
Diagnosing Czech Patients with Inherited Platelet Disorders

 , , , , ,
, , , , ,
Abstract
1. Introduction
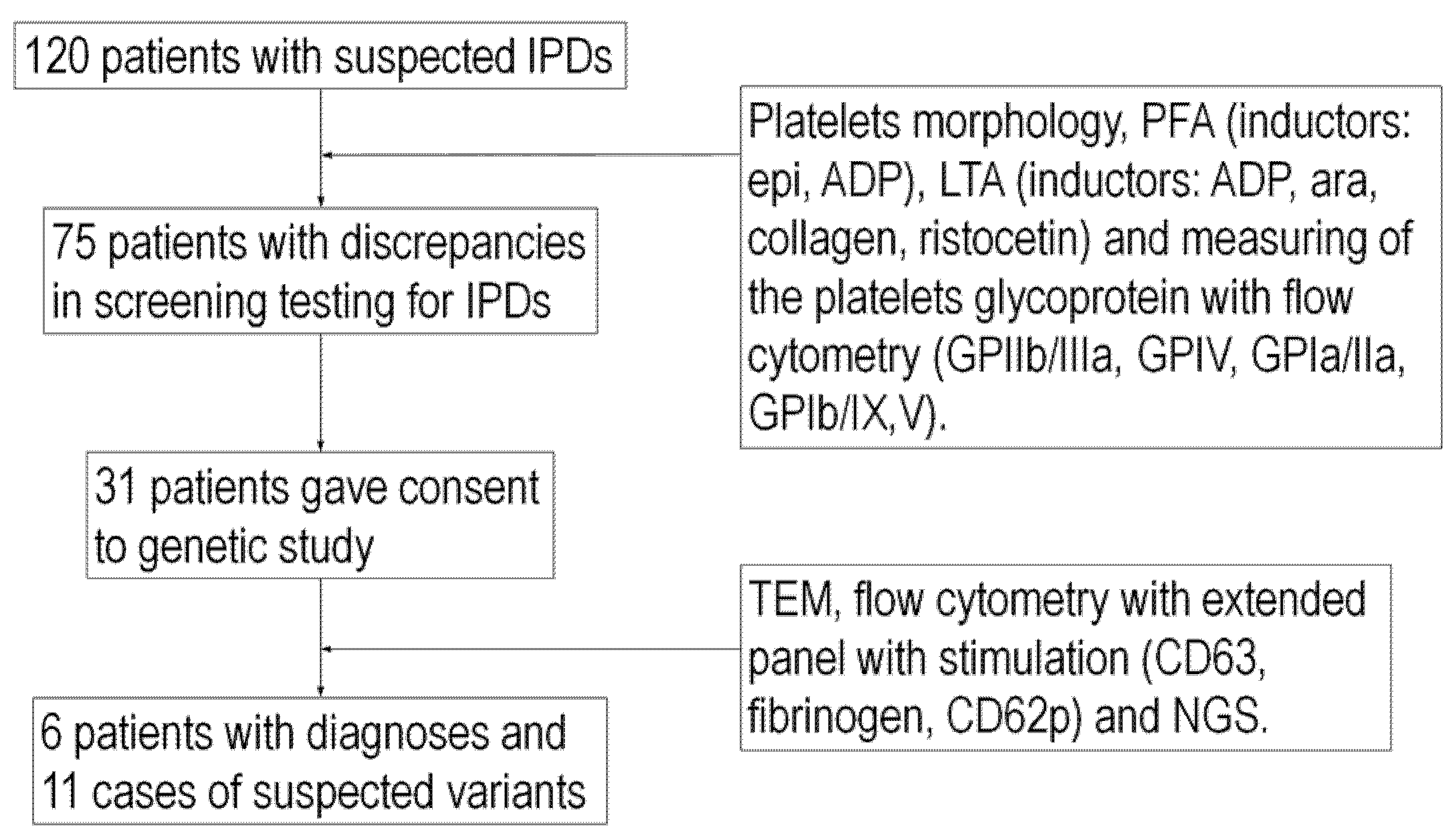
2. Results
2.1. Patient Characteristics
2.2. Bleeding Assessment Tool
2.3. Platelet Morphology
2.4. Platelet Function Analyzer
2.5. Light-Transmission Aggregometry
2.6. Flow Cytometry
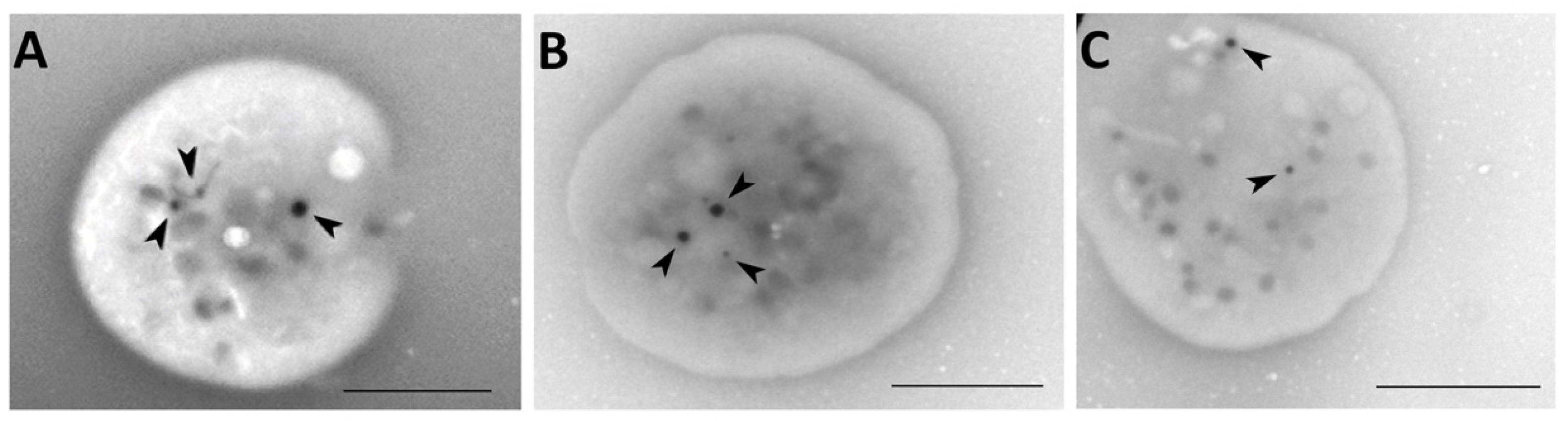
2.7. Whole Mount Transmission Electron Microscopy
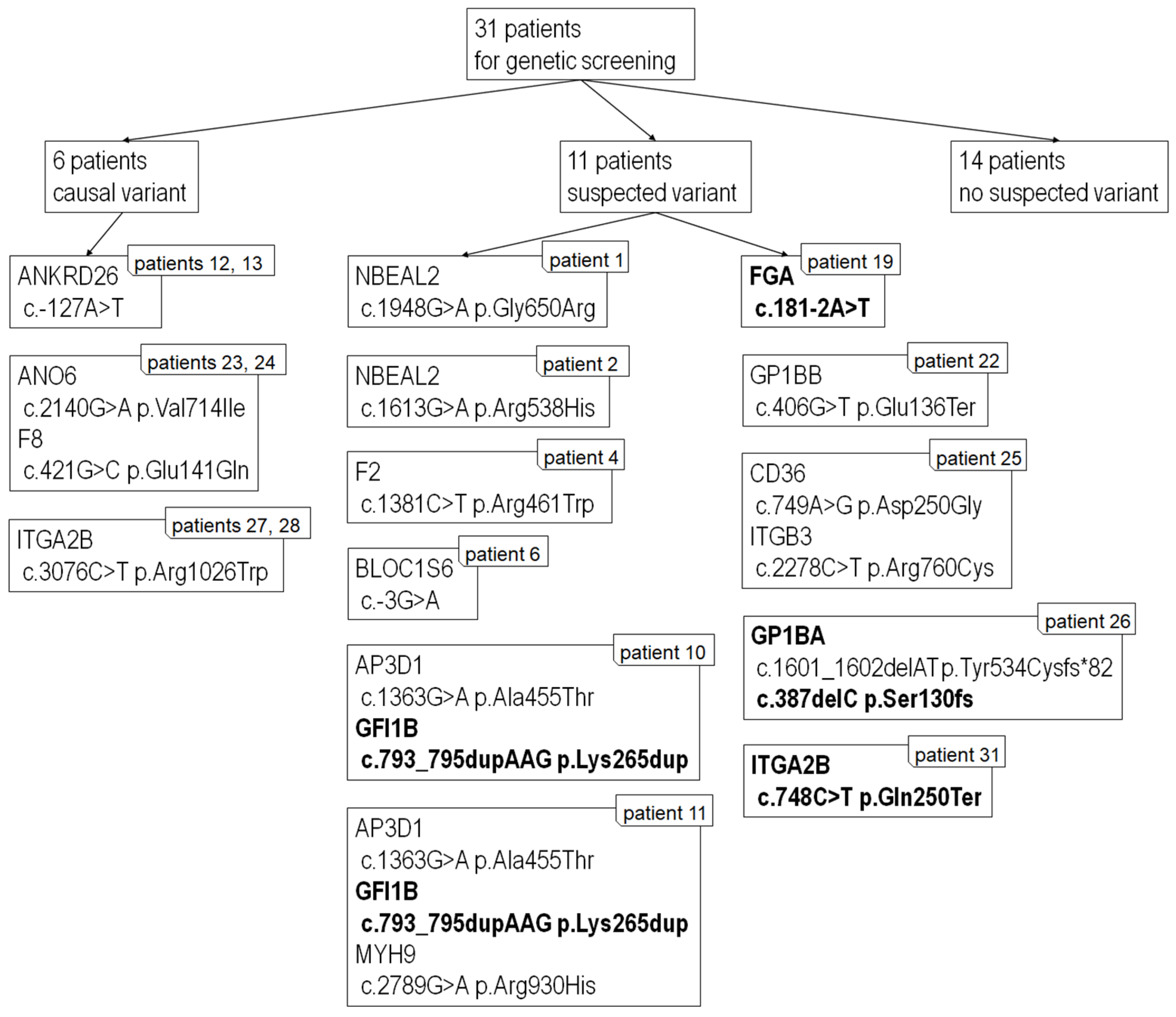
2.8. Next-Generation Sequencing
3. Discussion
3.1. Morphology
3.2. Functional Testing (PFA, LTA, FACS)
3.3. Transmission Electron Microscopy
3.4. Next-Generation Sequencing
4. Materials and Methods
4.1. Patient Group
4.2. Materials
4.3. Bleeding Assessment Tool
4.4. Platelet Function Assay
4.5. Light-Transmission Aggregometry
4.6. Flow Cytometry
4.7. Whole-Mount Transmission Electron Microscopy
4.8. Next-Generation Sequencing Analysis and Variant Classification
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palma-Barqueros, V.; Revilla, N.; Sánchez, A.; Zamora Cánovas, A.; Rodriguez-Alén, A.; Marín-Quílez, A.; González-Porras, J.R.; Vicente, V.; Lozano, M.L.; Bastida, J.M.; et al. Inherited Platelet Disorders: An Updated Overview. Int. J. Mol. Sci. 2021, 22, 4521. [Google Scholar] [CrossRef] [PubMed]
- Al-Huniti, A.; Kahr, W.H.A. Inherited Platelet Disorders: Diagnosis and Management. Transfus. Med. Rev. 2020, 34, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Lambert, M.P. Inherited Platelet Disorders: A Modern Approach to Evaluation and Treatment. Hematol. Clin. 2019, 33, 471–487. [Google Scholar] [CrossRef]
- Noris, P.; Pecci, A. Hereditary thrombocytopenias: A growing list of disorders. Hematol. Am. Soc. Hematol. Educ. Progr. 2017, 2017, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Eekels, J.J.M. Diagnosis of hereditary platelet disorders in the era of next-generation sequencing: “primum non nocere”. J. Thromb. Haemost. 2019, 17, 551–554. [Google Scholar] [CrossRef]
- Fasulo, M.R.; Biguzzi, E.; Abbattista, M.; Stufano, F.; Pagliari, M.T.; Mancini, I.; Gorski, M.M.; Cannavò, A.; Corgiolu, M.; Peyvandi, F.; et al. The ISTH Bleeding Assessment Tool and the risk of future bleeding. J. Thromb. Haemost. 2018, 16, 125–130. [Google Scholar] [CrossRef]
- Pecci, A.; Balduini, C.L. Inherited thrombocytopenias: An updated guide for clinicians. Blood Rev. 2021, 48, 100784. [Google Scholar] [CrossRef]
- Bolton-Maggs, P.H.B.; Chalmers, E.A.; Collins, P.W.; Harrison, P.; Kitchen, S.; Liesner, R.J.; Minford, A.; Mumford, A.D.; Parapia, L.A.; Perry, D.J.; et al. A review of inherited platelet disorders with guidelines for their management on behalf of the UKHCDO. Br. J. Haematol. 2006, 135, 603–633. [Google Scholar] [CrossRef]
- Lambert, M.P. Updates in diagnosis of the inherited platelet disorders. Curr. Opin. Hematol. 2020, 27, 333–340. [Google Scholar] [CrossRef]
- Harrison, P.; Mackie, I.; Mumford, A.; Briggs, C.; Liesner, R.; Winter, M.; Machin, S. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br. J. Haematol. 2011, 155, 30–44. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Pabinger, I.; Ragni, M.; Abdul-Kadir, R.; Berntorp, E.; Blanchette, V.; Bodó, I.; Casini, A.; Gresele, P.; Lassila, R.; et al. Fundamentals for a Systematic Approach to Mild and Moderate Inherited Bleeding Disorders: An EHA Consensus Report. HemaSphere 2019, 3, e286. [Google Scholar] [CrossRef]
- Gresele, P. Diagnosis of inherited platelet function disorders: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Megy, K.; Downes, K.; Morel-Kopp, M.-C.; Bastida, J.M.; Brooks, S.; Bury, L.; Leinoe, E.; Gomez, K.; Morgan, N.V.; Othman, M.; et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: Communication from the ISTH SSC Subcommittee on Genomics in Thrombosis and Hemostasis. J. Thromb. Haemost. 2021, 19, 2612–2617. [Google Scholar] [CrossRef]
- Elbaz, C.; Sholzberg, M. An illustrated review of bleeding assessment tools and common coagulation tests. Res. Pract. Thromb. Haemost. 2020, 4, 761–773. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lillicrap, D. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef] [PubMed]
- Elbatarny, M.; Mollah, S.; Grabell, J.; Bae, S.; Deforest, M.; Tuttle, A.; Hopman, W.; Clark, D.S.; Mauer, A.C.; Bowman, M.; et al. Normal range of bleeding scores for the ISTH-BAT: Adult and pediatric data from the merging project. Haemophilia 2014, 20, 831–835. [Google Scholar] [CrossRef]
- Gresele, P.; Orsini, S.; Noris, P.; Falcinelli, E.; Alessi, M.C.; Bury, L.; Borhany, M.; Santoro, C.; Glembotsky, A.C.; Cid, A.R.; et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: A communication from the Platelet Physiology SSC. J. Thromb. Haemost. 2020, 18, 732–739. [Google Scholar] [CrossRef]
- Cattaneo, M.; Cerletti, C.; Harrison, P.; Hayward, C.P.M.; Kenny, D.; Nugent, D.; Nurden, P.; Rao, A.K.; Schmaier, A.H.; Watson, S.P.; et al. Recommendations for the Standardization of Light Transmission Aggregometry: A Consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. J. Thromb. Haemost. 2013. [Google Scholar] [CrossRef] [PubMed]
- Rubak, P.; Nissen, P.H.; Kristensen, S.D.; Hvas, A.-M. Investigation of platelet function and platelet disorders using flow cytometry. Platelets 2016, 27, 66–74. [Google Scholar] [CrossRef]
- Gunning, W.T., 3rd; Raghavan, M.; Calomeni, E.P.; Turner, J.N.; Roysam, B.; Roysam, S.; Smith, M.R.; Kouides, P.A.; Lachant, N.A. A Morphometric Analysis of Platelet Dense Granules of Patients with Unexplained Bleeding: A New Entity of Delta-Microgranular Storage Pool Deficiency. J. Clin. Med. 2020, 9, 1734. [Google Scholar] [CrossRef]
- Zaninetti, C.; Greinacher, A. Diagnosis of Inherited Platelet Disorders on a Blood Smear. J. Clin. Med. 2020, 9, 539. [Google Scholar] [CrossRef] [PubMed]
- Hayward, C.P.M.; Harrison, P.; Cattaneo, M.; Ortel, T.L.; Rao, A.K. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J. Thromb. Haemost. 2006, 4, 312–319. [Google Scholar] [CrossRef]
- van Asten, I.; Schutgens, R.E.G.; Baaij, M.; Zandstra, J.; Roest, M.; Pasterkamp, G.; Huisman, A.; Korporaal, S.J.A.; Urbanus, R.T. Validation of flow cytometric analysis of platelet function in patients with a suspected platelet function defect. J. Thromb. Haemost. 2018, 16, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Lombardi, A.M.; Putti, M.C.; Bertomoro, A.; Cortella, I.; Barzon, I.; Girolami, A.; Fabris, F. Spectrum of 5’UTR mutations in ANKRD26 gene in patients with inherited thrombocytopenia: C.-140C>G mutation is more frequent than expected. Platelets 2017, 28, 621–624. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef] [PubMed]
- Perez Botero, J.; Dugan, S.N.; Anderson, M.W. ANKRD26-Related Thrombocytopenia. In GeneReviews® [Internet]; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993–2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507664/ (accessed on 25 October 2022).
- Galera, P.; Dulau-Florea, A.; Calvo, K.R. Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. Int. J. Lab. Hematol. 2019, 41 (Suppl. S1), 131–141. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Khoriaty, R.; Ozel, A.B.; Ramdas, S.; Ross, C.; Desch, K.; Shavit, J.A.; Everett, L.; Siemieniak, D.; Li, J.Z.; Ginsburg, D. Genome-wide linkage analysis and whole-exome sequencing identifies an ITGA2B mutation in a family with thrombocytopenia. Br. J. Haematol. 2019, 186, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, S.; Kashiwagi, H.; Otsu, M.; Takayama, N.; Eto, K.; Onodera, M.; Miyajima, Y.; Takamatsu, Y.; Suzumiya, J.; Matsubara, K.; et al. Heterozygous ITGA2B R995W mutation inducing constitutive activation of the αIIbβ3 receptor affects proplatelet formation and causes congenital macrothrombocytopenia. Blood 2011, 117, 5479–5484. [Google Scholar] [CrossRef]
- Kashiwagi, H.; Kunishima, S.; Kiyomizu, K.; Amano, Y.; Shimada, H.; Morishita, M.; Kanakura, Y.; Tomiyama, Y. Demonstration of novel gain-of-function mutations of αIIbβ3: Association with macrothrombocytopenia and glanzmann thrombasthenia-like phenotype. Mol. Genet. Genom. Med. 2013, 1, 77–86. [Google Scholar] [CrossRef]
- Morais, S.; Oliveira, J.; Lau, C.; Pereira, M.; Gonçalves, M.; Monteiro, C.; Gonçalves, A.R.; Matos, R.; Sampaio, M.; Cruz, E.; et al. αIIbβ3 variants in ten families with autosomal dominant macrothrombocytopenia: Expanding the mutational and clinical spectrum. PLoS ONE 2020, 15, e0235136. [Google Scholar] [CrossRef]
- Romano, A.A.; Allanson, J.E.; Dahlgren, J.; Gelb, B.D.; Hall, B.; Pierpont, M.E.; Roberts, A.E.; Robinson, W.; Takemoto, C.M.; Noonan, J.A. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics 2010, 126, 746–759. [Google Scholar] [CrossRef]
- Pluthero, F.G.; Di Paola, J.; Carcao, M.D.; Kahr, W.H.A. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets 2018, 29, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Aarts, C.E.M.; Downes, K.; Hoogendijk, A.J.; Sprenkeler, E.G.G.; Gazendam, R.P.; Favier, R.; Favier, M.; Tool, A.T.J.; van Hamme, J.L.; Kostadima, M.A.; et al. Neutrophil specific granule and NETosis defects in gray platelet syndrome. Blood Adv. 2021, 5, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Perez Botero, J.; Higgins, R.A.; Medina, E.A. Gray Platelet Syndrome Presenting With Pancytopenia, Splenomegaly, and Bone Marrow Fibrosis. Am. J. Clin. Pathol. 2021, 156, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica 2018, 103, 148–162. [Google Scholar] [CrossRef]
- Andres, O.; König, E.-M.; Althaus, K.; Bakchoul, T.; Bugert, P.; Eber, S.; Knöfler, R.; Kunstmann, E.; Manukjan, G.; Meyer, O.; et al. Use of Targeted High-Throughput Sequencing for Genetic Classification of Patients with Bleeding Diathesis and Suspected Platelet Disorder. Open Compan. J. Thromb. Haemost. 2018, 2, e445–e454. [Google Scholar] [CrossRef]
- Johns, M.B.J.; Paulus-Thomas, J.E. Purification of human genomic DNA from whole blood using sodium perchlorate in place of phenol. Anal. Biochem. 1989, 180, 276–278. [Google Scholar] [CrossRef]
- Kalina, T.; Flores-Montero, J.; van der Velden, V.H.J.; Martin-Ayuso, M.; Böttcher, S.; Ritgen, M.; Almeida, J.; Lhermitte, L.; Asnafi, V.; Mendonça, A.; et al. EuroFlow standardization of flow cytometer instrument settings and immunophenotyping protocols. Leukemia 2012, 26, 1986–2010. [Google Scholar] [CrossRef]
- Chen, D.; Uhl, C.B.; Bryant, S.C.; Krumwiede, M.; Barness, R.L.; Olson, M.C.; Gossman, S.C.; Erdogan Damgard, S.; Gamb, S.I.; Cummins, L.A.; et al. Diagnostic laboratory standardization and validation of platelet transmission electron microscopy. Platelets 2018, 29, 574–582. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patient No. | Age | Sex | BAT Score | Thrombocytopenia | Size of Platelet |
|---|---|---|---|---|---|
| 1 | 34 | F | 4 | No | Macro |
| 2 | 53 | F | 14 | No | Normal |
| 3 | 63 | F | 9 | No | Normal |
| 4 | 31 | F | 6 | No | Normal |
| 5 | 36 | F | 9 | No | Normal |
| 6 | 32 | F | 8 | No | Normal |
| 7 | 37 | M | 8 | Yes | Macro |
| 8 | 45 | F | 8 | No | Macro |
| 9 | 28 | F | 6 | No | Normal |
| 10 a | 42 | F | 11 | No | Macro |
| 11 a | 8 | M | 5 | NA | NA |
| 12 b | 44 | F | 11 | Yes | Macro |
| 13 b | 49 | F | 6 | Yes | Normal |
| 14 c | 48 | F | 10 | No | Normal |
| 15 c | 25 | F | 1 | No | Normal |
| 16 | 48 | F | 5 | No | Normal |
| 17 | 29 | F | 7 | No | Normal |
| 18 | 32 | F | 10 | No | Normal |
| 19 | 59 | F | 4 | No | Normal |
| 20 | 20 | F | 21 | No | Normal |
| 21 | 62 | F | 3 | No | Normal |
| 22 | 41 | M | 2 | Yes | Macro |
| 23 d | 53 | F | 8 | No | Normal |
| 24 d | 25 | F | 6 | No | Normal |
| 25 | 66 | F | 5 | No | Macro |
| 26 | 27 | F | 8 | Yes | Macro |
| 27 e | 61 | M | 4 | Yes | Normal |
| 28 e | 40 | M | 4 | Yes | Normal |
| 29 | 30 | F | 5 | Yes | Macro |
| 30 | 28 | F | 4 | No | Normal |
| 31 | 35 | F | 8 | No | Normal |
| Patient No. | PFA | LTA (ADP, Arachidonic Acid, Collagen, Ristocetin) | Flow Cytometry |
|---|---|---|---|
| 1 | pathological (epi, ADP) | Normal | Normal |
| 2 | Normal | low nonspecific | low GPIIb/IIIa |
| 3 | pathological (epi) | low nonspecific | low GPIIb/IIIa, GPIb/IX/V a GPVI |
| 4 | Normal | Normal | low GPVI |
| 5 | pathological (epi) | low nonspecific | NA |
| 6 | pathological (epi, ADP) | low nonspecific | Normal |
| 7 | pathological (epi) | Normal | low GPVI, GPIb/IX/V, GPIa/IIa |
| 8 | pathological (epi, ADP) | slightly low | low GPIIb/IIIa |
| 9 | pathological (epi) | Normal | slightly low GPVI |
| 10 a | Normal | slightly low ADP | low GPVI |
| 11 a | NA | low ADP, ara | NA |
| 12 b | pathological (epi, ADP) | NA | low GPIIb/IIIa, GPIa/IIa, GPVI |
| 13 b | pathological (epi, ADP) | NA | low GPIIb/IIIa, GPIa/IIa, GPVI |
| 14 c | pathological (epi) | low nonspecific | Normal |
| 15 c | pathological (epi, ADP) | slightly low nonspecific | Normal |
| 16 | pathological (epi, ADP) | low nonspecific | low GPVI, GPIIb/IIIa |
| 17 | pathological (epi, ADP) | Normal | NA |
| 18 | Normal | slightly low nonspecific | Normal |
| 19 | Normal | Normal | low GPVI, GPIIb/ IIIa |
| 20 | Normal | slightly low nonspecific | NA |
| 21 | pathological (epi) | slightly low | low GPIIb/IIIa and GPVI, slightly low GPIa/IIa |
| 22 | pathological (ADP) | Normal | Normal |
| 23 d | pathological (epi, ADP) | slightly low nonspecific | low GPVI, GPIIb/IIIa |
| 24 d | Normal | Borderline | low GPVI, GPIb/IX/V, GPllb/IIIa |
| 25 | Normal | low mainly ADP, ara | low GPVI, GPIa/IIa |
| 26 | pathological (epi, ADP) | low, mainly ristocetine | low GPIb/IX/V |
| 27 e | pathological (epi, ADP) | very low ara, slightly low ADP | low GPIIb/IIIa, GPVI |
| 28 e | pathological (epi, ADP) | slightly low ADP, ara | low GPIIb/IIIa a GPVI |
| 29 | pathological (epi, ADP) | NA | low GPVI |
| 30 | pathological (epi) | Normal | slightly low GPVI |
| 31 | pathological (epi, ADP) | slightly low, mainly ADP | low GPIIb/IIIa, GPVI |
| Patient 4 | Patient 5 | Controls (n = 5) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Stimulant/Marker | CD62p | CD63 | Bound Fbg | CD62p | CD63 | Bound Fbg | CD62p * | CD63 * | Bound Fbg * |
| EDTA | 1.30 | 2.55 | 3.37 | 2.16 | 8.77 | 3.68 | 1.11 | 0.99 | 0.02 |
| (0.46–1.75) | (0.92–1.06) | (0–0.04) | |||||||
| ADP | 55.60 | 32.40 | 31.50 | 51.40 | 38.40 | 32.50 | 66.64 | 26.74 | 45.66 |
| (56.47–78.81) | (21.24–32.24) | (37.72–53.6) | |||||||
| Ara | 7.79 | 57.60 | 60.00 | 21.20 | 55.70 | 57.20 | 60.04 | 26.10 | 32.26 |
| (45.5–74.58) | (13.86–38.34) | (11.27–53.25) | |||||||
| Ristocetin | 93.30 | 8.84 | 98.90 | 88.40 | 13.70 | 98.90 | 99.66 | 39.82 | 99.76 |
| (99.47–99.85) | (23.86–55.78) | (99.59–99.93) | |||||||
| Trap-6 | 90.30 | 80.80 | 66.20 | 46.20 | 45.50 | 15.50 | 87.82 | 77.96 | 40.34 |
| (82.15–93.49) | (67.19–88.73) | (30.19–50.49) | |||||||
| Patient No. | Number of Dense Granules * |
|---|---|
| 4 | 2.00 ± 1.91 |
| 5 | 2.39 ± 1.95 |
| 10 | 2.89 ± 2.24 |
| 11 | 2.08 ± 1.49 |
| Patient No. | Gene | Variant | Reference Sequence | Disease Associated with the Gene | NCBI | |
|---|---|---|---|---|---|---|
| 1 | NBEAL2 | het.c.1948G>A | p.Gly650Arg | NM_015175.3 | Gray platelet sy | rs201373710 |
| 2 | NBEAL2 | het.c.1613G>A | p.Arg538His | NM_015175.3 | Gray platelet sy | rs368310677 |
| 4 | ||||||
| F2 | het.c.1381C>T | p.Arg461Trp | NM_000506.5 | FII deficiency | rs121918478 | |
| 6 | BLOC1S6 | het.c.-3G>A | - | NM_012388.4 | Hermansky-Pudlak sy type 9 | rs746824320 ¥ |
| 10 a | AP3D1 | het.c.1363G>A | p.Ala455Thr | NM_004188.8 | Hermansky-Pudlak sy | rs200459002 |
| GFI1B | het.c.793_795dupAAG | p.Lys265dup | NM_004188.8 | Gray platelet sy | --- | |
| 11 a | AP3D1 | het.c.1363G>A | p.Ala455Thr | NM_003938.8 | Hermansky-Pudlak sy | rs200459002 |
| GFI1B | het.c.793_795dupAAG | p.Lys265dup | NM_004188.8 | Gray platelet sy | --- | |
| MYH9 | het.c.2789G>A | p.Arg930His | MYH9RD | rs727504740 | ||
| 12 b | ANKRD26 | het.c.-127A>T | - | NM_014915.3 | - | --- |
| 13 b | ANKRD26 | het.c.-127A>T | - | NM_014915.3 | - | --- |
| 19 | FGA | het.c.181-2A>T | - | NM_000508.5 | dysfibrinogenemia | --- |
| 22 | GP1BB | het.c.406G>T | p.Glu136Ter | NM_000407.5 | Bernard-Soulier sy | rs953345181 ¥ |
| 23 d | ANO6 | het.c.2140G>A | p.Val714Ile | NM_001025356.3 | Scott sy | rs765525912 ¥ |
| F8 | het.c.421G>C | p.Glu141Gln | NM_000132.3 | Hemophilia A | --- | |
| 24 d | ANO6 | het.c.2140G>A | p.Val714Ile | NM_001025356.3 | Scott sy | rs765525912 ¥ |
| F8 | het.c.421G>C | p.Glu141Gln | NM_000132.3 | Hemophilia A | --- | |
| 25 | CD36 | het.c.749A>G | p.Asp250Gly | NM_000072.3 | GPIV deficiency | rs1375879584 ¥ |
| ITGB3 | het.c.2278C>T | p.Arg760Cys | NM_000212.3 | Glanzmann thrombasthenia | --- | |
| 26 | GP1BA | het.c.1601_1602delAT | p.Tyr534Cysfs*82 | NM_000173.7 | Bernard-Soulier sy | rs763978422 |
| GP1BA | het.c.387delC | p.Ser130fs | NM_000173.7 | Bernard-Soulier sy | --- | |
| 27 e | ITGA2B | het.c.3076C>T | p.Arg1026Trp | NM_000419.5 | Glanzmann thrombasthenia | rs147265794 |
| 28 e | ITGA2B | het.c.3076C>T | p.Arg1026Trp | NM_000419.5 | Glanzmann thrombasthenia | rs147265794 |
| 31 | ITGA2B | het.c.748C>T | p.Gln250Ter | NM_000419.5 | Glanzmann thrombasthenia | --- |
| 3, 5, 7, 8, 9, 14 c, 15 c, 16, 17, 18, 20, 21, 29, 30-no suspected causal variant detected | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louzil, J.; Stikarova, J.; Provaznikova, D.; Hrachovinova, I.; Fenclova, T.; Musil, J.; Radek, M.; Kaufmanova, J.; Geierova, V.; Ceznerova, E.; et al. Diagnosing Czech Patients with Inherited Platelet Disorders. Int. J. Mol. Sci. 2022, 23, 14386. https://doi.org/10.3390/ijms232214386
Louzil J, Stikarova J, Provaznikova D, Hrachovinova I, Fenclova T, Musil J, Radek M, Kaufmanova J, Geierova V, Ceznerova E, et al. Diagnosing Czech Patients with Inherited Platelet Disorders. International Journal of Molecular Sciences. 2022; 23(22):14386. https://doi.org/10.3390/ijms232214386
Chicago/Turabian StyleLouzil, Jan, Jana Stikarova, Dana Provaznikova, Ingrid Hrachovinova, Tereza Fenclova, Jan Musil, Martin Radek, Jirina Kaufmanova, Vera Geierova, Eliska Ceznerova, and et al. 2022. "Diagnosing Czech Patients with Inherited Platelet Disorders" International Journal of Molecular Sciences 23, no. 22: 14386. https://doi.org/10.3390/ijms232214386
APA StyleLouzil, J., Stikarova, J., Provaznikova, D., Hrachovinova, I., Fenclova, T., Musil, J., Radek, M., Kaufmanova, J., Geierova, V., Ceznerova, E., Salaj, P., & Kotlin, R. (2022). Diagnosing Czech Patients with Inherited Platelet Disorders. International Journal of Molecular Sciences, 23(22), 14386. https://doi.org/10.3390/ijms232214386