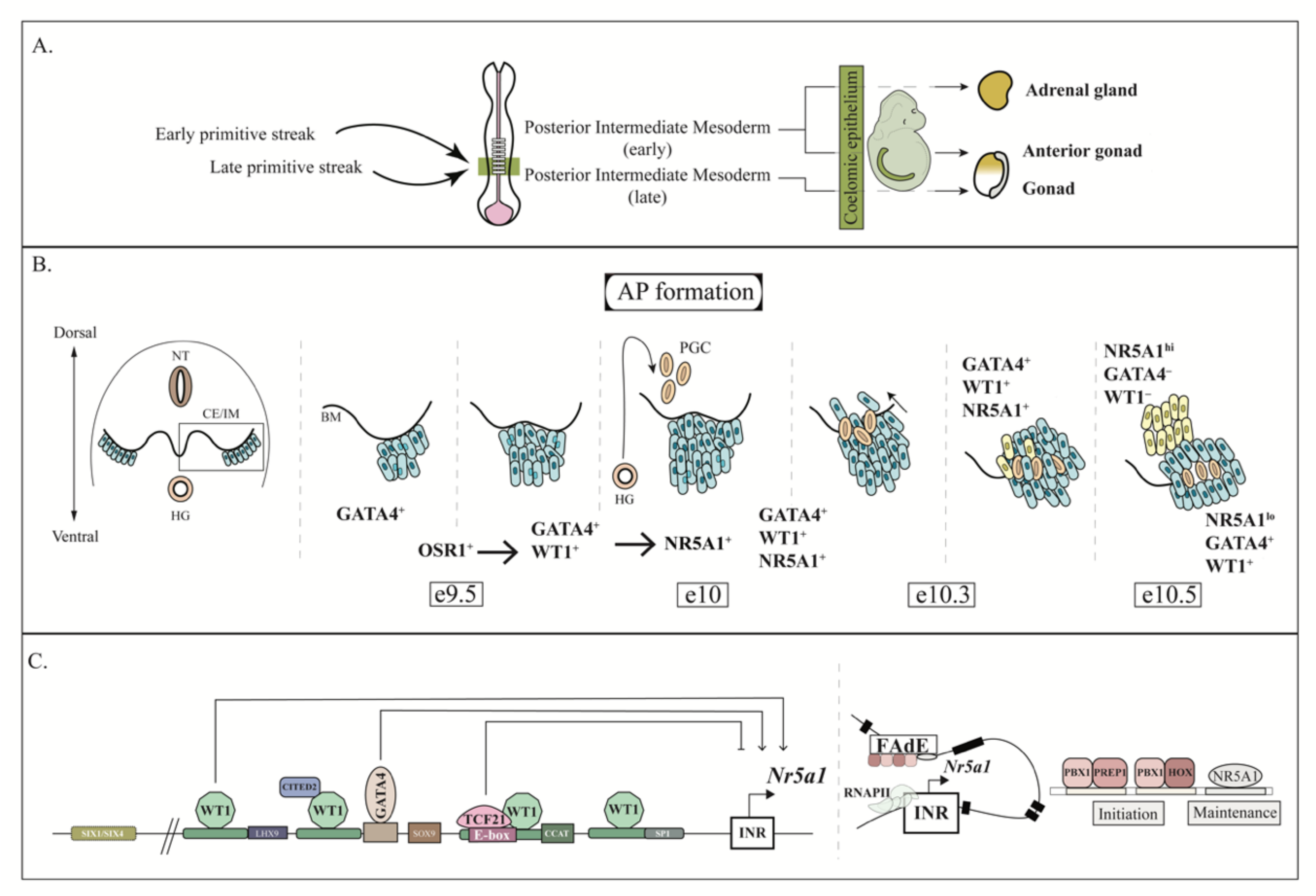
1. Introduction
2. Mouse Models Used to Study the Adrenal Cortex Development and Maintenance
2.1. Mouse Strains to Study AGP Formation
2.2. Mouse Strains Using Nr5a1 Regulatory Sequences to Drive Cre Expression
2.3. Mouse Strains Using Regulatory Sequence of Genes Coding for Steroidogenic Enzymes to Drive Cre Expression
2.4. Other Mouse Strains Used to Target the Adrenal Cortex
3. AGP Development
4. AP and Fetal Adrenal Development
5. Fate of the Fetal Cortex
6. Encapsulation of the Adrenal Cortex
7. Development and Maintenance of the Definitive Cortex: The Key Role of Hedgehog and Canonical WNT Signaling Pathways
8. Establishment of Zonation: The Opposing Roles of WNT and PKA Signaling Cascades
9. Sexual Hormones Play an Important Role in the Maintenance of the Adrenal Cortex
10. Emergence of Spindle-Shaped Cells in Aging Mice
11. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abcb1b | ATP-binding cassette, sub-family B (MDR/TAP), member 1B |
| Akr1b7 | aldo-keto reductase family 1, member B7 |
| Amhr2 | anti-mullerian hormone type 2 receptor |
| Apc | adenomatosis polyposis coli |
| Ar | androgen receptor |
| Axin2 | axin2 |
| Cbx2 | chromobox 2 |
| Cited2 | Glu/Asp-rich carboxy-terminal domain 2 |
| Ctnnb1 | beta-catenin |
| Cyb5 | cytochrome b5 |
| Cyp2f2 | cytochrome P450, family 2, subfamily f, polypeptide 2 |
| Cyp11a1 | cytochrome P450 side chain cleavage enzyme 11a1 |
| Cyp11b1 | cytochrome P450, family 11, subfamily b, polypeptide 1 |
| Cyp11b2/As | cytochrome P450, family 11, subfamily b, polypeptide 2/aldosterone synthase |
| Cyp17a1 | cytochrome P450, family 17, subfamily a, polypeptide 1 |
| Cyp21a1 | cytochrome P450, family 21, subfamily a, polypeptide 1 |
| Dennd1a | DENN/MADD domain containing 1A |
| Dhcr24 | 24-dehydrocholesterol reductase |
| Emx2 | empty spiracles homeobox 2 |
| Ezh2 | zeste homolog 2 |
| FAdE/Nr5a1 | Fetal adrenal enhancer of nuclear receptor subfamily 5, Group A, member 1 |
| Foxl2 | forkhead box L2 |
| Fgfr2 | fibroblast growth factor receptor 2 |
| Frzb | Frizzled-related protein |
| Gata4 | GATA binding protein 4 |
| Gata6 | GATA binding protein 6 |
| Gli1 | GLI-Kruppel family member 1 |
| Gli2 | GLI-Kruppel family member 2 |
| Gli3 | GLI-Kruppel family member 3 |
| Hoxb9 | homeobox B9 |
| IGF | insulin growth factor |
| Igf1r | insulin-like growth factor 1 receptor |
| Inha | inhibin a |
| Insr | insulin receptor |
| Lats1 | large tumor suppressor 1 |
| Lats2 | large tumor suppressor 2 |
| Lhcgr | luteinizing hormone/choriogonadotropin receptor |
| Lhx9 | LIM homeobox protein 9 |
| MAPK | mitogen-activate kinase protein |
| Mc2r | melanocortin 2 receptor |
| Mrap | melanocortin 2 receptor accessory protein |
| Nes | nestin |
| Nr0b1 | nuclear receptor subfamily 0, group B, member 1 |
| Nr2f2 | nuclear receptor subfamily 2, group F, member 2 |
| Nr5a1 | nuclear receptor subfamily 5, group A, member 1 |
| Osr1 | odd-skipped related transcription factor 1 |
| Pbx1 | pre B cell leukemia homeobox 1 |
| Pde1b | phosphodiesterase 1B, Ca2+ calmodulin dependent |
| Pde2a | phosphodiesterase 2A, cGMP-stimulated |
| Pde3a | phosphodiesterase 3A, cGMP-inhibited |
| Pde7b | phosphodiesterase 7B |
| Pde8b | phosphodiesterase 8B |
| Pde11a | phosphodiesterase 11A |
| PKA | protein kinase A |
| Porcn | porcupine homolog |
| Pou5f1 | POU domain, class 5, transcription factor 1 |
| Prkar1a | protein kinase, cAMP dependent regulatory, type 1, alpha |
| Prep1 | Pbx-knotted 1 homeobox |
| Rspo3 | R-spondin 3 homolog |
| Shh | sonic hedgehog |
| Siah1a | seven in absentia 1A |
| Six1 | sine oculis-related homeobox-1 |
| Six4 | sine oculis-related homeobox-4 |
| Sfrp2 | Secreted frizzled-related protein 2 |
| Smo | smoothened homolog |
| Taz | transcriptional co-activator with PDZ-binding motif |
| Tcf21 | transcription factor 21 |
| Tbx18 | T-box18 |
| Wnt4 | wingless-type MMTV integration site family, member 4 |
| Wnt2b | wingless-type MMTV integration site family, member 2 |
| Wt1 | wilms tumor 1 |
| Yap | yes-associated protein |
| Znrf3 | zinc and ring finger 3 |
References
- Gottschau, M. Struktur und Embryonale Entwickelung der Nebennieren bei Saugetieren. Archiv fur Anatomie und Entwickelungsgeschichte. Anat. Abt. 1883, 9, 412–458. [Google Scholar]
- Zwemer, R.L. A study of adrenal cortex morphology. Am. J. Pathol. 1936, 84, 107–114.1. [Google Scholar] [CrossRef] [PubMed]
- Salmon, T.N.; Zwemer, R.L. A study of the life history of cortico-adrenal gland cells of the rat by means of trypan blue injections. Anat. Rec. 1941, 80, 421–429. [Google Scholar] [CrossRef]
- Hayashi, S.; McMahon, A.P. Efficient Recombination in Diverse Tissues by a Tamoxifen-Inducible Form of Cre: A Tool for Temporally Regulated Gene Activation/Inactivation in the Mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef]
- Hu, Y.-C.; Okumura, L.M.; Page, D.C. Gata4 Is Required for Formation of the Genital Ridge in Mice. PLoS Genet. 2013, 9, e1003629. [Google Scholar] [CrossRef]
- Vidal, V.; Sacco, S.; Rocha, A.S.; da Silva, F.; Panzolini, C.; Dumontet, T.; Doan, T.M.P.; Shan, J.; Rak-Raszewska, A.; Bird, T.; et al. The adrenal capsule is a signaling center controlling cell renewal and zonation through Rspo3. Genes Dev. 2016, 30, 1389–1394. [Google Scholar] [CrossRef]
- Mugford, J.W.; Sipilä, P.; McMahon, J.A.; McMahon, A.P. Osr1 expression demarcates a multi-potent population of intermediate mesoderm that undergoes progressive restriction to an Osr1-dependent nephron progenitor compartment within the mammalian kidney. Dev. Biol. 2008, 324, 88–98. [Google Scholar] [CrossRef]
- Sasaki, K.; Oguchi, A.; Cheng, K.; Murakawa, Y.; Okamoto, I.; Ohta, H.; Yabuta, Y.; Iwatani, C.; Tsuchiya, H.; Yamamoto, T.; et al. The embryonic ontogeny of the gonadal somatic cells in mice and monkeys. Cell Rep. 2021, 35, 109075. [Google Scholar] [CrossRef]
- Trowe, M.-O.; Shah, S.; Petry, M.; Airik, R.; Schuster-Gossler, K.; Kist, R.; Kispert, A. Loss of Sox9 in the periotic mesenchyme affects mesenchymal expansion and differentiation, and epithelial morphogenesis during cochlea development in the mouse. Dev. Biol. 2010, 342, 51–62. [Google Scholar] [CrossRef]
- Häfner, R.; Bohnenpoll, T.; Rudat, C.; Schultheiss, T.M.; Kispert, A. Fgfr2 is required for the expansion of the early adrenocortical primordium. Mol. Cell. Endocrinol. 2015, 413, 168–177. [Google Scholar] [CrossRef]
- Sankoda, N.; Tanabe, W.; Tanaka, A.; Shibata, H.; Woltjen, K.; Chiba, T.; Haga, H.; Sakai, Y.; Mandai, M.; Yamamoto, T.; et al. Epithelial expression of Gata4 and Sox2 regulates specification of the squamous–columnar junction via MAPK/ERK signaling in mice. Nat. Commun. 2021, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; Von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, R.; Vidal, V.P.; Motamedi, F.J.; Clarkson, M.; Sahut-Barnola, I.; von Gise, A.; Pu, W.T.; Hohenstein, P.; Martinez, A.; Schedl, A. WT1 Maintains Adrenal-Gonadal Primordium Identity and Marks a Population of AGP-like Progenitors within the Adrenal Gland. Dev. Cell 2013, 27, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Bingham, N.C.; Verma-Kurvari, S.; Parada, L.F.; Parker, K.L. Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis 2006, 44, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Heaton, J.H.; Wood, M.A.; Kim, A.C.; Lima, L.O.; Barlaskar, F.M.; Almeida, M.Q.; Fragoso, M.C.; Kuick, R.; Lerario, A.M.; Simon, D.P.; et al. Progression to Adrenocortical Tumorigenesis in Mice and Humans through Insulin-Like Growth Factor 2 and β-Catenin. Am. J. Pathol. 2012, 181, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.C.; Reuter, A.L.; Zubair, M.; Else, T.; Serecky, K.; Bingham, N.C.; Lavery, G.G.; Parker, K.L.; Hammer, G.D. Targeted disruption of beta-catenin in Sf1-expressing cells impairs development and maintenance of the adrenal cortex. Development 2008, 135, 2593–2602. [Google Scholar] [CrossRef]
- Huang, C.-C.J.; Liu, C.; Yao, H.H.-C. Investigating the role of adrenal cortex in organization and differentiation of the adrenal medulla in mice. Mol. Cell. Endocrinol. 2012, 361, 165–171. [Google Scholar] [CrossRef]
- Krill, K.T.; Gurdziel, K.; Heaton, J.H.; Simon, D.P.; Hammer, G.D. Dicer Deficiency Reveals MicroRNAs Predicted to Control Gene Expression in the Developing Adrenal Cortex. Mol. Endocrinol. 2013, 27, 754–768. [Google Scholar] [CrossRef]
- Mathieu, M.; Drelon, C.; Rodriguez, S.; Tabbal, H.; Septier, A.; Damon-Soubeyrand, C.; Dumontet, T.; Berthon, A.; Sahut-Barnola, I.; Djari, C.; et al. Steroidogenic differentiation and PKA signaling are programmed by histone methyltransferase EZH2 in the adrenal cortex. Proc. Natl. Acad. Sci. USA 2018, 115, E12265–E12274. [Google Scholar] [CrossRef]
- Kim, Y.; Bingham, N.; Sekido, R.; Parker, K.L.; Lovell-Badge, R.; Capel, B. Fibroblast growth factor receptor 2 regulates proliferation and Sertoli differentiation during male sex determination. Proc. Natl. Acad. Sci. USA 2007, 104, 16558–16563. [Google Scholar] [CrossRef]
- Tevosian, S.G.; Jimenez, E.; Hatch, H.; Jiang, T.; Morse, D.A.; Fox, S.C.; Padua, M.B. Adrenal Development in Mice Requires GATA4 and GATA6 Transcription Factors. Endocrinology 2015, 156, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Padua, M.B.; Jiang, T.; Morse, D.A.; Fox, S.C.; Hatch, H.M.; Tevosian, S.G. Combined Loss of the GATA4 and GATA6 Transcription Factors in Male Mice Disrupts Testicular Development and Confers Adrenal-Like Function in the Testes. Endocrinology 2015, 156, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Basham, K.J.; Rodriguez, S.; Turcu, A.F.; Lerario, A.M.; Logan, C.Y.; Rysztak, M.R.; Gomez-Sanchez, C.E.; Breault, D.T.; Koo, B.-K.; Clevers, H.; et al. A ZNRF3-dependent Wnt/β-catenin signaling gradient is required for adrenal homeostasis. Genes Dev. 2019, 33, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, T.; Sahut-Barnola, I.; Dufour, D.; Lefrançois-Martinez, A.; Berthon, A.; Montanier, N.; Ragazzon, B.; Djari, C.; Pointud, J.; Roucher-Boulez, F.; et al. Hormonal and spatial control of SUMOylation in the human and mouse adrenal cortex. FASEB J. 2019, 33, 10218–10230. [Google Scholar] [CrossRef] [PubMed]
- Drelon, C.; Berthon, A.; Sahut-Barnola, I.; Mathieu, M.; Dumontet, T.; Rodriguez, S.; Batisse-Lignier, M.; Tabbal, H.; Tauveron, I.; Lefrançois-Martinez, A.-M.; et al. PKA inhibits WNT signalling in adrenal cortex zonation and prevents malignant tumour development. Nat. Commun. 2016, 7, 12751. [Google Scholar] [CrossRef]
- Sahut-Barnola, I.; de Joussineau, C.; Val, P.; Lambert-Langlais, S.; Damon, C.; Lefrançois-Martinez, A.M.; Pointud, J.C.; Marceau, G.; Sapin, V.; Tissier, F.; et al. Cushing’s syndrome and fetal features resurgence in adrenal cortex-specific Prkar1a knockout mice. PLoS Genet. 2010, 6, e1000980. [Google Scholar] [CrossRef]
- Huang, C.-C.J.; Miyagawa, S.; Matsumaru, D.; Parker, K.L.; Yao, H.H.-C. Progenitor Cell Expansion and Organ Size of Mouse Adrenal Is Regulated by Sonic Hedgehog. Endocrinology 2010, 151, 1119–1128. [Google Scholar] [CrossRef]
- King, P.; Paul, A.; Laufer, E. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc. Natl. Acad. Sci. USA 2009, 106, 21185–21190. [Google Scholar] [CrossRef]
- Ching, S.; Vilain, E. Targeted disruption of Sonic Hedgehog in the mouse adrenal leads to adrenocortical hypoplasia. Genesis 2009, 47, 628–637. [Google Scholar] [CrossRef]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin Directly Activates SF1 Neurons in the VMH, and This Action by Leptin Is Required for Normal Body-Weight Homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef]
- Pihlajoki, M.; Gretzinger, E.; Cochran, R.; Kyrönlahti, A.; Schrade, A.; Hiller, T.; Sullivan, L.; Shoykhet, M.; Schoeller, E.L.; Brooks, M.D.; et al. Conditional Mutagenesis of Gata6 in SF1-Positive Cells Causes Gonadal-Like Differentiation in the Adrenal Cortex of Mice. Endocrinology 2013, 154, 1754–1767. [Google Scholar] [CrossRef] [PubMed]
- Levasseur, A.; St-Jean, G.; Paquet, M.; Boerboom, D.; Boyer, A. Targeted Disruption of YAP and TAZ Impairs the Maintenance of the Adrenal Cortex. Endocrinology 2017, 158, 3738–3753. [Google Scholar] [CrossRef] [PubMed]
- Ménard, A.; Nader, N.A.; Levasseur, A.; St-Jean, G.; Roy, M.L.G.-L.; Boerboom, D.; Benoit-Biancamano, M.-O.; Boyer, A. Targeted Disruption of Lats1 and Lats2 in Mice Impairs Adrenal Cortex Development and Alters Adrenocortical Cell Fate. Endocrinology 2020, 161, bqaa052. [Google Scholar] [CrossRef] [PubMed]
- Nader, N.A.; Blais, É.; St-Jean, G.; Boerboom, D.; Zamberlam, G.; Boyer, A. Effect of inactivation of Mst1 and Mst2 in the mouse adrenal cortex. J. Endocr. Soc. 2022, 7, 143. [Google Scholar] [CrossRef]
- Zubair, M.; Parker, K.L.; Morohashi, K.-I. Developmental Links between the Fetal and Adult Zones of the Adrenal Cortex Revealed by Lineage Tracing. Mol. Cell. Biol. 2008, 28, 7030–7040. [Google Scholar] [CrossRef]
- Wood, M.A.; Acharya, A.; Finco, I.; Swonger, J.M.; Elston, M.J.; Tallquist, M.D.; Hammer, G.D. Fetal adrenal capsular cells serve as progenitor cells for steroidogenic and stromal adrenocortical cell lineages in M. musculus. Development 2013, 140, 4522–4532. [Google Scholar] [CrossRef]
- Dumontet, T.; Sahut-Barnola, I.; Septier, A.; Montanier, N.; Plotton, I.; Roucher-Boulez, F.; Ducros, V.; Lefrançois-Martinez, A.-M.; Pointud, J.-C.; Zubair, M.; et al. PKA signaling drives reticularis differentiation and sexually dimorphic adrenal cortex renewal. JCI Insight 2018, 3, e98394. [Google Scholar] [CrossRef]
- Wu, H.-S.; Lin, H.-T.; Wang, C.-K.L.; Chiang, Y.-F.; Chu, H.-P.; Hu, M.-C. HumanCYP11A1 promoter drives Cre recombinase expression in the brain in addition to adrenals and gonads. Genesis 2007, 45, 59–65. [Google Scholar] [CrossRef]
- Neirijnck, Y.; Calvel, P.; Kilcoyne, K.R.; Kühne, F.; Stévant, I.; Griffeth, R.J.; Pitetti, J.-L.; Andric, S.A.; Hu, M.-C.; Pralong, F.; et al. Insulin and IGF1 receptors are essential for the development and steroidogenic function of adult Leydig cells. FASEB J. 2018, 32, 3321–3335. [Google Scholar] [CrossRef]
- Buaas, F.W.; Gardiner, J.R.; Clayton, S.; Val, P.; Swain, A. In vivo evidence for the crucial role of SF1 in steroid-producing cells of the testis, ovary and adrenal gland. Development 2012, 139, 4561–4570. [Google Scholar] [CrossRef]
- Francis, J.C.; Gardiner, J.R.; Renaud, Y.; Chauhan, R.; Weinstein, Y.; Gomez-Sanchez, C.; Lefrançois-Martinez, A.M.; Bertherat, J.; Val, P.; Swain, A. HOX genes promote cell proliferation and are potential therapeutic targets in adrenocortical tumours. Br. J. Cancer 2021, 124, 805–816. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, L.; York, J.P.; Zhang, P.; Smith, L.B. Targeting of GFP-Cre to the Mouse Cyp11a1 Locus Both Drives Cre Recombinase Expression in Steroidogenic Cells and Permits Generation of Cyp11a1 Knock Out Mice. PLoS ONE 2014, 9, e84541. [Google Scholar] [CrossRef] [PubMed]
- Gannon, A.-L.; O’Hara, L.; Mason, I.J.; Jørgensen, A.; Frederiksen, H.; Curley, M.; Milne, L.; Smith, S.; Mitchell, R.T.; Smith, L.B. Androgen Receptor Is Dispensable for X-Zone Regression in the Female Adrenal but Regulates Post-Partum Corticosterone Levels and Protects Cortex Integrity. Front. Endocrinol. 2021, 11, 599869. [Google Scholar] [CrossRef] [PubMed]
- Gannon, A.-L.; O’Hara, L.; Mason, J.I.; Jørgensen, A.; Frederiksen, H.; Milne, L.; Smith, S.; Mitchell, R.T.; Smith, L.B. Androgen receptor signalling in the male adrenal facilitates X-zone regression, cell turnover and protects against adrenal degeneration during ageing. Sci. Rep. 2019, 9, 10457. [Google Scholar] [CrossRef]
- Lambert-Langlais, S.; Val, P.; Guyot, S.; Ragazzon, B.; Sahut-Barnola, I.; De Haze, A.; Lefrançois-Martinez, A.-M.; Martinez, A. A transgenic mouse line with specific Cre recombinase expression in the adrenal cortex. Mol. Cell. Endocrinol. 2009, 300, 197–204. [Google Scholar] [CrossRef]
- Berthon, A.; Sahut-Barnola, I.; Lambert-Langlais, S.; de Joussineau, C.; Damon-Soubeyrand, C.; Louiset, E.; Taketo, M.M.; Tissier, F.; Bertherat, J.; Lefrançois-Martinez, A.M.; et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 2010, 19, 1561–1576. [Google Scholar] [CrossRef]
- Drelon, C.; Berthon, A.; Ragazzon, B.; Tissier, F.; Bandiera, R.; Sahut-Barnola, I.; De Joussineau, C.; Batisse-Lignier, M.; Lefrançois-Martinez, A.-M.; Bertherat, J.; et al. Analysis of the Role of Igf2 in Adrenal Tumour Development in Transgenic Mouse Models. PLoS ONE 2012, 7, e44171. [Google Scholar] [CrossRef]
- Freedman, B.D.; Kempna, P.B.; Carlone, D.L.; Shah, M.S.; Guagliardo, N.A.; Barrett, P.Q.; Gomez-Sanchez, C.E.; Majzoub, J.A.; Breault, D.T. Adrenocortical Zonation Results from Lineage Conversion of Differentiated Zona Glomerulosa Cells. Dev. Cell 2013, 26, 666–673. [Google Scholar] [CrossRef]
- Leng, S.; Pignatti, E.; Khetani, R.S.; Shah, M.S.; Xu, S.; Miao, J.; Taketo, M.M.; Beuschlein, F.; Barrett, P.Q.; Carlone, D.L.; et al. β-Catenin and FGFR2 regulate postnatal rosette-based adrenocortical morphogenesis. Nat. Commun. 2020, 11, 1680. [Google Scholar] [CrossRef]
- Pignatti, E.; Leng, S.; Yuchi, Y.; Borges, K.S.; Guagliardo, N.A.; Shah, M.S.; Ruiz-Babot, G.; Kariyawasam, D.; Taketo, M.M.; Miao, J.; et al. Beta-Catenin Causes Adrenal Hyperplasia by Blocking Zonal Transdifferentiation. Cell Rep. 2020, 31, 107524. [Google Scholar] [CrossRef]
- Zhang, N.-N.; Wang, C.-N.; Ni, X. Construction of transgenic mice with specific Cre recombinase expression in the zona fasciculata in adrenal cortex. Acta Physiol. Sin. 2020, 72, 148–156. [Google Scholar]
- Ahn, S.; Joyner, A.L. Dynamic Changes in the Response of Cells to Positive Hedgehog Signaling during Mouse Limb Patterning. Cell 2004, 118, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Finco, I.; Lerario, A.M.; Hammer, G.D. Sonic Hedgehog and WNT Signaling Promote Adrenal Gland Regeneration in Male Mice. Endocrinology 2017, 159, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Grabek, A.; Dolfi, B.; Klein, B.; Jian-Motamedi, F.; Chaboissier, M.-C.; Schedl, A. The Adult Adrenal Cortex Undergoes Rapid Tissue Renewal in a Sex-Specific Manner. Cell Stem Cell 2019, 25, 290–296.e2. [Google Scholar] [CrossRef]
- Dörner, J.; Rodriguez, V.M.; Ziegler, R.; Röhrig, T.; Cochran, R.S.; Götz, R.M.; Levin, M.D.; Pihlajoki, M.; Heikinheimo, M.; Wilson, D.B. GLI1+ progenitor cells in the adrenal capsule of the adult mouse give rise to heterotopic gonadal-like tissue. Mol. Cell. Endocrinol. 2017, 441, 164–175. [Google Scholar] [CrossRef]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C.J. Evidence for an Expansion-Based Temporal Shh Gradient in Specifying Vertebrate Digit Identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef]
- Van Amerongen, R.; Bowman, A.N.; Nusse, R. Developmental stage and time dictate the fate of Wnt/β-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 2012, 11, 387–400. [Google Scholar] [CrossRef]
- Kobayashi, A.; Valerius, M.T.; Mugford, J.W.; Carroll, T.J.; Self, M.; Oliver, G.; McMahon, A.P. Six2 Defines and Regulates a Multipotent Self-Renewing Nephron Progenitor Population throughout Mammalian Kidney Development. Cell Stem Cell 2008, 3, 169–181. [Google Scholar] [CrossRef]
- Burns, K.A.; Ayoub, A.E.; Breunig, J.J.; Adhami, F.; Weng, W.L.; Colbert, M.C.; Rakic, P.; Kuan, C.Y. Nestin-CreER mice reveal DNA synthesis by nonapoptotic neurons following cerebral ischemia hypoxia. Cereb. Cortex 2007, 17, 2585–2592. [Google Scholar] [CrossRef]
- Steenblock, C.; de Celis, M.F.R.; Silva, L.F.D.; Pawolski, V.; Brennand, A.; Werdermann, M.; Berger, I.; Santambrogio, A.; Peitzsch, M.; Andoniadou, C.L.; et al. Isolation and characterization of adrenocortical progenitors involved in the adaptation to stress. Proc. Natl. Acad. Sci. USA 2018, 115, 12997–13002. [Google Scholar] [CrossRef]
- Kreidberg, J.A.; Sariola, H.; Loring, J.M.; Maeda, M.; Pelletier, J.; Housman, D.; Jaenisch, R. WT-1 is required for early kidney development. Cell 1993, 74, 679–691. [Google Scholar] [CrossRef]
- Pilon, N.; Raiwet, D.; Viger, R.S.; Silversides, D.W. Novel pre- and post-gastrulation expression of Gata4 within cells of the inner cell mass and migratory neural crest cells. Dev. Dyn. 2008, 237, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Schachterle, W.; Xu, S.-M.; Martín, F.; Black, B.L. Direct transcriptional regulation of Gata4 during early endoderm specification is controlled by FoxA2 binding to an intronic enhancer. Dev. Biol. 2010, 346, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Vidal, V.P.; Chaboissier, M.-C.; Lützkendorf, S.; Cotsarelis, G.; Mill, P.; Hui, C.-C.; Ortonne, N.; Ortonne, J.-P.; Schedl, A. Sox9 Is Essential for Outer Root Sheath Differentiation and the Formation of the Hair Stem Cell Compartment. Curr. Biol. 2005, 15, 1340–1351. [Google Scholar] [CrossRef]
- Del Monte, G.; Casanova, J.C.; Guadix, J.A.; MacGrogan, D.; Burch, J.B.; Pérez-Pomares, J.M.; de la Pompa, J.L. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 2011, 108, 824–836. [Google Scholar] [CrossRef]
- So, P.L.; Danielian, P.S. Cloning and expression analysis of a mouse gene related to Drosophila odd-skipped. Mech. Dev. 1999, 84, 157–160. [Google Scholar] [CrossRef]
- Hata, K.; Kusumi, M.; Yokomine, T.; Li, E.; Sasaki, H. Meiotic and epigenetic aberrations inDnmt3L-deficient male germ cells. Mol. Reprod. Dev. 2005, 73, 116–122. [Google Scholar] [CrossRef]
- Armstrong, J.F.; Pritchard-Jones, K.; Bickmore, W.A.; Hastie, N.D.; Bard, J.B. The expression of the Wilms’ tumour gene, WT1, in the developing mammalian embryo. Mech. Dev. 1993, 40, 85–97. [Google Scholar] [CrossRef]
- Bayne, S.; Jones, M.E.; Li, H.; Pinto, A.R.; Simpson, E.R.; Liu, J.-P. Estrogen deficiency leads to telomerase inhibition, telomere shortening and reduced cell proliferation in the adrenal gland of mice. Cell Res. 2008, 18, 1141–1150. [Google Scholar] [CrossRef]
- Doroszko, M.; Chrusciel, M.; Stelmaszewska, J.; Slezak, T.; Rivero-Muller, A.; Padzik, A.; Anisimowicz, S.; Wolczynski, S.; Huhtaniemi, I.; Toppari, J.; et al. Luteinizing Hormone and GATA4 Action in the Adrenocortical Tumorigenesis of Gonadectomized Female Mice. Cell. Physiol. Biochem. 2017, 43, 1064–1076. [Google Scholar] [CrossRef]
- Chrusciel, M.; Vuorenoja, S.; Mohanty, B.; Rivero-Müller, A.; Li, X.; Toppari, J.; Huhtaniemi, I.; Rahman, N.A. Transgenic GATA-4 expression induces adrenocortical tumorigenesis in C57Bl/6 mice. J. Cell Sci. 2013, 126, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Suemaru, S.; Darlington, D.N.; Akana, S.F.; Cascio, C.S.; Dallman, M.F. Ventromedial Hypothalamic Lesions Inhibit Corticosteroid Feedback Regulation of Basal ACTH during the Trough of the Circadian Rhythm. Neuroendocrinology 1995, 61, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Sahut-Barnola, I.; Lefrançois-Martinez, A.-M.; Dufour, D.; Botto, J.-M.; Kamilaris, C.; Faucz, F.R.; Stratakis, C.A.; Val, P.; Martinez, A. Steroidogenic Factor-1 Lineage Origin of Skin Lesions in Carney Complex Syndrome. J. Investig. Dermatol. 2022, 142, 2949–2957.e9. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Seita, Y.; Moriwaki, T.; Noshiro, K.; Sakata, Y.; Hwang, Y.S.; Torigoe, T.; Saitou, M.; Tsuchiya, H.; Iwatani, C.; et al. The developmental origin and the specification of the adrenal cortex in humans and cynomolgus monkeys. Sci. Adv. 2022, 8, eabn8485. [Google Scholar] [CrossRef] [PubMed]
- Saito, D.; Tamura, K.; Takahashi, Y. Early segregation of the adrenal cortex and gonad in chicken embryos. Dev. Growth Differ. 2017, 59, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lan, Y.; Cho, E.-S.; Maltby, K.M.; Jiang, R. Odd-skipped related 1 (Odd1) is an essential regulator of heart and urogenital development. Dev. Biol. 2005, 288, 582–594. [Google Scholar] [CrossRef]
- Moore, A.; McInnes, L.; Kreidberg, J.; Hastie, N.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar] [CrossRef]
- Miyamoto, N.; Yoshida, M.; Kuratani, S.; Matsuo, I.; Aizawa, S. Defects of urogenital development in mice lacking Emx2. Development 1997, 124, 1653–1664. [Google Scholar] [CrossRef]
- Kusaka, M.; Katoh-Fukui, Y.; Ogawa, H.; Miyabayashi, K.; Baba, T.; Shima, Y.; Sugiyama, N.; Sugimoto, Y.; Okuno, Y.; Kodama, R.; et al. Abnormal Epithelial Cell Polarity and Ectopic Epidermal Growth Factor Receptor (EGFR) Expression Induced in Emx2 KO Embryonic Gonads. Endocrinology 2010, 151, 5893–5904. [Google Scholar] [CrossRef]
- Birk, O.S.; Casiano, D.E.; Wassif, C.A.; Cogliati, T.; Zhao, L.; Zhao, Y.; Grinberg, A.; Huang, S.; Kreidberg, J.A.; Parker, K.L.; et al. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature 2000, 403, 909–913. [Google Scholar] [CrossRef]
- Katoh-Fukui, Y.; Owaki, A.; Toyama, Y.; Kusaka, M.; Shinohara, Y.; Maekawa, M.; Toshimori, K.; Morohashi, K.-I. Mouse Polycomb M33 is required for splenic vascular and adrenal gland formation through regulating Ad4BP/SF1 expression. Blood 2005, 106, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.; Bragança, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.; Kranc, K.R.; Farza, H.; Henderson, D.; Hurst, H.C.; Bhattacharya, S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Val, P.; Martinez-Barbera, J.P.; Swain, A. Adrenal development is initiated by Cited2 and Wt1 through modulation of Sf-1 dosage. Development 2007, 134, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Teves, M.E.; Modi, B.P.; Kulkarni, R.; Han, A.X.; Marks, J.S.; Subler, M.A.; Windle, J.; Newall, J.M.; McAllister, J.M.; Strauss, J.F., 3rd. Human DENND1A.V2 Drives Cyp17a1 Expression and Androgen Production in Mouse Ovaries and Adrenals. Int. J. Mol. Sci. 2020, 21, 2545. [Google Scholar] [CrossRef]
- Guasti, L.; Sze, W.C.; McKay, T.; Grose, R.; King, P.J. FGF signalling through Fgfr2 isoform IIIb regulates adrenal cortex development. Mol. Cell. Endocrinol. 2013, 371, 182–188. [Google Scholar] [CrossRef]
- Krachulec, J.; Vetter, M.; Schrade, A.; Löbs, A.-K.; Bielinska, M.; Cochran, R.; Kyrönlahti, A.; Pihlajoki, M.; Parviainen, H.; Jay, P.Y.; et al. GATA4 Is a Critical Regulator of Gonadectomy-Induced Adrenocortical Tumorigenesis in Mice. Endocrinology 2012, 153, 2599–2611. [Google Scholar] [CrossRef][Green Version]
- Böse, J.; Grotewold, L.; Rüther, U. Pallister-Hall syndrome phenotype in mice mutant for Gli3. Hum. Mol. Genet. 2002, 11, 1129–1135. [Google Scholar] [CrossRef]
- Laufer, E.; Kesper, D.; Vortkamp, A.; King, P. Sonic hedgehog signaling during adrenal development. Mol. Cell. Endocrinol. 2012, 351, 19–27. [Google Scholar] [CrossRef]
- Pitetti, J.-L.; Calvel, P.; Romero, Y.; Conne, B.; Truong, V.; Papaioannou, M.D.; Schaad, O.; Docquier, M.; Herrera, P.L.; Wilhelm, D.; et al. Insulin and IGF1 Receptors Are Essential for XX and XY Gonadal Differentiation and Adrenal Development in Mice. PLoS Genet. 2013, 9, e1003160. [Google Scholar] [CrossRef]
- Chida, D.; Nakagawa, S.; Nagai, S.; Sagara, H.; Katsumata, H.; Imaki, T.; Suzuki, H.; Mitani, F.; Ogishima, T.; Shimizu, C.; et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18205–18210. [Google Scholar] [CrossRef]
- Novoselova, T.V.; Hussain, M.; King, P.J.; Guasti, L.; Metherell, L.A.; Charalambous, M.; Clark, A.J.L.; Chan, L.F. MRAP deficiency impairs adrenal progenitor cell differentiation and gland zonation. FASEB J. 2018, 32, 6186–6196. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Morohashi, K.-I.; Ingraham, H.A.; Hammer, G.D. Timing of adrenal regression controlled by synergistic interaction between Sf1 SUMOylation and Dax1. Development 2017, 144, 3798–3807. [Google Scholar] [CrossRef] [PubMed]
- Scheys, J.O.; Heaton, J.H.; Hammer, G.D. Evidence of Adrenal Failure in Aging Dax1-Deficient Mice. Endocrinology 2011, 152, 3430–3439. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ikeda, Y.; Parker, K.L. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 1994, 77, 481–490. [Google Scholar] [CrossRef]
- Bland, M.L.; Jamieson, C.A.; Akana, S.F.; Bornstein, S.R.; Eisenhofer, G.; Dallman, M.F.; Ingraham, H.A. Haploinsufficiency of steroidogenic factor-1 in mice disrupts adrenal development leading to an impaired stress response. Proc. Natl. Acad. Sci. USA 2000, 97, 14488–14493. [Google Scholar] [CrossRef]
- Bland, M.L.; Fowkes, R.C.; Ingraham, H.A. Differential Requirement for Steroidogenic Factor-1 Gene Dosage in Adrenal Development Versus Endocrine Function. Mol. Endocrinol. 2004, 18, 941–952. [Google Scholar] [CrossRef]
- Zubair, M.; Oka, S.; Parker, K.L.; Morohashi, K.-I. Transgenic Expression ofAd4BP/SF-1in Fetal Adrenal Progenitor Cells Leads to Ectopic Adrenal Formation. Mol. Endocrinol. 2009, 23, 1657–1667. [Google Scholar] [CrossRef]
- Lee, F.Y.; Faivre, E.J.; Suzawa, M.; Lontok, E.; Ebert, D.; Cai, F.; Belsham, D.D.; Ingraham, H.A. Eliminating SF-1 (NR5A1) Sumoylation In Vivo Results in Ectopic Hedgehog Signaling and Disruption of Endocrine Development. Dev. Cell 2011, 21, 315–327. [Google Scholar] [CrossRef]
- Doghman, M.; Karpova, T.; Rodrigues, G.A.; Arhatte, M.; de Moura, J.; Cavalli, L.R.; Virolle, V.; Barbry, P.; Zambetti, G.P.; Figueiredo, B.C.; et al. Increased Steroidogenic Factor-1 Dosage Triggers Adrenocortical Cell Proliferation and Cancer. Mol. Endocrinol. 2007, 21, 2968–2987. [Google Scholar] [CrossRef]
- Schnabel, C.A.; Selleri, L.; Cleary, M.L. Pbx1 is essential for adrenal development and urogenital differentiation. Genesis 2003, 37, 123–130. [Google Scholar] [CrossRef]
- Lichtenauer, U.D.; Duchniewicz, M.; Kolanczyk, M.; Hoeflich, A.; Hahner, S.; Else, T.; Bicknell, A.B.; Zemojtel, T.; Stallings, N.R.; Schulte, M.M.; et al. Pre-B-Cell Transcription Factor 1 and Steroidogenic Factor 1 Synergistically Regulate Adrenocortical Growth and Steroidogenesis. Endocrinology 2007, 148, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Tsai, L.C.; Shimizu-Albergine, M.; Beavo, J.A. The high-affinity cAMP-specific phosphodiesterase 8B controls steroidogenesis in the mouse adrenal gland. Mol. Pharmacol. 2011, 79, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Leal, L.F.; Szarek, E.; Berthon, A.; Nesterova, M.; Faucz, F.R.; London, E.; Mercier, C.; Abu-Asab, M.; Starost, M.F.; Dye, L.; et al. Pde8b haploinsufficiency in mice is associated with modest adrenal defects, impaired steroidogenesis, and male infertility, unaltered by concurrent PKA or Wnt activation. Mol. Cell. Endocrinol. 2020, 522, 111117. [Google Scholar] [CrossRef] [PubMed]
- Levy, I.; Szarek, E.; Maria, A.G.; Starrost, M.; Sierra, M.D.L.L.; Faucz, F.R.; Stratakis, C.A. A phosphodiesterase 11 (Pde11a) knockout mouse expressed functional but reduced Pde11a: Phenotype and impact on adrenocortical function. Mol. Cell. Endocrinol. 2020, 520, 111071. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Jiang, X.; Cong, F. Control of Wnt Receptor Turnover by R-spondin-ZNRF3/RNF43 Signaling Module and Its Dysregulation in Cancer. Cancers 2016, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Berthon, A.; Settas, N.; Giannakou, A.; Garcia, G.; Li, J.-L.; James, B.; Liddington, R.C.; Vilches-Moure, J.G.; Stratakis, C.A.; et al. The E3 ubiquitin ligase Siah1 regulates adrenal gland organization and aldosterone secretion. JCI Insight 2017, 2, e97128. [Google Scholar] [CrossRef] [PubMed]
- Berthon, A.; Drelon, C.; Ragazzon, B.; Boulkroun, S.; Tissier, F.; Amar, L.; Samson-Couterie, B.; Zennaro, M.-C.; Plouin, P.-F.; Skah, S.; et al. WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum. Mol. Genet. 2013, 23, 889–905. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Tanaka, S.S.; Yamaguchi, Y.L.; Kobayashi, H.; Kuroki, S.; Tachibana, M.; Shinomura, M.; Kanai, Y.; Morohashi, K.-I.; Kawakami, K.; et al. Homeoproteins Six1 and Six4 Regulate Male Sex Determination and Mouse Gonadal Development. Dev. Cell 2013, 26, 416–430. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kawakami, K.; Asashima, M.; Nishinakamura, R. Six1 and Six4 are essential for Gdnf expression in the metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mech. Dev. 2007, 124, 290–303. [Google Scholar] [CrossRef]
- Heikkilä, M.; Peltoketo, H.; Leppäluoto, J.; Ilves, M.; Vuolteenaho, O.; Vainio, S. Wnt-4 Deficiency Alters Mouse Adrenal Cortex Function, Reducing Aldosterone Production. Endocrinology 2002, 143, 4358–4365. [Google Scholar] [CrossRef]
- Wilmouth, J.J.; Olabe, J.; Garcia-Garcia, D.; Lucas, C.; Guiton, R.; Roucher-Boulez, F.; Dufour, D.; Damon-Soubeyrand, C.; Sahut-Barnola, I.; Pointud, J.-C.; et al. Sexually dimorphic activation of innate antitumor immunity prevents adrenocortical carcinoma development. Sci. Adv. 2022, 8, eadd0422. [Google Scholar] [CrossRef] [PubMed]
- Heikinheimo, M.; Scandrett, J.M.; Wilson, D. Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Dev. Biol. 1994, 164, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Lin, Q.; Duncan, S.A.; Olson, E.N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997, 11, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.T.; Morrisey, E.E.; Anandappa, R.; Sigrist, K.; Lu, M.M.; Parmacek, M.S.; Soudais, C.; Leiden, J.M. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997, 11, 1048–1060. [Google Scholar] [CrossRef]
- Moore, A.W.; Schedl, A.; McInnes, L.; Doyle, M.; Hecksher-Sorensen, J.; Hastie, N.D. YAC transgenic analysis reveals Wilms’ Tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mech. Dev. 1998, 79, 169–184. [Google Scholar] [CrossRef]
- Furuhata, A.; Murakami, M.; Ito, H.; Gao, S.; Yoshida, K.; Sobue, S.; Kikuchi, R.; Iwasaki, T.; Takagi, A.; Kojima, T.; et al. GATA-1 and GATA-2 binding to 3′ enhancer of WT1 gene is essential for its transcription in acute leukemia and solid tumor cell lines. Leukemia 2009, 23, 1270–1277. [Google Scholar] [CrossRef]
- Klattig, J.; Sierig, R.; Kruspe, D.; Makki, M.; Englert, C. WT1-Mediated Gene Regulation in Early Urogenital Ridge Development. Sex. Dev. 2007, 1, 238–254. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Taniguchi, H.; Hamel, F.; Silversides, D.W.; Viger, R.S. A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol. Biol. 2008, 9, 44. [Google Scholar] [CrossRef]
- Wilhelm, D.; Englert, C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 2002, 16, 1839–1851. [Google Scholar] [CrossRef]
- Tremblay, J.J.; Viger, R.S. GATA Factors Differentially Activate Multiple Gonadal Promoters through Conserved GATA Regulatory Elements. Endocrinology 2001, 142, 977–986. [Google Scholar] [CrossRef]
- Dupont, J.; Holzenberger, M. Biology of insulin-like growth factors in development. Birth Defects Res. C Embryo Today 2003, 69, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Hammer, G.D.; Krylova, I.; Zhang, Y.; Darimont, B.D.; Simpson, K.; Weigel, N.L.; Ingraham, H.A. Phosphorylation of the nuclear receptor SF-1 modulates cofactor recruitment: Integration of hormone signaling in reproduction and stress. Mol. Cell 1999, 3, 521–526. [Google Scholar] [CrossRef]
- França, M.M.; Ferraz-De-Souza, B.; Santos, M.G.; Lerario, A.M.; Fragoso, M.C.B.V.; Latronico, A.C.; Kuick, R.D.; Hammer, G.D.; Lotfi, C.F. POD-1 binding to the E-box sequence inhibits SF-1 and StAR expression in human adrenocortical tumor cells. Mol. Cell. Endocrinol. 2013, 371, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Kanno, Y.; Chuma, S.; Saito, T.; Nakatsuji, N. Pod-1/Capsulin shows a sex- and stage-dependent expression pattern in the mouse gonad development and represses expression of Ad4BP/SF-1. Mech. Dev. 2001, 102, 135–144. [Google Scholar] [CrossRef]
- Garcia-Moreno, S.A.; Lin, Y.-T.; Futtner, C.R.; Salamone, I.M.; Capel, B.; Maatouk, D.M. CBX2 is required to stabilize the testis pathway by repressing Wnt signaling. PLoS Genet. 2019, 15, e1007895. [Google Scholar] [CrossRef]
- Katoh-Fukui, Y.; Miyabayashi, K.; Komatsu, T.; Owaki, A.; Baba, T.; Shima, Y.; Kidokoro, T.; Kanai, Y.; Schedl, A.; Wilhelm, D.; et al. Cbx2, a Polycomb Group Gene, Is Required for Sry Gene Expression in Mice. Endocrinology 2012, 153, 913–924. [Google Scholar] [CrossRef]
- Katoh-Fukui, Y.; Tsuchiya, R.; Shiroishi, T.; Nakahara, Y.; Hashimoto, N.; Noguchi, K.; Higashinakagawa, T. Male-to-female sex reversal in M33 mutant mice. Nature 1998, 393, 688–692. [Google Scholar] [CrossRef]
- Kiiveri, S.; Liu, J.; Westerholm-Ormio, M.; Narita, N.; Wilson, D.B.; Voutilainen, R.; Heikinheimo, M. Transcription factors gata-4 and gata-6 during mouse and human adrenocortical development. Endocr. Res. 2002, 28, 647–650. [Google Scholar] [CrossRef]
- Kiiveri, S.; Liu, J.; Westerholm-Ormio, M.; Narita, N.; Wilson, D.B.; Voutilainen, R.; Heikinheimo, M. Differential expression of GATA-4 and GATA-6 in fetal and adult mouse and human adrenal tissue. Endocrinology 2002, 143, 3136–3143. [Google Scholar] [CrossRef]
- Chia, C.Y.; Madrigal, P.; Denil, S.; Martinez, I.; Garcia-Bernardo, J.; El-Khairi, R.; Chhatriwala, M.; Shepherd, M.H.; Hattersley, A.T.; Dunn, N.R.; et al. GATA6 Cooperates with EOMES/SMAD2/3 to Deploy the Gene Regulatory Network Governing Human Definitive Endoderm and Pancreas Formation. Stem Cell Rep. 2019, 12, 57–70. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, H.; Xiang, M.; Zhou, L.; Wu, B.; Moskowitz, I.; Zhang, K.; Xie, L. Gata4 regulates hedgehog signaling and Gata6 expression for outflow tract development. PLoS Genet. 2019, 15, e1007711. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, T.; Yokura, Y.; Ohashi, K.; Yamamoto, H.; Maeda, M. Conserved GC-boxes, E-box and GATA motif are essential for GATA-4 gene expression in P19CL6 cells. Biochem. Biophys. Res. Commun. 2011, 413, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Zubair, M.; Ishihara, S.; Oka, S.; Okumura, K.; Morohashi, K.-I. Two-Step Regulation of Ad4BP/SF-1 Gene Transcription during Fetal Adrenal Development: Initiation by a Hox-Pbx1-Prep1 Complex and Maintenance via Autoregulation by Ad4BP/SF-1. Mol. Cell. Biol. 2006, 26, 4111–4121. [Google Scholar] [CrossRef] [PubMed]
- Bielohuby, M.; Herbach, N.; Wanke, R.; Maser-Gluth, C.; Beuschlein, F.; Wolf, E.; Hoeflich, A. Growth analysis of the mouse adrenal gland from weaning to adulthood: Time- and gender-dependent alterations of cell size and number in the cortical compartment. Am. J. Physiol. Metab. 2007, 293, E139–E146. [Google Scholar] [CrossRef] [PubMed]
- Howard-Miller, E. A transitory zone in the adrenal cortex which shows age and sex relationships. Am. J. Anat. 1927, 40, 251–293. [Google Scholar] [CrossRef]
- Furlan, A.; Dyachuk, V.; Kastriti, M.E.; Calvo-Enrique, L.; Abdo, H.; Hadjab, S.; Chontorotzea, T.; Akkuratova, N.; Usoskin, D.; Kamenev, D.; et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science 2017, 357, eaal3753. [Google Scholar] [CrossRef]
- Hanemaaijer, E.S.; Margaritis, T.; Sanders, K.; Bos, F.L.; Candelli, T.; Al-Saati, H.; van Noesel, M.M.; Meyer-Wentrup, F.A.G.; van de Wetering, M.; Holstege, F.C.P.; et al. Single-cell atlas of developing murine adrenal gland reveals relation of Schwann cell precursor signature to neuroblastoma phenotype. Proc. Natl. Acad. Sci. USA 2021, 118, e2022350118. [Google Scholar] [CrossRef]
- Kameneva, P.; Artemov, A.V.; Kastriti, M.E.; Faure, L.; Olsen, T.K.; Otte, J.; Erickson, A.; Semsch, B.; Andersson, E.R.; Ratz, M.; et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat. Genet. 2021, 53, 694–706. [Google Scholar] [CrossRef]
- Britsch, S.; Li, L.; Kirchhoff, S.; Theuring, F.; Brinkmann, V.; Birchmeier, C.; Riethmacher, D. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998, 12, 1825–1836. [Google Scholar] [CrossRef]
- Tomooka, Y.; Yasui, T. Electron microscopic study of the response of the adrenocortical X-zone in mice treated with sex steroids. Cell Tissue Res. 1978, 194, 269–277. [Google Scholar] [CrossRef]
- Holmes, P.V.; Dickson, A.D. X-zone degeneration in the adrenal glands of adult and immature female mice. J. Anat. 1971, 108, 159–168. [Google Scholar] [PubMed]
- Janat, M.F.; Shire, J.G. The adrenal X-zone of mice: Genetic analysis of its development with recombinant-inbred strains. Exp. Biol. 1987, 46, 217–221. [Google Scholar] [PubMed]
- Ungar, F.; Stabler, T.A. 20α-Hydroxysteroid dehydrogenase activity and the x-zone of the female mouse adrenal. J. Steroid Biochem. 1980, 13, 23–28. [Google Scholar] [CrossRef]
- Shibata, H.; Kurihara, I.; Kobayashi, S.; Yokota, K.; Suda, N.; Saito, I.; Saruta, T. Regulation of differential COUP-TF-coregulator interactions in adrenal cortical steroidogenesis. J. Steroid Biochem. Mol. Biol. 2003, 85, 449–456. [Google Scholar] [CrossRef]
- Hall, J.G.; Pallister, P.D.; Clarren, S.K.; Beckwith, J.B.; Wiglesworth, F.W.; Fraser, F.C.; Cho, S.; Benke, P.J.; Reed, S.D. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly—A new syndrome? Part I: Clinical, causal, and pathogenetic considerations. Am. J. Med. Genet. 1980, 7, 47–74. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.B.; Snel, B.; Clevers, H.C. The R-spondin protein family. Genome Biol. 2012, 13, 242. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, A.; Zhang, S.; Ruusunen, T.; Kreidberg, J.A.; Peltoketo, H.; Drummond, I.; Vainio, S. Induction of ureter branching as a response to Wnt-2b signaling during early kidney organogenesis. Dev. Dyn. 2001, 222, 26–39. [Google Scholar] [CrossRef]
- Lopez, J.P.; Brivio, E.; Santambrogio, A.; De Donno, C.; Kos, A.; Peters, M.; Rost, N.; Czamara, D.; Brückl, T.M.; Roeh, S.; et al. Single-cell molecular profiling of all three components of the HPA axis reveals adrenal ABCB1 as a regulator of stress adaptation. Sci. Adv. 2021, 7, eabe4497. [Google Scholar] [CrossRef]
- Lyu, Q.; Wang, H.; Kang, Y.; Wu, X.; Zheng, H.S.; Laprocina, K.; Junghans, K.; Ding, X.; Huang, C.-C.J. RNA-Seq Reveals Sub-Zones in Mouse Adrenal Zona Fasciculata and the Sexually Dimorphic Responses to Thyroid Hormone. Endocrinology 2020, 161, bqaa126. [Google Scholar] [CrossRef]
- Tee, M.K.; Speek, M.; Legeza, B.; Modi, B.; Teves, M.E.; McAllister, J.M.; Strauss, J.F., 3rd; Miller, W.L. Alternative splicing of DENND1A, a PCOS candidate gene, generates variant 2. Mol. Cell. Endocrinol. 2016, 434, 25–35. [Google Scholar] [CrossRef]
- McAllister, J.M.; Modi, B.; Miller, B.A.; Biegler, J.; Bruggeman, R.; Legro, R.S.; Strauss, J.F. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc. Natl. Acad. Sci. USA 2014, 111, E1519–E1527. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, R.; Teves, M.E.; Han, A.X.; McAllister, J.M.; Strauss, J.F. Colocalization of Polycystic Ovary Syndrome Candidate Gene Products in Theca Cells Suggests Novel Signaling Pathways. J. Endocr. Soc. 2019, 3, 2204–2223. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.M.; Legro, R.S.; Modi, B.; Strauss, J.F. Functional genomics of PCOS: From GWAS to molecular mechanisms. Trends Endocrinol. Metab. 2015, 26, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.A.; Ratnayake, U.; Dickinson, H.; Nguyen, T.-H.; McIntosh, M.; Castillo-Melendez, M.; Conley, A.J.; Walker, D.W. Ontogeny of the Adrenal Gland in the Spiny Mouse, With Particular Reference to Production of the Steroids Cortisol and Dehydroepiandrosterone. Endocrinology 2013, 154, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Zhang, A.; Hernandez, S.; Marck, B.T.; Zhang, X.; Tamae, D.; Biehl, H.E.; Tretiakova, M.S.; Bartlett, J.; Burns, J.F.; et al. Contribution of Adrenal Glands to Intratumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 426–439. [Google Scholar] [CrossRef]
- Yaglova, N.V.; Obernikhin, S.S.; Nazimova, S.V.; Yaglov, V.V. Role of Transcription Factor Oct4 in Postnatal Development and Function of the Adrenal Cortex. Bull. Exp. Biol. Med. 2019, 167, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Moog, F.; Bennett, C.J.; Dean, C.M., Jr. Growth and cytochemistry of the adrenal gland of the mouse from birth to maturity. Anat. Rec. 1954, 120, 873–891. [Google Scholar] [CrossRef]
- Callow, R.K.; Deanesly, R. Effect of androsterone and of male hormone concentrates on the accessory reproductive organs of castrated rats, mice and guinea-pigs. Biochem. J. 1935, 29, 1424–1445. [Google Scholar] [CrossRef]
- Starkey, W.F.; Schmidt, E.C.H. THE EFFECT OF TESTOSTERONE-PROPIONATE ON THE X-ZONE OF THE MOUSE ADRENAL. Endocrinology 1938, 23, 339–344. [Google Scholar] [CrossRef]
- Mukai, T.; Kusaka, M.; Kawabe, K.; Goto, K.; Nawata, H.; Fujieda, K.; Morohashi, K.-I. Sexually dimorphic expression of Dax-1 in the adrenal cortex. Genes Cells 2002, 7, 717–729. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Alptekin, A.; Lewis, M.J.; Garraway, I.P.; Cinar, B. YAP1 and AR interactions contribute to the switch from androgen-dependent to castration-resistant growth in prostate cancer. Nat. Commun. 2015, 6, 8126. [Google Scholar] [CrossRef] [PubMed]
- Audenet, F.; Méjean, A.; Chartier-Kastler, E.; Rouprêt, M. Adrenal tumours are more predominant in females regardless of their histological subtype: A review. World J. Urol. 2013, 31, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Jasim, S.; Feng, L.; Ejaz, S.; Deniz, F.; Busaidy, N.; Waguespack, S.G.; Naing, A.; Sircar, K.; Wood, C.G.; et al. Adrenocortical carcinoma: Clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur. J. Endocrinol. 2013, 169, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Scollo, C.; Russo, M.; Trovato, M.A.; Sambataro, D.; Giuffrida, D.; Manusia, M.; Sapuppo, G.; Malandrino, P.; Vigneri, R.; Pellegriti, G. Prognostic Factors for Adrenocortical Carcinoma Outcomes. Front. Endocrinol. 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Dolfi, B.; Gallerand, A.; Firulyova, M.M.; Xu, Y.; Merlin, J.; Dumont, A.; Castiglione, A.; Vaillant, N.; Quemener, S.; Gerke, H.; et al. Unravelling the sex-specific diversity and functions of adrenal gland macrophages. Cell Rep. 2022, 39, 110949. [Google Scholar] [CrossRef]
- Boyle, M.H.; Paranjpe, M.G.; Creasy, D.M. High Background Incidence of Spontaneous Subcapsular Adrenal Gland Hyperplasia of Tg.rasH2 Mice Used in 26-week Carcinogenicity Studies. Toxicol. Pathol. 2018, 46, 444–448. [Google Scholar] [CrossRef]
- Petterino, C.; Naylor, S.; Mukaratirwa, S.; Bradley, A. Adrenal Gland Background Findings in CD-1 (Crl:CD-1(ICR)BR) Mice from 104-week Carcinogenicity Studies. Toxicol. Pathol. 2015, 43, 816–824. [Google Scholar] [CrossRef]
- Yoshida, A.; Maita, K.; Shirasu, Y. Subcapsular cell hyperplasia in the mouse adrenal glands. Jpn. J. Vet. Sci. 1986, 48, 719–728. [Google Scholar] [CrossRef]
- Bielinska, M.; Kiiveri, S.; Parviainen, H.; Mannisto, S.; Heikinheimo, M.; Wilson, D.B. Gonadectomy-induced Adrenocortical Neoplasia in the Domestic Ferret (Mustela putorius furo) and Laboratory Mouse. Vet. Pathol. 2006, 43, 97–117. [Google Scholar] [CrossRef]
- Bielinska, M.; Parviainen, H.; Porter-Tinge, S.B.; Kiiveri, S.; Genova, E.; Rahman, N.; Huhtaniemi, I.T.; Muglia, L.J.; Heikinheimo, M.; Wilson, D. Mouse Strain Susceptibility to Gonadectomy-Induced Adrenocortical Tumor Formation Correlates with the Expression of GATA-4 and Luteinizing Hormone Receptor. Endocrinology 2003, 144, 4123–4133. [Google Scholar] [CrossRef][Green Version]
- Bielinska, M.; Genova, E.; Boime, I.; Parviainen, H.; Kiiveri, S.; Leppäluoto, J.; Rahman, N.; Heikinheimo, M.; Wilson, D.B. Gonadotropin-Induced Adrenocortical Neoplasia in NU/J Nude Mice. Endocrinology 2005, 146, 3975–3984. [Google Scholar] [CrossRef] [PubMed]
- Looyenga, B.; Hammer, G.D. Origin and Identity of Adrenocortical Tumors in Inhibin Knockout Mice: Implications for Cellular Plasticity in the Adrenal Cortex. Mol. Endocrinol. 2006, 20, 2848–2863. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Strains | Ref (Mouse Dev.) | Targeted Tissues/Cells during Adrenal Development and Maintenance | Targeted Genes (Inactivated)/Tracing Cell Populations | Ref |
|---|---|---|---|---|
| CAG-CreER | [4] | Global inactivation | Gata4 | [5] |
| Rspo3 | [6] | |||
| Osr1eGFP-CreERt2 | [7] | Coelomic epithelium | Gata4 | [5] |
| Tracing intermediate mesoderm descendants | [8] | |||
| Tbx18Cre | [9] | Coelomic epithelium | Fgfr2 | [10] |
| Gata4CreERt2 | [11] | Coelomic epithelium/AGP | ||
| Wt1CreERt2/+ | [12] | Coelomic epithelium/AGP Subpopulation of capsular cells | Gata4 | [5] |
| Tracing (capsular population) | [13] | |||
| Tracing intermediate mesoderm descendants | [8] | |||
| Nr5a1-Crehigh (High transgene copy numbers) | [14] | AGP/fetal cortex, definitive cortex | Apc | [15] |
| Ctnnb1 | [16,17] | |||
| Ctnnb1ex3 | [17] | |||
| Dicer | [17,18] | |||
| Ezh2 | [19] | |||
| Fgfr2 | [20] | |||
| Gata4 | [21,22] | |||
| Gata6 | [21,22] | |||
| Porcn | [23] | |||
| Prkar1a | [24,25,26] | |||
| Rnf43 | [23] | |||
| Shh | [17,27,28,29] | |||
| Smo | [28] | |||
| Wnt4 | [6,25] | |||
| Wt1 (activation) | [13] | |||
| Znrf3 | [23] | |||
| Nr5a1-Crelow (low transgene copy numbers) | [14] | AGP/fetal cortex, definitive cortex, few cells affected | Apc | [15] |
| Ctnnb1 | [15,16] | |||
| H19 | [15] | |||
| Nr5a1-Cre | [30] | AGP/fetal cortex, definitive cortex | Gata6 | [31] |
| Yap/Taz | [32] | |||
| Lats1/Lats2 | [33] | |||
| Mst1/Mst2 | [34] | |||
| FAdE/Nr5a1-Cre | [35] | Fetal cortex | Tracing fetal adrenocortical cells descendants | [35,36] |
| FAdE/Nr5a1-CreERT2 | [35] | Fetal cortex | Prkar1a | [37] |
| Tracing fetal adrenocortical cells descendants | [35,37] | |||
| Nr5a1 eGFP-CreERt2 | - | AGP/fetal cortex, definitive cortex | ||
| hCyp11a1-iCre | [38] | Fetal cortex/definitive cortex | Insr/Igf1r | [39] |
| mCyp11a1-iCre | [40] | Fetal cortex/definitive cortex | Ctnnb1ex3 | [41] |
| Nr5a1 | [40] | |||
| Cyp11a1Gfp,Cre/+ | [42] | Fetal cortex/definitive cortex | AR | [43,44] |
| Akr1b7-Cre | [45] | Fetal cortex/definitive cortex | Ctnnb1ex3 | [24,25,46,47] |
| Prkar1a | [25,26] | |||
| Prkaca | [25] | |||
| Cyp11b2Cre/ASCre | [48] | Aldosterone producing zG cells and their zF descendants | Ctnnb1 | [49] |
| Ctnnb1ex3 | [49,50] | |||
| Ffg2r | [49] | |||
| Prkar1a | [37] | |||
| Nr5a1 | [48] | |||
| Nr0b1 | [48] | |||
| Znrf3 | [23] | |||
| Tracing zG cell descendants | [23,37,48,50] | |||
| Cyp11b1eGFP-Cre | [51] | zF cells | Cth | [51] |
| Gli1CreERt2 | [52] | Capsular stem cells | Rspo3 | [6] |
| Smo (activation) | [53] | |||
| Tracing capsular stem cell descendants | [27,28,36,53,54,55] | |||
| ShhCre | [56] | Subcapsular progenitor zG cells | Tracing subcapsular progenitor cell descendants | [28] |
| ShhCreERt2 | [56] | Subcapsular progenitor zG cells | Tracing subcapsular progenitor cell descendants | [28,53] |
| Axin2CreERt2 | [57] | WNT signaling activated zG cells | Ctnnb1 | [53] |
| Tracing zG cells descendants (including subcapsular progenitor cells) | [53,54] | |||
| Wnt4 CreERt2 | [58] | WNT signaling activated zG cells | Tracing zG cells descendants (including subcapsular progenitor cells) | [54] |
| Nes-CreERt2 | [59] | Stress induced adrenocortical progenitor cells | Tracing stress induced progenitor cell descendants | [60] |
| Genes | Mouse Models | Phenotype | Ref |
|---|---|---|---|
| Apc | Apc flox/flox; Nr5a1-Crehigh | Adrenal hypoplasia | [15] |
| Apc flox/flox; Nr5a1-Crelow | Adrenal Hyperplasia, rare adenoma in older animals | [15] | |
| Apc flox/flox; Ctnnb1 flox/flox; Nr5a1-Crelow | Rescue of the hyperplasia | [15] | |
| Apc flox/flox; H19 floxDMD/floxDMD; Nr5a1-Crelow | Adrenal Hyperplasia with higher incidence of adenoma | [15] | |
| AR | Ar flox/Y; Cyp11a1 Gfp,Cre/+ | Abnormal retention of the X-zone, Subcapsular spindle-shaped cell hyperplasia | [44] |
| Ar flox/flox; Cyp11a1 Gfp,Cre/+ | Reduced expression of the zF markers AKR1B7, Subcapsular spindle-shaped cell hyperplasia | [43] | |
| Cbx2 | Cbx2−/− | Mild hypoplastic adrenal gland at e18.5 | [81] |
| Cited2 | Cited2−/− | Adrenal agenesis | [82,83] |
| Cited2+/−; Wt1+/− | Adrenal and gonadal hypoplasia | [83] | |
| Ctnnb1 | Ctnnb1 flox/flox; Nr5a1-Crehigh | Adrenal aplasia | [16,17] |
| Ctnnb1 flox/flox; Nr5a1-Crelow | Age-dependent adrenal cortex degeneration | [16] | |
| Ctnnb1 flox/flox; Axin2CreERt2/+ | Inefficient regeneration of the adrenal cortex | [53] | |
| Ctnnb1 flox/flox; AS Cre/+ | Impaired rosette formation in the zG | [49] | |
| Ctnnb1ex3/+; Nr5a1-Crehigh | Adrenal agenesis (right adrenal), adrenal hypoplasia (left adrenal) | [17,25] | |
| Ctnnb1ex3/+; Prkar1a flox/flox; Nr5a1-Crehigh | Partial rescue of the adrenal hypoplasia. | [25] | |
| Ctnnb1ex3/+; Akr1b7-Cre | Ectopic expression of zG cells at the expense of zF cells, hyperaldosteronism, Subcapsular spindle-shaped cell hyperplasia, rare adenoma in older animals | [46] | |
| Increased SUMOylation in the zF | [24] | ||
| Ctnnb1ex3/+; Prkar1a flox/flox; Akr1b7-Cre | Decreased WNT induced hyperproliferation and ectopic zG differentiation | [25] | |
| Ctnnb1ex3/+; Prkaca +/−; Akr1b7-Cre | Accelerated WNT induced tumorigenesis | [25] | |
| Ctnnb1ex3/+; Akr1b7-Cre, Akr1b7-Igf2 | Same phenotype as the Ctnnb1ex3/+; Akr1b7-Cre mice | [47] | |
| Ctnnb1ex3/+; mCyp11a1-iCre | Adenoma (Dab2+) | [41] | |
| Nr5a1-Hoxb9; Ctnnb1ex3/+; mCyp11a1-iCre | Adenoma, increase adrenal size in male compared to activation of CTNNB1 alone | [41] | |
| Ctnnb1ex3/+; AS Cre/+ | Hyperaldosteronism, increased rosette frequency in the zG, block differentiation of zG to zF cells | [49,50] | |
| Dennd1a.V2 | pCMV-BAM hDenndia.V2 (overexpression of the human V2 isoform) | Overexpression of Cyp17a1, phenotype not evaluated | [84] |
| Dicer | Dicer flox/flox; Nr5a1-Crehigh | Adrenal hypoplasia at e16.5 and adrenal failure at birth | [17,18] |
| Ezh2 | Ezh2 flox/flox; Nr5a1-Crehigh | Aberrant zonal differentiation, loss of PKA activity in the zF, expansion of the zG, appearance of subcapsular spindle-shaped cells, phenotype more pronounced in males | [19] |
| Fgfr2 | Fgfr2 flox/flox;Tbx-Cre | Adrenal hypoplasia | [10] |
| Fgfr2 flox/flox;Nr5a1-Crehigh | Adrenal hypoplasia | [20] | |
| Fgfr2 IIIb flox/−;K5Cre/+ (global inactivation via recombination in germ cells) | Adrenal hypoplasia at e15.5 | [85] | |
| Fgfr2 flox/flox; AS Cre/+ | Impaired rosette formation in the zG | [49] | |
| Gata4/ Gata6 | Gata4 flox/flox; Wt1CreERt2/+ | Disruption of coelomic epithelium thickening | [5] |
| Gata4 flox/flox; Osr1eGFP-CreERt2/+ | Disruption of coelomic epithelium thickening | [5] | |
| Gata4 flox/flox; Osr1eGFP-CreERt2/+; Wt1CreERt2/+ | Disruption of coelomic epithelium thickening | [5] | |
| Gata4 flox/flox; CAG-CreER | Disruption of coelomic epithelium thickening | [5] | |
| Gata4+/− | Reduced subcapsular spindle-shaped cell hyperplasia following gonadectomy | [86] | |
| Cyp21a1-Gata4 | Subcapsular spindle-shaped cell hyperplasia | [71] | |
| Gata6 flox/flox; Nr5a1-Cre | Adrenal hypoplasia, absence of an X-zone in postnatal adrenal, Subcapsular spindle-shaped cell hyperplasia | [31] | |
| Gata4 flox/flox; Gata6 flox/flox; Nr5a1-Crehigh | Adrenocortical like cells in the testes | [22] | |
| Adrenal agenesis, Adrenocortical like cells in the testes | [21] | ||
| Gli3 | Gli3Δ699/ Δ699 | Adrenal aplasia | [87] |
| Normal adrenals | [88] | ||
| Hoxb9 | Nr5a1-Hoxb9 | Large X-zone | [41] |
| Nr5a1-Hoxb9; Ctnnb1ex3/+; mCyp11a1-iCre | Adrenal tumor formation | [41] | |
| Igf2 | H19 floxDMD/floxDMD; Nr5a1-Crelow | Normal adrenal | [15] |
| Akr1b7-Igf2 | Subcapsular spindle-shaped cell hyperplasia | [47] | |
| Insr/ Igf1r | Insr−/−; Igfr1−/− (via recombination of Insrflox/flox; Igfr1flox/flox in germ cells) | Adrenal agenesis and gonadal hypoplasia | [89] |
| Insrflox/flox; Igfr1flox/flox; hCyp11a1-iCre | Abnormal hypoplastic adrenal | [39] | |
| Lats1/ Lats2 | Lats1 flox/flox; Lats2 flox/flox; Nr5a1-Cre | Transdifferentiation of adrenocortical cells into myofibroblast like cells | [33] |
| Lhcgr | Lhcgr−/− | Prevention of GATA4 induction and tumor formation in inhα/Tag mice | [70] |
| Mc2r | Mc2r−/− | Adrenal hypoplasia limited to the zF, zG still present | [90] |
| Mst1/ Mst2 | Mst1 flox/flox; Mst2 flox/flox; Nr5a1-Cre | Premature subcapsular spindle-shaped cell hyperplasia | [34] |
| Mrap | Mrap−/− | Adrenal hypoplasia limited to the zF (following corticosterone replacement therapy), expansion of WNT/CTNNB1 signaling in the cortex | [91] |
| Nr0b1 | Nr0b1−/Y | Delayed regression of the X-zone | [92] |
| Enhanced subcapsular proliferation in young animals followed by progressive adrenal cortex degeneration in male | [93] | ||
| Nr0b1flox/Y; AS Cre/+ | No effect on the differentiation of zG cells into zF cells | [48] | |
| Nr5a1 | Nr5a1−/− | Gonadal and adrenal agenesis | [94] |
| Nr5a1+/− | Adrenal hypoplasia | [95,96] | |
| Nr5a1flox/flox; mCyp11a1-iCre | Morphological changes in the shape of steroidogenic cells of the fetal cortex, Nr5a1- cells never observed in the definitive cortex | [40] | |
| FAdE-Nr5a1 | Hyperplastic adrenal, ectopic thoracic adrenal tissue, incomplete separation of the AP and GP | [97] | |
| Nr5a12KR/2KR | Delayed regression of the X-zone | [92] | |
| Expansion of SHH+ cells in the zF, presence of Sox9+ (Sertoli-like cells?) in the cortex, delayed regression of the X-zone | [98] | ||
| Nr5a1 flox/flox; ASCre/+ | Loss of zG (and zF maintenance independent of the zG) | [48] | |
| Nr5a1-TR (overexpression of rat Nr5a1) | Subcapsular spindle-shaped cell hyperplasia and nodule formation | [99] | |
| Osr1 | Osr1−/− | Gonadal and adrenal agenesis | [66,76] |
| Pbx1 | Pbx1−/− | Adrenal agenesis | [100] |
| Pbx1+/− | Adrenal hypoplasia and smaller X-zone | [101] | |
| Pde8b | Pde8b−/− | Elevated urinary corticosterone | [102] |
| Elevated basal serum corticosterone level in female, Subcapsular spindle-shaped cell hyperplasia | [103] | ||
| Pde11a | Pde11a-/- | Persistence or resurgence of the X-zone, higher cAMP levels, higher incidence of subcapsular spindle-shaped cell hyperplasia, milder phenotype in males | [104] |
| Porcn | Porcn flox/flox; Nr5a1-Crehigh | Normal adrenal | [23] |
| Prkar1a | Prkar1a flox/flox; Akr1b7-Cre | Adrenal hyperplasia, increased PKA signaling, hypercorticosteronemia, appearance of subcapsular spindle-shaped cells, resurgence of an X-zone/presumptive zR (origin not evaluated), milder phenotype in males | [26] |
| Prkar1a flox/flox; Nr5a1-Crehigh | Expansion of the zF at the expense of the zG | [25] | |
| Repress SUMOylation | [24] | ||
| Prkar1a flox/flox; FadE/Nr5a1-CreERT2 | Normal adrenal (tamoxifen induction at e14.5) | [37] | |
| Prkar1a flox/flox; AS Cre/+ | Hypercorticosteronemia, differentiation of lower zF into a presumptive zR, DHEA secretion | [37] | |
| Rnfr3 | Rnfr3 flox/flox; Nr5a1-Crehigh | Normal adrenal | [105] |
| Rspo3 | Rspo3 flox/flox; CAG-CreER | Progressive adrenal cortex degeneration, loss of zG markers | [6] |
| Rspo3 flox/flox; Gli1CreERt2/+ | Progressive adrenal cortex degeneration, loss of zG markers | [6] | |
| Siahi1a | Siahi1a−/− | Smaller X-zone and dysregulation of the zG | [106] |
| Sfrp2 | Sfrp2−/− | Ectopic expression of CTNNB1+ cells in the zF | [107] |
| Shh | Shh flox/flox; Nr5a1-Crehigh | Adrenal hypoplasia (more severe on the right side) | [17,27,28,29] |
| Six1/ Six4 | Six1−/−; Six4−/− | Potential marginal hypoplastic adrenal gland at 1dpp (unconfirmed, suggested in [107]) | [108,109] |
| Smo | Smo flox/flox; Nr5a1-Crehigh | Normal adrenal | [28] |
| RosaSmoM2; Gli1CreERt2/+ | Enhanced subcapsular WNT/CTNNB1 signaling | [53] | |
| Tcf21 | Tcf21LacZ/LacZ | Improper separation of the AP and GP | [36] |
| Wnt4 | Wnt4−/− | Reduced aldosterone secretion | [110] |
| Wnt4 flox/flox; Nr5a1-Crehigh | Reduction in zG markers | [6] | |
| Expansion of the zF at the expense of the zG | [25] | ||
| Wt1 | Wt1−/− | Gonadal and adrenal agenesis | [61] |
| Wt1−/−; WT280 (WT1 complementation) | Rudimentary hypoplastic adrenal gland at e15.5 | [77] | |
| Cited2+/−; Wt1+/− | Adrenal hypoplasia | [83] | |
| Rosa26Wt1+KTS/Wt1+KTS; Nr5a1-Crehigh | Adrenal hypoplasia, subcapsular spindle-shaped cell hyperplasia | [13] | |
| Yap/Taz | Yap flox/flox; Taz flox/flox; Nr5a1-Cre | Progressive adrenal cortex degeneration in male | [32] |
| Znrf3 | Znrf3 flox/flox; Nr5a1-Crehigh | Adrenal hyperplasia, expansion of the zF, disrupted adrenal organization | [23] |
| Development of adrenocortical carcinoma in 78 weeks-old females, activation of androgen-dependent innate antitumor immunity in males | [111] | ||
| Znrf3 flox/flox; Rnfr3 flox/flox; Nr5a1-Crehigh | Same as the Znrf3 flox/flox; Nr5a1-Crehigh | [23] | |
| Znrf3 flox/flox; Porcn flox/flox; Nr5a1-Crehigh | Rescue the phenotype observed in Znrf3 flox/flox; Nr5a1-Crehigh | [23] | |
| Znrf3 flox/flox; AS Cre/+ | Adrenal hyperplasia, expansion of the zF, disrupted adrenal organization, moderate increased WNT/CTNNB1 signaling in the upper zF | [23] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abou Nader, N.; Zamberlam, G.; Boyer, A. Transgenic Mouse Models to Study the Development and Maintenance of the Adrenal Cortex. Int. J. Mol. Sci. 2022, 23, 14388. https://doi.org/10.3390/ijms232214388
Abou Nader N, Zamberlam G, Boyer A. Transgenic Mouse Models to Study the Development and Maintenance of the Adrenal Cortex. International Journal of Molecular Sciences. 2022; 23(22):14388. https://doi.org/10.3390/ijms232214388
Chicago/Turabian StyleAbou Nader, Nour, Gustavo Zamberlam, and Alexandre Boyer. 2022. "Transgenic Mouse Models to Study the Development and Maintenance of the Adrenal Cortex" International Journal of Molecular Sciences 23, no. 22: 14388. https://doi.org/10.3390/ijms232214388
APA StyleAbou Nader, N., Zamberlam, G., & Boyer, A. (2022). Transgenic Mouse Models to Study the Development and Maintenance of the Adrenal Cortex. International Journal of Molecular Sciences, 23(22), 14388. https://doi.org/10.3390/ijms232214388