
Targeting the Complement–Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy


 , and
, and 
Abstract
1. Introduction

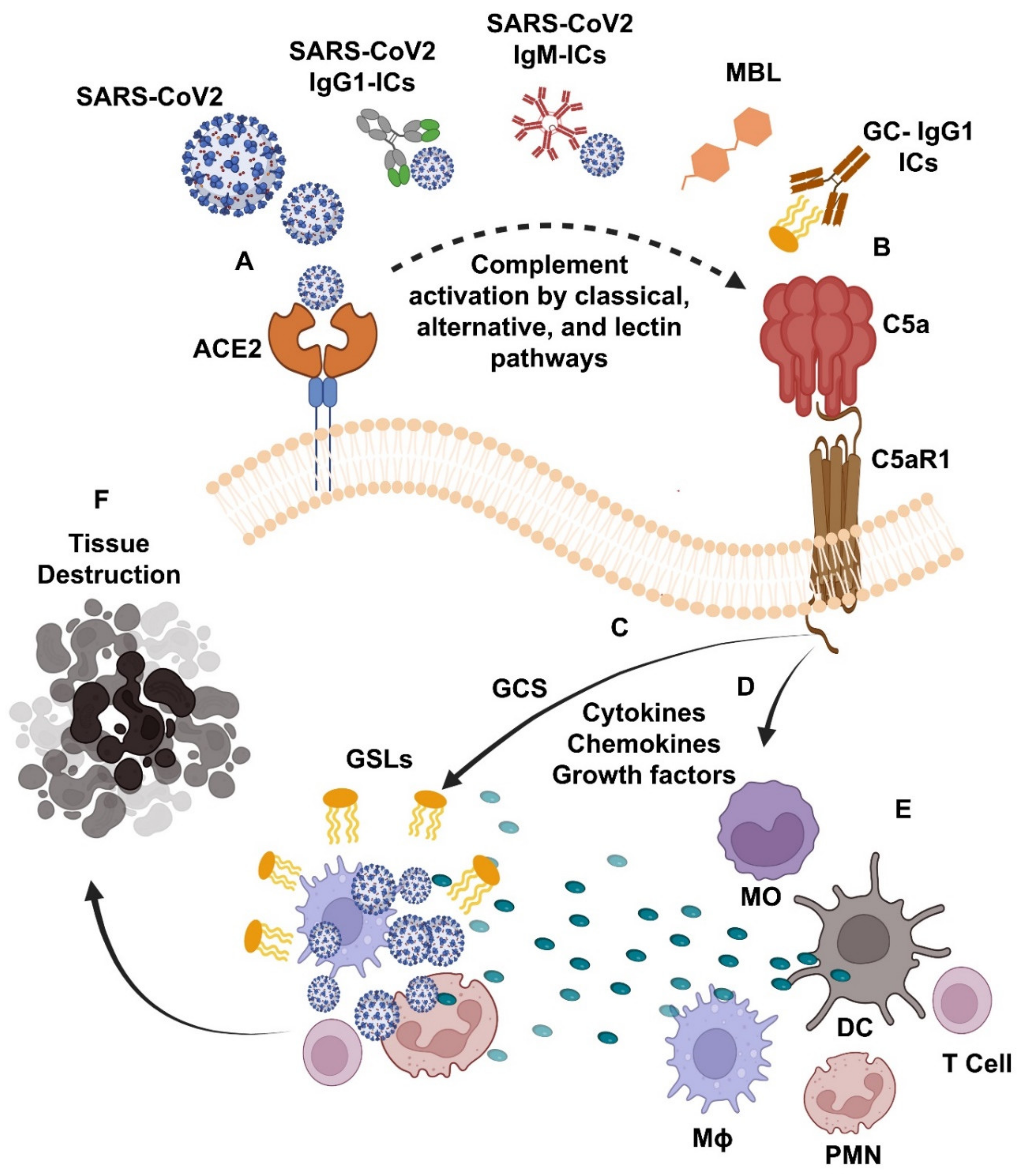{kind=link}
| Coronavirus | Immune Cell Involvement | Source | Changes in Complement Products, Cytokines and Chemokines | References |
|---|---|---|---|---|
| SARS-CoV-2 | Leucocytes, PMNs PCs Endothelial cells | Blood Sera Lungs | C5a +++ C5aR1 +++ MAC +++ | [24,25,26,27,28] |
| SARS-CoV-2 | Type-II pneumocytes Pulmonary cells Platelets | Blood Sera Lungs | C1q P+++ C3b-regulatory factor H (FH) P+++ C3 P+++ | [26,29,30,31] |
| SARS-CoV-2 | Type-II pneumocytes PBMCs | Blood Sera Lung | C3a P+++ C3aR P +++ C3b-CD46 P+++ | [26,31,32,33,34] |
| SARS-CoV-2 | PBMCs | Blood Sera | sC5b-9 P+++ | [27,35,36,37] |
| SARS-2 | Respiratory specimen cells Alveolar cells | Blood Sera Lung | C4d P+++ | [35,38] |
| SARS-CoV-2 | Respiratory specimen cells | Blood Sera | C3bBbP P+++ | [35] |
| SARS-CoV-2 | Respiratory specimen cells | Blood Sera | C3bc P+++ | [35] |
| SARS-CoV-2 | PBMCs Glomeruli Cardiac Microthrombi and Alveolar cells | Blood Sera Lung Heart Kidney | C5b-C9 P+++ | [36,37,38,39,40] |
| SARS-CoV-2 | Pulmonary cells | Lung | C1r P+++ | [29] |
| SARS-CoV-1 | Pulmonary cells | Lung | iC3b P+++ | [31] |
| SARS-CoV-1 | Pulmonary cells | Lung | C3c P++ | [31] |
| SARS-CoV-1 | Pulmonary cells | Lung | C3dg P+++ | [31] |
| MERS | MOs/Mɸs T cells mDCs pDCs PMNs | Liver SPL Lung Kidney Heart Serum | IL12 M+++ IL8 M+++ TNFα M+++ IL-6 M&P+++ IFNλ M+++ CXCL10 M&P+++ CCL2 M+++ CCL3 M+++ CCL5 M+++ IFNα M+ & P+++ IFNβ M+ & P+++ | [41,42,43,44,45,46,47] |
| SARS-CoV-1 | Mos Mɸs DCs Cord blood cells | Lung Blood | IFNβ M+ IFNα M+ TNFα M+++ IFNλ M+++ IL8 M+++ TNFα M+++ IL6 M+++ CCL2 M+++ CCL3 M+++ CCL5 M+++ CXCL10 M+ | [42,48,49,50] |
| SARS-CoV-2 | MOs Mɸs | Blood | IL6 P+++ IL8 P+++ IL10 P+++ TNFα P+++ | [51] |
| SARS-CoV-2 | CD8+ cells NK+ cells | PBMCs | IL2 P+ TNFα P+ IFNγ P+++ Granzyme B P+++ | [52,53] |
| SARS-CoV-2 | CD8+ cells CD4+ cells | Liver Lung Heart | Cytokines NR | [53] |
| SARS-CoV-2 | PMNs | Lung | Cytokines NR | [54] |
| MERS | CD4+ T cells CD8+ T cells | Lymph Nodes Spleen Tonsils PBMCs | Caspase-3 P+++ | [55] |
| SARS-CoV-1 | CD4+ cells CD8+ cells CD45RO+ and CD27+ cells | PBMCs | IL2 P++ TNFα P++ IFNγ P+++ IL4 PNS CXCL10 PNS | [56,57] |
| SARS-CoV-2 | CD4+ T cells CD8+ T cells Regulatory T cells | PBMCs | CCR6 P+++ Perforin P+++ IL6 P+++ IL2 M+++ IL7 M+++ | [51,58,59,60,61,62,63] |
| MERS | Epithelial cells | Lung | IL1β M+++ IL6 M+++ IL8 M+++ IFNα M+ CCL2 M+ CXCL10 M+ | [64,65] |
| SARS-CoV-1 | Epithelial cells | Lung | TNFα M+++ IFNβ M+++ CXCL10 M+++ | [64] |
1.1. COVID-19
| Coronavirus | Source | Cytokines | Chemokines | Growth Factors | References |
|---|---|---|---|---|---|
| MERS | Sera | IFNα P+++ IL6 P+++ IL8 P+++ | CCL5 P+++ CXCL10 P+++ | [45,47,104] | |
| SARS-CoV-1 | Sera | IFNα P+++ IFNγ P+++ IL1 P+++ IL2 P+++ IL6 P+++ IL8 P+++ IL10 P+ IL12 P+++ | CCL2 P+++ CXCL9 P+++ CXCL10 P+++ | TGFβ P+++ | [104,105,106,107,108,109,110,111,112,113,114,115] |
| SARS-CoV-2 | Sera | IFNγ P+++ TNFα P+++ IL1b P+++ IL1RA P+++ IL2 P++ IL2R P++ IL4 P++ IL5 P++ IL6 P+++ IL7 P+++ IL8 P+++ IL9 P+++ IL10 NS IL12 P+++ | CCL2 P+ CCL3 P+ CXCL10 P+ | G-CSF P+++ GMCSF P+++ VEGF P+++ FGF P+++ PDGF P+++ | [57,59,62,104,116,117,118,119,120] |
| SARS-CoV-2 | Sera | CCL2 P+++ | [115,120] | ||
| SARS-CoV-2 | Sera | CCL3 NS | [115] | ||
| SARS-CoV-2 | Sera | CXCL8 P+++ | [120] | ||
| SARS-CoV-2 | Sera | CXCL10 P++ | [115,120] |
| Mouse Model of GD | GD Patients | |||
|---|---|---|---|---|
| Immune Cells | Tissue Recruitment | References | Immune Cells | References |
| MOs | Blood +++ | [121] | Blood - | [122,123] |
| Mɸs | Blood +++, Liver +++, Spleen +++, Lung +++ | [66,121,124,125] | Lymph node +++ | [126] |
| mDCs | Blood +++, Liver +++, Spleen +++, Lung +++ | [66,121,124,125] | Blood - | [122,123,127,128] |
| pDCs | Blood - | [122,123,127] | ||
| PMNs | Blood +++, Liver +++, Spleen +++, Bone Marrow +++ | [66,121] | ||
| CD4 + TCells | Liver +++, Spleen +++, Lung +++ | [66,124,125] | Blood +++ | [127,128] |
| CD8 + T Cells | Thymus +++, Spleen +++ | [124,125] | Blood +++ | [128,129] |
| NK Cells | Blood - | [127,129] | ||
1.2. COVID-19 Vaccines and Alternative Treatments
1.3. SARS-CoV-2 Reinfection
1.4. Gaucher Disease
1.5. Complement Activation in COVID-19 and Gaucher Disease
| Immunological Signature | Mouse Model of GD | Human GD | ||
|---|---|---|---|---|
| Source | References | Source | References | |
| C1qα M+++ | Lung | [261] | ||
| C1qβ M+++ | Lung | [261] | Lymph node | [126] |
| C1qc M+++ | Lung | [261] | ||
| C3 M+++ | Lymph node | [126] | ||
| Clec4d M+++ Clec4n M+++ Clec5a M+++ | Lung | [261] | ||
| Clec7 M+++ | Liver | [261] | ||
| MR P+ | Lymph node, Mɸs | [126] | ||
| C3aR1 M+++ | Lung | [261] | ||
| C4b P++ | Lymph node | [126] | ||
| C5a M/P+++ | Mɸs, DCs, Sera | [258] | Lymph node, CBE-induced GCase-targeted Mɸs, iPSCs-derived Mɸs of human Gaucher disease, Sera | [79,126,258] |
| C5aR1 P+++ | Mɸs and DCs | [258] | Lymph node, Mɸs | [126,258] |
| IFNβ P+++ | Neurons | [266] | ||
| IFNγ P+++ | Anti-CD3 and CD28-stimulated liver, spleen, and lung derived CD4+ T cells, GC-stimulated liver, spleen, and lung-derived DCs, CD4+ T cells, sera, plasma, microglial cells | [66,267] | Sera/Plasma and GC-ICs stimulated CBE-induced GCase targeted Mɸs | [66,67,267] |
| TNFα P+++ | Anti-CD3 and CD28-stimulated Liver, spleen, and lung-derived CD4+ T cells, GC-stimulated liver, spleen, and lung-derived DCs, CD4+ T cells, sera, plasma, microglial cells, whole brain | [66,267] | Sera/Plasma, GC-ICs stimulated CBE-induced GCase-targeted Mɸs, and iPSCs-derived Mɸs of human Gaucher disease | [66,67,79,267] |
| IL1α M/P +++ | Sera/Plasma and whole brain | [258] | ||
| IL1β P+++ | Anti-CD3 and CD28-stimulated liver, spleen, and lung-derived CD4+ T cells, GC-stimulated liver, spleen, and lung-derived DCs, CD4+ T cells, sera, plasma, and whole brain | [66,258,267] | Sera/Plasma, GC-ICs stimulated CBE-induced GCase-targeted Mɸs | [66,67,267] |
| IL6 M/P+++ | Anti-CD3 and CD28-stimulated liver, spleen, and lung- derived CD4+ T cells, GC-stimulated liver, spleen, and lung derived DCs, CD4+ T cells, sera, plasma, microglial cells, and whole brain | [66,267] | Sera/Plasma and GC-ICs stimulated CBE-induced GCase-targeted Mɸs | [66,67,267] |
| IL8 P++ | Sera | [127] | ||
| IL12 P+++ | Anti-CD3 and CD28-stimulated liver, spleen, and lung- derived CD4+ T cells, GC-stimulated liver, spleen, and lung derived DCs, CD4+ T cells, sera, and plasma | [66,267] | Sera/Plasma and GC-ICs stimulated CBE-induced GCase-targeted Mɸs | [66,67,267] |
| IL17 P+++ | Anti-CD3 and CD28-stimulated liver, spleen, and lung- derived CD4+ T cells, GC-stimulated liver, spleen, and lung-derived DCs, CD4+ T cells, β-GC 22:0 (βGL1-22)-stimulated type II natural killer+ CD19+ B cells, sera, and plasma | [66,67,267,268] | Sera/Plasma, peripheral blood-derived MOs, β-GC 22:0 (βGL1-22)- stimulated type II natural killer cells, CD19+ B cells, and GC-ICs stimulated CBE-induced GCase- targeted Mɸs | [66,67,267,268] |
| IL18 P+++ | Sera | Sera/Plasma | [127] | |
| IL21 P+++ | β-GC 22:0 (βGL1-22)-stimulated type II natural killer+ CD19+ B cell | [67,267,268] | Peripheral blood-derived Mos, β-GC 22:0 (βGL1-22)- stimulated type II natural killer cells, and CD19+ B cell | [67,267,268] |
| IL23 P+++ | Anti-CD3 and CD28 stimulated liver, spleen, and lung-derived CD4+ T cells, GC-stimulated liver, spleen, and lung-derived DCs, CD4+ T cells Sera/Plasma | [66,267] | Sera/Plasma and GC-ICs stimulated CBE-induced GCase-targeted Mɸs | [66,67,267] |
| CCL1 P+++ | Sera/Plasma | [258] | ||
| CCL2 M/P+++ | Sera/Plasma Lung tissues | [161,258] | Sera/Plasma, Lymph node and Spleen tissues | [67,126,161] |
| CCL3 M/P+++ | Sara/Plasma Lung tissues | [161,258] | Sera/Plasma | [67,161] |
| CCL4 P+++ | Sera/Plasma | [161,258] | Sera/Plasma | [67,161] |
| CCL5 P+++ | Sera/Plasma | [161,258] | Sera/Plasma | [67,161] |
| CCL6 M +++ | Lung tissues | [161] | ||
| CCL9 M +++ | Lung tissues | [161] | ||
| CCL17 M +++ | Lung tissues | [161] | ||
| CCL18 P+++ | Sera/Plasma | [161] | Sera/Plasma and Spleen tissues | [67,161] |
| CCL22 M+++ | Lung tissues | [67,161] | ||
| CXCL1 M/P+++ | Sera/Plasma Lung tissues | [161] | ||
| CXCL2 P+++ | Sera/Plasma | [161,258] | ||
| CXCL8 P+++ | Sera/Plasma | [161] | Sera/Plasma | [67,161] |
| CXCL9 M/P+++ | Sera/Plasma Lung macrophages | [121,258] | ||
| CXCL10 P+++ | Sera/Plasma | [121,258] | ||
| CXCL11 P+++ | Sera/Plasma | [121,258] | ||
| CXCL12 M+++ | Lung tissues | [67,161] | ||
| CXCL13 P+++ | Sera/Plasma | [258] | ||
| TGFβ1 P+++ | Sera/Plasma | [67,161] | Lymph node | [126] |
| HGF P+++ | Sera/Plasma | [67,161] | ||
| MCSF P+++ | Sera/Plasma | [67,161,258] | Sera/Plasma | [67,161] |
| GCSF P+++ | Sera/Plasma | [67,161] | Sera/Plasma | [67,161] |
| GMCSF P+++ | Sera/Plasma | [67,161,258] | ||
| Vaccines/Alternative Treatment | Treatment Efficacy | Effectiveness Time Period, Notes | Possible Side Effects | References |
|---|---|---|---|---|
| mRNA Vaccines | 48–>90% | 5–6 months | Severe anaphylaxis, myocarditis/pericarditis, lymphadenopathy, appendicitis, herpes-zoster infection, viral facial palsy, transverse myelitis, Guillain-Barré syndrome, encephalopathy, thromboembolism, and thrombosis-thrombocytopenia syndrome | [130,132,133] |
| Viral vector vaccines (Sputnik) | 65–91.6% | |||
| Inactivated Vaccines | 50–91.6% | |||
| Protein-based Vaccines | 90% (80% in adults over 65) | |||
| Lopinavir and Ritonavir (protease inhibitor | No significant difference with the standard of care | NA | Could prevent the secondary immune burst without causing severe adverse events at early stages of COVID-19 and are therefore not recommended for COVID-19 at severe stages of disease | [135,136,137] |
| Favipiravir (Nucleoside precursor) | Improved the clinical condition of COVID-19 patients | No significant differences in hospitalization length and clinical recovery | Possible safety concerns for favipiravir include increased blood uric acid and teratogenicity, as the medication is secreted in semen and breast milk and has demonstrated toxicity in animal studies | [138,139,140,141] |
| Remdesivir (Nucleoside analog) | Improved recovery and clinical outcomes of hospitalized COVID-19 patients, though its effects in reducing mortality are still uncertain | Favorable influence on length of hospital stay but does not confer any mortality benefit | Nausea, hypokalemia, and headaches; mortality rate is not different from placebo | [138,139,140,142,143,144] |
| IFNγ | Rapid decline in SARS-CoV-2 load and a positive-to-negative viral culture conversion. Four patients recovered, and no signs of hyperinflammation were observed | Administration within 5–6 days of symptom onset/diagnosis may improve outcomes in patients with SARS-CoV-2, particularly time to viral clearance and time to clinical improvement | Nausea, temporary digestion issues | [150] |
| Statins | Partially associated with altered mortality | Reduced in-hospital mortality, mainly in patients with coronary disease | 18% increased risk of severe COVID-19 infection | [269,270,271,272] |
| RAAS inhibitors | Partially associated with altered mortality | RAAS inhibitors should be continued in patients with COVID-19 | Does not negatively affect clinical course of COVID-19 in hypertensive patients | [273,274] |
| ACE inhibitors (e.g., Lisinopril) | Efficiently treated COVID-19 vaccination- induced development of hyaluronic acid delayed inflammatory reaction | Reduced risk of COVID-19 positivity | Cough, hyperkalemia, fatigue, dizziness, headache, dysgeusia | [275,276] |
| IL-1α/β inhibitor (Anakinra) | Prevented severe respiratory failure of COVID-19 and decreased the mortality | Significantly reduced the risk of worse clinical outcome at day 28 | Immunosuppression with anakinra may facilitate sepsis, necessitating continuous screening for bacterial superinfections | [277] |
| IL-6R antagonists (tocilizumab and sarilumab) | Controlled the disease severity and improved survival in critically ill patients with COVID-19 | Significantly reduced the risk of worse clinical outcome at day 28 | Bacterial, viral, fungal, and opportunistic infections | [278,279] |
| JAK inhibitors (ruxolitinib and baricitinib) | Decreased use of invasive mechanical ventilation; borderline impact on intensive care unit rates | JAK-inhibitors did not decrease length of hospitalization but exhibited a lower 28-day mortality rate | Viral (herpes, influenza), fungal, mycobacterial infections; musculoskeletal and connective tissue disorders, embolism and thrombosis, neoplasms | [280,281,282,283] |
| Anti-TNFα antibody (adalimumab) | Not shown; no alterations in mortality or mechanical ventilation requirements | No significant differences with the standard of care | Cutaneous swelling, pain, nose and throat sinus infections, headache, stomach pain, muscle pain | [148,284] |
| GMCSF (sargramostim) | Increased anti- viral antibody titers, lowered mean lung viral titers, and increased survival in SARS-CoV-2 infected human ACE2 transgenic mouse model of COVID-19 | NA | Flu-like symptoms, leukocytosis and capillary leak syndrome, acute lung injury in rare cases | [285] |
| Anti-GMCSF monoclonal antibody (gimsilumab) | No improvement in mortality or other key clinical outcomes, i.e., pneumonia and evidence of systemic inflammation in patients with COVID-19 | All-cause mortality did not vary between gimsilumab and placebo at day 43 | Infections and sepsis, renal failure, infusion-related reactions | [286] |
| IVIGs | Improved the lung symptoms, cutaneous vasculitis, and acute encephalopathy in patients with COVID-19 | IVIGs reduced mortality and increased the hospitalization length in severe COVID-19 infection | Thromboembolic events | [287,288] |
| Corticosteroids | Reduce mortality in patients with COVID-19 | Acute trial | Hyperglycemia. No increase of neuromuscular weakness, gastrointestinal bleeding, or superinfections | [289] |
| SARS-CoV-2 Protein | IgG and Isotypes Antibodies to SARS-CoV-2 Proteins | IgM Antibodies to SARS-CoV-2 Proteins | IgA Antibodies to SARS-CoV-2 Proteins |
|---|---|---|---|
| S (S1 and/or S2) | IgG++ [260,290,291,292,293,294,295,296,297,298,299,300,301,302,303] +++IgG1 > IgG2 > IgG3 [260] | IgM+ [260,292,293,298,299,300,301,302] | IgA+ [260,295,299,301,302] |
| RBD | IgG++ [251,300] and IgG1+++ [251] | IgM++ [251] | |
| N | IgG+++ [251,290,291,296,300,303,304,305,306] | IgM+ [293,306] | |
| E | IgM+ [292] |
1.6. Complement Activation Is Linked to the Increased Synthesis of Glucosylceramide Synthase Enzymes and Excess Production of Sphingolipids in COVID-19 and GD
| Complement Activation Products | Cells | Enzyme Secretion | References |
|---|---|---|---|
| IgG and IgM-IC | PMNs R | Alkaline phosphatase, Acid phosphatase, and β -Glucuronidase | [327] |
| BSA-anti-BSA | Mɸs R | β-glucuronidase | [328] |
| Rheumatoid factor complex (IgG/IgM) | Leuko H | β-glucuronidase | [329] |
| C3 | Mɸs M | β-glucuronidase | [330] |
| C3a | Mɸs M | β-glucuronidase, | [330] |
| C3b | Mɸs M | β-galactosidase, β-glucuronidase, N-acetyl-β-D-glucosaminidase | [330,331] |
| C3d | PMN H | Elastase | [332] |
| C5a | PMN H | β-glucuronidase, Lysozyme | [333,334] |
| GD Patients | Treatment | GD Patients with COVID-19 | SARS-CoV-2 Positivity | Symptoms | References |
|---|---|---|---|---|---|
| 181 | SRT, ERT | 45/181 | 18 | Fever, dyspnea, cough, fatigue, chills, night sweats, taste and smell loss, chest pain, feeling of short breath, COVID-19 toes, extreme exhaustion, weakness, and no death | [335] |
| 8 | SRT, ERT | 7/8 | 2 | Fever, dyspnea, cough, fatigue, bilateral pneumonia, and multi-organ failure 50% death | [336] |
| 550 | NR | 1 | 1 | Mild and short clinical course managed by quarantine for 14 days | [337] |
| 39 | NR | 39 | Hypertension, Type 2 diabetes mellitus, dyslipidemia, asthma or chronic obstructive pulmonary disease, chronic kidney disease, coronary artery disease, heart failure, cancer | [338] | |
| 1 | ERT | 1 | 1 | Fever, cough, severe neutropenia, and cavitation in the chest | [339] |
| 1471 | ERT, SRT | 82 | 0 | Asymptomatic, mildly affected but 2/82 exhibited severe/critical infection | [340] |
| 2 | ERT, Ambroxol | 0 | 0 |
Mood changes, cognitive and
motor deterioration | [341] |
1.7. Complement Activation Is Linked to the Increased Production of Chemokines, Growth Factors, Immune Cells Infiltration, and Pro-Inflammatory Cytokine Production in COVID-19 and GD
2. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, D.; Shu, T.; Yang, X.; Song, J.-X.; Zhang, M.; Yao, C.; Liu, W.; Huang, M.; Yu, Y.; Yang, Q.; et al. Plasma metabolomic and lipidomic alterations associated with COVID-19. Natl. Sci. Rev. 2020, 7, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Avraham, R.; Politi, B.; Melamed, S.; Israely, T. Elevation in sphingolipid upon SARS-CoV-2 infection: Possible implications for COVID-19 pathology. Life Sci. Alliance 2021, 5, e202101168. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.M. Inferring a causal relationship between ceramide levels and COVID-19 respiratory distress. Sci. Rep. 2021, 11, 20866. [Google Scholar] [CrossRef] [PubMed]
- Torretta, E.; Garziano, M.; Poliseno, M.; Capitanio, D.; Biasin, M.; Santantonio, T.A.; Clerici, M.; Caputo, S.L.; Trabattoni, D.; Gelfi, C. Severity of COVID-19 Patients Predicted by Serum Sphingolipids Signature. Int. J. Mol. Sci. 2021, 22, 10198. [Google Scholar] [CrossRef]
- Russo, F.P.; Burra, P.; Zanetto, A. COVID-19 and liver disease: Where are we now? Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 277–278. [Google Scholar] [CrossRef]
- Tahtabasi, M.; Hosbul, T.; Karaman, E.; Akın, Y.; Konukoglu, O.; Sahiner, F. Does COVID-19 cause an increase in spleen dimensions? Possible effects of immune activation, hematopoietic suppression and microthrombosis. Clin. Imaging 2021, 79, 104–109. [Google Scholar] [CrossRef]
- Suwaidi, A.S.; Alakasheh, B.J.; Al-Ozaibi, L.S. Splenic Infarction in a COVID-19 Patient without Respiratory Symptoms. Dubai Med. J. 2022, 5, 74–77. [Google Scholar] [CrossRef]
- Shaukat, I.; Khan, R.; Diwakar, L.; Kemp, T.; Bodasing, N. Atraumatic splenic rupture due to COVID-19 infection. Clin. Infect. Pract. 2020, 10, 100042. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, E.; Bowe, B.; Al-Aly, Z. Long-term cardiovascular outcomes of COVID-19. Nat. Med. 2022, 28, 583–590. [Google Scholar] [CrossRef]
- Abbasi, J. Even Mild COVID-19 May Change the Brain. JAMA 2022, 327, 1321–1322. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.L.R.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Haj, E.M.; Altintas, E.; Chapelet, G.; Kapogiannis, D.; Gallouj, K. High depression and anxiety in people with Alzheimer’s disease living in retirement homes during the COVID-19 crisis. Psychiatry Res. 2020, 291, 113294. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.J.; Movassaghi, M.; Gordy, D.; Olson, M.G.; Zhang, T.; Khurana, M.S.; Chen, Z.; Perez-Rosendahl, M.; Thammachantha, S.; Singer, E.J.; et al. Neuropathology of COVID-19 (neuro-COVID): Clinicopathological update. Free Neuropathol. 2021, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; McHugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The continuing 2019-nCoV epidemic threat of novel coronaviruses to global health—The latest 2019 novel coronavirus outbreak in Wuhan, China. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef]
- Luk, H.K.H.; Li, X.; Fung, J.; Lau, S.K.P.; Woo, P.C.Y. Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infect. Genet. Evol. 2019, 71, 21–30. [Google Scholar] [CrossRef]
- Ramadan, N.; Shaib, H. Middle East respiratory syndrome coronavirus (MERS-CoV): A review. Germs 2019, 9, 35–42. [Google Scholar] [CrossRef]
- Menachery, V.D.; Yount, B.L., Jr.; Debbink, K.; Agnihothram, S.; Gralinski, L.E.; Plante, J.A.; Graham, R.L.; Scobey, T.; Ge, X.-Y.; Donaldson, E.F.; et al. A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nat. Med. 2015, 21, 1508–1513. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Hemida, M.G.; Ali, A.M.; Alnaeem, A. The Middle East respiratory syndrome coronavirus (MERS-CoV) nucleic acids detected in the saliva and conjunctiva of some naturally infected dromedary camels in Saudi Arabia-2019. Zoonoses Public Health 2021, 68, 353–357. [Google Scholar] [CrossRef]
- Hemida, M.G.; Alhammadi, M.; Almathen, F.; Alnaeem, A. Lack of detection of the Middle East respiratory syndrome coronavirus (MERS-CoV) nucleic acids in some Hyalomma dromedarii infesting some Camelus dromedary naturally infected with MERS-CoV. BMC Res. Notes 2021, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Benito-Pascual, B.; Gegúndez, A.J.; Díaz-Valle, D.; Arriola-Villalobos, P.; Carreño, E.; Culebras, E.; Rodríguez-Avial, I.; Benitez-Del-Castillo, J.M. Panuveitis and Optic Neuritis as a Possible Initial Presentation of the Novel Coronavirus Disease 2019 (COVID-19). Ocul. Immunol. Inflamm. 2020, 28, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K. Pre-existing humoral immune comebacks control the development of the severe form of coronavirus disease 2019 in Gaucher patients. Clin. Transl. Discov. 2022, 2, e96. [Google Scholar] [CrossRef]
- Carvelli, J.; Demaria, O.; Vély, F.; Batista, L.; Benmansour, N.C.; Fares, J.; Carpentier, S.; Thibult, M.-L.; Morel, A.; Remark, R.; et al. Association of COVID-19 inflammation with activation of the C5a–C5aR1 axis. Nature 2020, 588, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sahu, S.K.; Cano, M.; Kuppuswamy, V.; Bajwa, J.; McPhatter, J.; Pine, A.; Meizlish, M.L.; Goshua, G.; Chang, C.H.; et al. Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. Sci. Immunol. 2021, 6, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Afzali, B.; Noris, M.; Lambrecht, B.N.; Kemper, C. The state of complement in COVID-19. Nat. Rev. Immunol. 2021, 22, 77–84. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Panigada, M.; Aliberti, S.; Blasi, F.; Tedesco, F.; et al. Complement activation in patients with COVID-19: A novel therapeutic target. J. Allergy Clin. Immunol. 2020, 146, 215–217. [Google Scholar] [CrossRef]
- Sinkovits, G.; Mező, B.; Réti, M.; Müller, V.; Iványi, Z.; Gál, J.; Gopcsa, L.; Reményi, P.; Szathmáry, B.; Lakatos, B.; et al. Complement Overactivation and Consumption Predicts In-Hospital Mortality in SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 663187. [Google Scholar] [CrossRef]
- Rockx, B.; Baas, T.; Zornetzer, G.A.; Haagmans, B.; Sheahan, T.; Frieman, M.; Dyer, M.D.; Teal, T.H.; Proll, S.; Brand, J.V.D.; et al. Early Upregulation of Acute Respiratory Distress Syndrome-Associated Cytokines Promotes Lethal Disease in an Aged-Mouse Model of Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2009, 83, 7062–7074. [Google Scholar] [CrossRef]
- Perico, L.; Morigi, M.; Galbusera, M.; Pezzotta, A.; Gastoldi, S.; Imberti, B.; Perna, A.; Ruggenenti, P.; Donadelli, R.; Benigni, A.; et al. SARS-CoV-2 Spike Protein 1 Activates Microvascular Endothelial Cells and Complement System Leading to Platelet Aggregation. Front. Immunol. 2022, 13, 827146. [Google Scholar] [CrossRef]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef]
- Ali, Y.M.; Ferrari, M.; Lynch, N.J.; Yaseen, S.; Dudler, T.; Gragerov, S.; Demopulos, G.; Heeney, J.L.; Schwaeble, W.J. Lectin Pathway Mediates Complement Activation by SARS-CoV-2 Proteins. Front. Immunol. 2021, 12, 714511. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Chan, K.H.; Law, H.K.-W.; Tso, G.H.W.; Kong, E.K.P.; Wong, H.S.W.; To, Y.F.; Yung, R.W.H.; Chow, E.Y.; Au, K.L.; et al. Mannose-Binding Lectin in Severe Acute Respiratory Syndrome Coronavirus Infection. J. Infect. Dis. 2005, 191, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Yi, X.; Sun, Y.; Bi, X.; Du, J.; Zhang, C.; Quan, S.; Zhang, F.; Sun, R.; Qian, L.; et al. Proteomic and Metabolomic Characterization of COVID-19 Patient Sera. Cell 2020, 182, 59–72.e15. [Google Scholar] [CrossRef] [PubMed]
- Holter, J.C.; Pischke, S.E.; de Boer, E.; Lind, A.; Jenum, S.; Holten, A.R.; Tonby, K.; Barratt-Due, A.; Sokolova, M.; Schjalm, C.; et al. Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc. Natl. Acad. Sci. USA 2020, 117, 25018–25025. [Google Scholar] [CrossRef] [PubMed]
- Syrimi, E.; Fennell, E.; Richter, A.; Vrljicak, P.; Stark, R.; Ott, S.; Murray, P.G.; Al-Abadi, E.; Chikermane, A.; Dawson, P.; et al. The immune landscape of SARS-CoV-2-associated Multisystem Inflammatory Syndrome in Children (MIS-C) from acute disease to recovery. iScience 2021, 24, 103215. [Google Scholar] [CrossRef] [PubMed]
- Romanova, E.S.; Vasilyev, V.V.; Startseva, G.; Karev, V.; Rybakova, M.G.; Platonov, P.G. Cause of death based on systematic post-mortem studies in patients with positive SARS-CoV-2 tissue PCR during the COVID-19 pandemic. J. Intern. Med. 2021, 290, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Pfister, F.; Vonbrunn, E.; Ries, T.; Jäck, H.-M.; Überla, K.; Lochnit, G.; Sheriff, A.; Herrmann, M.; Büttner-Herold, M.; Amann, K.; et al. Complement Activation in Kidneys of Patients With COVID-19. Front. Immunol. 2020, 11, 594849. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Coleman, C.M.; Sisk, J.M.; Halasz, G.; Zhong, J.; Beck, S.E.; Matthews, K.L.; Venkataraman, T.; Rajagopalan, S.; Kyratsous, C.A.; Frieman, M.B. CD8+ T Cells and Macrophages Regulate Pathogenesis in a Mouse Model of Middle East Respiratory Syndrome. J. Virol. 2017, 91, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Tynell, J.; Westenius, V.; Rönkkö, E.; Munster, V.; Melén, K.; Österlund, P.; Julkunen, I. Middle East respiratory syndrome coronavirus shows poor replication but significant induction of antiviral responses in human monocyte-derived macrophages and dendritic cells. J. Gen. Virol. 2016, 97, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chu, H.; Li, C.; Wong, B.H.-Y.; Cheng, Z.-S.; Poon, V.K.-M.; Sun, T.; Lau, C.C.-Y.; Wong, K.K.-Y.; Chan, J.Y.-W.; et al. Active Replication of Middle East Respiratory Syndrome Coronavirus and Aberrant Induction of Inflammatory Cytokines and Chemokines in Human Macrophages: Implications for Pathogenesis. J. Infect. Dis. 2013, 209, 1331–1342. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.L.; Hosani, A.F.; Keating, M.K.; Gerber, S.I.; Jones, T.L.; Metcalfe, M.G.; Tong, S.; Tao, Y.; Alami, N.N.; Haynes, L.M.; et al. Clinicopathologic, Immunohistochemical, and Ultrastructural Findings of a Fatal Case of Middle East Respiratory Syndrome Coronavirus Infection in the United Arab Emirates, April 2014. Am. J. Pathol. 2016, 186, 652–658. [Google Scholar] [CrossRef]
- Min, C.-K.; Cheon, S.; Ha, N.-Y.; Sohn, K.M.; Kim, Y.; Aigerim, A.; Shin, H.M.; Choi, J.-Y.; Inn, K.-S.; Kim, J.H.; et al. Comparative and kinetic analysis of viral shedding and immunological responses in MERS patients representing a broad spectrum of disease severity. Sci. Rep. 2016, 6, 25359. [Google Scholar] [CrossRef]
- Scheuplein, V.A.; Seifried, J.; Malczyk, A.H.; Miller, L.; Höcker, L.; Vergara-Alert, J.; Dolnik, O.; Zielecki, F.; Becker, B.; Spreitzer, I.; et al. High Secretion of Interferons by Human Plasmacytoid Dendritic Cells upon Recognition of Middle East Respiratory Syndrome Coronavirus. J. Virol. 2015, 89, 3859–3869. [Google Scholar] [CrossRef]
- Kim, E.S.; Choe, P.G.; Park, W.B.; Oh, H.S.; Kim, E.J.; Nam, E.Y.; Na, S.H.; Kim, M.; Song, K.-H.; Bang, J.H.; et al. Clinical Progression and Cytokine Profiles of Middle East Respiratory Syndrome Coronavirus Infection. J. Korean Med. Sci. 2016, 31, 1717–1725. [Google Scholar] [CrossRef]
- Law, H.K.-W.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus–infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef]
- Cheung, C.Y.; Poon, L.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.-H.; Yuen, K.-Y.; Gordon, S.; Guan, Y.; et al. Cytokine Responses in Severe Acute Respiratory Syndrome Coronavirus-Infected Macrophages In Vitro: Possible Relevance to Pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef]
- Spiegel, M.; Schneider, K.; Weber, F.; Weidmann, M.; Hufert, F.T. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 2006, 87, 1953–1960. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Heide, R.S.V. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Zhou, J.; Wong, B.H.-Y.; Li, C.; Chan, J.F.-W.; Cheng, Z.-S.; Yang, D.; Wang, D.; Lee, A.C.-Y.; Li, C.; et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J. Infect. Dis. 2015, 213, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-Y.; Huang, Z.-T.; Li, L.; Wu, M.-H.; Yu, T.; Koup, R.A.; Bailer, R.T.; Wu, C.-Y. Characterization of SARS-CoV-specific memory T cells from recovered individuals 4 years after infection. Arch. Virol. 2009, 154, 1093–1099. [Google Scholar] [CrossRef]
- Li, C.K.-F.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.; Weiss, R.A.; Brenchley, J.M.; et al. T Cell Responses to Whole SARS Coronavirus in Humans. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients with Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus—Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Yao, L.; Wei, T.; Tian, F.; Jin, D.Y.; Chen, L.; Wang, M. Presumed Asymptomatic Carrier Transmission of COVID-19. JAMA 2020, 323, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.-H.; Li, C.P.Y.; Chen, H.; Jin, D.-Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.-Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef]
- Yen, Y.-T.; Liao, F.; Hsiao, C.-H.; Kao, C.-L.; Chen, Y.-C.; Wu-Hsieh, B.A. Modeling the Early Events of Severe Acute Respiratory Syndrome Coronavirus Infection In Vitro. J. Virol. 2006, 80, 2684–2693. [Google Scholar] [CrossRef]
- Pandey, M.K.; Rani, R.; Zhang, W.; Setchell, K.; Grabowski, G.A. Immunological cell type characterization and Th1–Th17 cytokine production in a mouse model of Gaucher disease. Mol. Genet. Metab. 2012, 106, 310–322. [Google Scholar] [CrossRef][Green Version]
- Pandey, M.K.; Grabowski, G.A. Immunological Cells and Functions in Gaucher Disease. Crit. Rev. Oncog. 2013, 18, 197–220. [Google Scholar] [CrossRef]
- Gigis, I.; Pitsilos, C.; Samoladas, E.; Pavlopoulos, C.; Hytiroglou, P.; Ditsios, K.; Papadopoulos, P. Gaucher Disease: An Unusual Cause of Knee Pain. JAAOS Glob. Res. Rev. 2022, 6, 1–7. [Google Scholar] [CrossRef]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher Disease: A Comprehensive Review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef]
- Dandana, A.; Khelifa, B.S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Carubbi, F.; Cappellini, M.D.; Fargion, S.; Fracanzani, A.L.; Nascimbeni, F. Liver involvement in Gaucher disease: A practical review for the hepatologist and the gastroenterologist. Dig. Liv. Dis. 2020, 52, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Arévalo, N.B.; Lamaizon, C.M.; Cavieres, V.A.; Burgos, P.V.; Álvarez, A.R.; Yañez, M.J.; Zanlungo, S. Neuronopathic Gaucher disease: Beyond lysosomal dysfunction. Front. Mol. Neurosci. 2022, 15, 934820. [Google Scholar] [CrossRef]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Motta, I.; Splenomegaly Gaucher Group; Consonni, D.; Stroppiano, M.; Benedetto, C.; Cassinerio, E.; Tappino, B.; Ranalli, P.; Borin, L.; Facchini, L.; et al. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci. Rep. 2021, 11, 2594. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.-S.; Yen, H.-J.; Niu, D.-M.; Hung, G.-Y.; Lee, C.-Y.; Yeh, Y.-C.; Chen, P.C.-H.; Chang, S.-K.; Yang, C.-F. Allogeneic hematopoietic stem cell transplantation for treating severe lung involvement in Gaucher disease. Mol. Genet. Metab. Rep. 2020, 25, 100652. [Google Scholar] [CrossRef] [PubMed]
- Mauhin, W.; Brassier, A.; London, J.; Subran, B.; Zeggane, A.; Besset, Q.; Jammal, C.; Montardi, C.; Mellot, C.; Strauss, C.; et al. Manifestations pulmonaires des maladies héréditaires du métabolisme. Rev. Mal. Respir. 2022, 39, 758–777. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Antommaria, A.H.; Kolodny, E.H.; Mistry, P.K. Gaucher disease: Basic and translational science needs for more complete therapy and management. Mol. Genet. Metab. 2020, 132, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Achdout, H.; Avraham, R.; Politi, B.; Cherry, L.; Tamir, H.; Yahalom-Ronen, Y.; Paran, N.; Melamed, S.; Erez, N.; et al. Glucosylceramide synthase inhibitors prevent replication of SARS-CoV-2 and influenza virus. J. Biol. Chem. 2021, 296, 100470. [Google Scholar] [CrossRef]
- Serfecz, J.C.; Saadin, A.; Santiago, C.P.; Zhang, Y.; Bentzen, S.M.; Vogel, S.N.; Feldman, R.A. C5a Activates a Pro-Inflammatory Gene Expression Profile in Human Gaucher iPSC-Derived Macrophages. Int. J. Mol. Sci. 2021, 22, 9912. [Google Scholar] [CrossRef]
- Laurence, J.; Mulvey, J.J.; Seshadri, M.; Racanelli, A.; Harp, J.; Schenck, E.J.; Zappetti, D.; Horn, E.M.; Magro, C.M. Anti-complement C5 therapy with eculizumab in three cases of critical COVID-19. Clin. Immunol. 2020, 219, 108555. [Google Scholar] [CrossRef]
- Lo, M.W.; Kemper, C.; Woodruff, T.M. COVID-19: Complement, Coagulation, and Collateral Damage. J. Immunol. 2020, 205, 1488–1495. [Google Scholar] [CrossRef]
- Mastellos, D.C.; da Silva, B.G.P.; Fonseca, B.A.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs. C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef]
- ElFiky, A.A.; Mahdy, S.M.; Elshemey, W.M. Quantitative structure-activity relationship and molecular docking revealed a potency of anti-hepatitis C virus drugs against human corona viruses. J. Med. Virol. 2017, 89, 1040–1047. [Google Scholar] [CrossRef]
- Chan, J.F.; Lau, S.K.; To, K.K.; Cheng, V.C.; Woo, P.C.; Yuen, K.Y. Middle East Respiratory Syndrome Coronavirus: Another Zoonotic Betacoronavirus Causing SARS-Like Disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Boehm, M.; Nabel, E.G. Angiotensin-Converting Enzyme 2—A New Cardiac Regulator. N. Engl. J. Med. 2002, 347, 1795–1797. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-Dos-Santos, A.J.; Costa, D.J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Marinho, P.M.; Marcos, A.A.A.; Romano, A.C.; Nascimento, H.; Belfort, R. Retinal findings in patients with COVID-19. Lancet 2020, 395, 1610. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020, 19, e13168. [Google Scholar] [CrossRef]
- Seah, I.; Agrawal, R. Can the Coronavirus Disease 2019 (COVID-19) Affect the Eyes? A Review of Coronaviruses and Ocular Implications in Humans and Animals. Ocul. Immunol. Inflamm. 2020, 28, 391–395. [Google Scholar] [CrossRef]
- Cardona, G.C.; Pájaro, L.D.Q.; Marzola, I.D.Q.; Villegas, Y.R.; Salazar, L.R.M. Neurotropism of SARS-CoV 2: Mechanisms and manifestations. J. Neurol. Sci. 2020, 412, 116824. [Google Scholar] [CrossRef]
- Lu, C.-W.; Liu, X.-F.; Jia, Z.-F. 2019-nCoV transmission through the ocular surface must not be ignored. Lancet 2020, 395, e39. [Google Scholar] [CrossRef]
- Seah, I.; Su, X.; Lingam, G. Revisiting the dangers of the coronavirus in the ophthalmology practice. Eye 2020, 34, 1155–1157. [Google Scholar] [CrossRef]
- Cheema, M.; Aghazadeh, H.; Nazarali, S.; Ting, A.; Hodges, J.; McFarlane, A.; Kanji, J.N.; Zelyas, N.; Damji, K.F.; Solarte, C. Keratoconjunctivitis as the initial medical presentation of the novel coronavirus disease 2019 (COVID-19). Can. J. Ophthalmol. 2020, 55, e125–e129. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. Histochem. J. 2020, 51, 613–628. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Camporota, L.; Cronin, J.N.; Busana, M.; Gattinoni, L.; Formenti, F. Pathophysiology of coronavirus-19 disease acute lung injury. Curr. Opin. Crit. Care. 2022, 28, 9–16. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br. J. Haematol. 2020, 189, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zawawi, A.; Naser, A.Y.; Alwafi, H.; Minshawi, F. Profile of Circulatory Cytokines and Chemokines in Human Coronaviruses: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 666223. [Google Scholar] [CrossRef]
- Chien, J.-Y.; Hsueh, P.-R.; Cheng, W.-C.; Yu, C.-J.; Yang, P.-C. Temporal changes in cytokine/chemokine profiles and pulmonary involvement in severe acute respiratory syndrome. Respirology 2006, 11, 715–722. [Google Scholar] [CrossRef]
- Wang, C.-H.; Liu, C.-Y.; Wan, Y.-L.; Chou, C.-L.; Huang, K.-H.; Lin, H.-C.; Lin, S.-M.; Lin, T.-Y.; Chung, K.F.; Kuo-Hsiung, H. Persistence of lung inflammation and lung cytokines with high-resolution CT abnormalities during recovery from SARS. Respir. Res. 2005, 6, 42. [Google Scholar] [CrossRef]
- Wong, C.K.; Lam, C.W.K.; Wu, A.K.L.; Ip, W.K.; Lee, N.L.S.; Chan, I.H.S.; Lit, L.C.W.; Hui, D.S.C.; Chan, M.H.M.; Chung, S.S.C.; et al. Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin. Exp. Immunol. 2004, 136, 95–103. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Zhan, Y.; Wu, L.; Yu, X.; Zhang, W.; Ye, L.; Xu, S.; Sun, R.; Wang, Y.; et al. Analysis of Serum Cytokines in Patients with Severe Acute Respiratory Syndrome. Infect. Immun. 2004, 72, 4410–4415. [Google Scholar] [CrossRef]
- Cameron, M.J.; Bermejo-Martin, J.F.; Danesh, A.; Muller, M.P.; Kelvin, D.J. Human immunopathogenesis of severe acute respiratory syndrome (SARS). Virus Res. 2008, 133, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.J.; Ran, L.; Xu, L.; Danesh, A.; Bermejo-Martin, J.F.; Cameron, C.M.; Muller, M.P.; Gold, W.L.; Richardson, S.E.; Poutanen, S.M.; et al. Interferon-Mediated Immunopathological Events Are Associated with Atypical Innate and Adaptive Immune Responses in Patients with Severe Acute Respiratory Syndrome. J. Virol. 2007, 81, 8692–8706. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-J.; Su, I.-J.; Theron, M.; Wu, Y.-C.; Lai, S.-K.; Liu, C.-C.; Lei, H.-Y. An interferon-?-related cytokine storm in SARS patients. J. Med. Virol. 2004, 75, 185–194. [Google Scholar] [CrossRef]
- Theron, M.; Huang, K.-J.; Chen, Y.-W.; Liu, C.-C.; Lei, H.-Y. A probable role for IFN-γ in the development of a lung immunopathology in SARS. Cytokine 2005, 32, 30–38. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, J.Y.; Yang, J.W.; Lee, K.H.; Effenberger, M.; Szpirt, W.; Kronbichler, A.; Shin, J.I. Immunopathogenesis and treatment of cytokine storm in COVID-19. Theranostics 2021, 11, 316–329. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Liu, C.; Su, L.; Zhang, D.; Fan, J.; Yang, Y.; Xiao, M.; Xie, J.; Xu, Y.; et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol. Med. 2020, 26, 97. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Conti, P.; Gallenga, C.E.; Tete, G.; Caraffa, A.; Ronconi, G.; Younes, A.; Toniato, E.; Ross, R.; Kritas, S.K. How to reduce the likelihood of coronavirus-19 (CoV-19 or SARS-CoV-2) infection and lung inflammation mediated by IL-1. J. Biol. Regul. Homeost. Agents 2020, 34, 333–338. [Google Scholar]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Choileain, N.O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Pandey, M.K.; Jabre, N.A.; Xu, Y.-H.; Zhang, W.; Setchell, K.D.; Grabowski, G.A. Gaucher disease: Chemotactic factors and immunological cell invasion in a mouse model. Mol. Genet. Metab. 2014, 111, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Zahran, A.M.; Saad, K.; Abdallah, A.-E.M.; Gad, E.F.; Abdel-Raheem, Y.F.; Zahran, Z.A.M.; Abdelsalam, E.M.N.; Elhoufey, A.; Alruwaili, T.; Mahmoud, K.H.; et al. Dendritic cells and monocyte subsets in children with Gaucher disease. Pediatr. Res. 2021, 90, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Micheva, I.; Marinakis, T.; Repa, C.; Kouraklis-Symeonidis, A.; Vlacha, V.; Anagnostopoulos, N.; Zoumbos, N. Dendritic cells in patients with type I Gaucher disease are decreased in number but functionally normal. Blood Cells Mol. Dis. 2006, 36, 298–307. [Google Scholar] [CrossRef]
- Magnusen, A.F.; Rani, R.; McKay, M.A.; Hatton, S.L.; Nyamajenjere, T.C.; Magnusen, D.N.A.; Köhl, J.; Grabowski, G.A.; Pandey, M.K. C-X-C Motif Chemokine Ligand 9 and Its CXCR3 Receptor Are the Salt and Pepper for T Cells Trafficking in a Mouse Model of Gaucher Disease. Int. J. Mol. Sci. 2021, 22, 12712. [Google Scholar] [CrossRef]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.-L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef]
- Kim, N.E.; Do, H.; Jeong, H.; Kim, T.; Heo, S.H.; Kim, Y.; Cheon, C.K.; Lee, Y.; Choi, Y.; Choi, I.H.; et al. Identification of a novel therapeutic target underlying atypical manifestation of Gaucher disease. Clin. Transl. Med. 2022, 12, e862. [Google Scholar] [CrossRef]
- Braudeau, C.; Graveleau, J.; Rimbert, M.; Néel, A.; Hamidou, M.; Grosbois, B.; Besançon, A.; Giraudet, S.; Terrien, C.; Josien, R.; et al. Altered innate function of plasmacytoid dendritic cells restored by enzyme replacement therapy in Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 281–288. [Google Scholar] [CrossRef]
- Sønder, S.U.; Limgala, R.P.; Ivanova, M.M.; Ioanou, C.; Plassmeyer, M.; Marti, G.E.; Alpan, O.; Goker-Alpan, O. Persistent immune alterations and comorbidities in splenectomized patients with Gaucher disease. Blood Cells Mol. Dis. 2016, 59, 8–15. [Google Scholar] [CrossRef]
- Zahran, A.M.; Saad, K.; Elsayh, K.I.; Abdou, M.A.A.; Abo-Elgheet, A.M.; Eloseily, E.M.; Khalaf, S.M.; Sror, S.; Ahmad, F.-A.; Elhoufey, A.; et al. Upregulation of Cytotoxic T-cells in pediatric patients with Gaucher disease. Sci. Rep. 2022, 12, 4977. [Google Scholar] [CrossRef]
- Fiolet, T.; Kherabi, Y.; MacDonald, C.-J.; Ghosn, J.; Peiffer-Smadja, N. Comparing COVID-19 vaccines for their characteristics, efficacy and effectiveness against SARS-CoV-2 and variants of concern: A narrative review. Clin. Microbiol. Infect. 2021, 28, 202–221. [Google Scholar] [CrossRef]
- Rubin, R. COVID-19 Vaccines vs Variants—Determining How Much Immunity Is Enough. JAMA 2021, 325, 1241. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Stephenson, K.E.; Sadoff, J.; Yu, J.; Chang, A.; Gebre, M.; McMahan, K.; Liu, J.; Chandarshekar, A.; Patel, S.; et al. Durable Humoral and Cellular Immune Responses Following Ad26.COV2.S Vaccination for COVID-19. N. Engl. J. Med. 2021, 385, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Gargano, J.W.; Wallace, M.; Hadler, S.C.; Langley, G.; Su, J.R.; Oster, M.E.; Broder, K.R.; Gee, J.; Weintraub, E.; Shimabukuro, T.; et al. Use of mRNA COVID-19 Vaccine After Reports of Myocarditis Among Vaccine Recipients: Update from the Advisory Committee on Immunization Practices—United States, June. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Chandwani, A.; Shuter, J. Lopinavir/ritonavir in the treatment of HIV-1 infection: A review. Ther. Clin. Risk Manag. 2008, 4, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Magro, P.; Zanella, I.; Pescarolo, M.; Castelli, F.; Quiros-Roldan, E. Lopinavir/ritonavir: Repurposing an old drug for HIV infection in COVID-19 treatment. Biomed. J. 2020, 44, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Jeon, S.; Shin, H.-Y.; Kim, M.J.; Seong, Y.M.; Lee, W.J.; Choe, K.-W.; Kang, Y.M.; Lee, B.; Park, S.-J. Case of the Index Patient Who Caused Tertiary Transmission of Coronavirus Disease 2019 in Korea: The Application of Lopinavir/Ritonavir for the Treatment of COVID-19 Pneumonia Monitored by Quantitative RT-PCR. J. Korean Med. Sci. 2020, 35, e79. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Rehman, S.U.; Yoo, H.H. COVID-19 challenges and its therapeutics. Biomed. Pharmacother. 2021, 142, 112015. [Google Scholar] [CrossRef]
- Qomara, W.F.; Primanissa, D.N.; Amalia, S.H.; Purwadi, F.V.; Zakiyah, N. Effectiveness of Remdesivir, Lopinavir/Ritonavir, and Favipiravir for COVID-19 Treatment: A Systematic Review. Int. J. Gen. Med. 2021, 14, 8557–8571. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, V.; Pepperrell, T.; Hill, A. A review of the safety of favipiravir—a potential treatment in the COVID-19 pandemic? J. Virus Erad. 2020, 6, 45–51. [Google Scholar] [CrossRef]
- Mahajan, L.; Singh, A. Gifty Clinical outcomes of using remdesivir in patients with moderate to severe COVID-19: A prospective randomised study. Indian J. Anaesth. 2021, 65, S41–S46. [Google Scholar] [CrossRef]
- Bistrovic, P.; Manola, S.; Lucijanic, M. Bradycardia during remdesivir treatment might be associated with improved survival in patients with COVID-19: A retrospective cohort study on 473 patients from a tertiary centre. Postgrad. Med. J. 2021, 98, 501–502. [Google Scholar] [CrossRef]
- Al-Abdouh, A.; Bizanti, A.; Barbarawi, M.; Jabri, A.; Kumar, A.; Fashanu, O.E.; Khan, S.U.; Zhao, D.; Antar, A.A.; Michos, E.D. Remdesivir for the treatment of COVID-19: A systematic review and meta-analysis of randomized controlled trials. Contemp. Clin. Trials 2021, 101, 106272. [Google Scholar] [CrossRef] [PubMed]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Kaly, L.; Rosner, I. Tocilizumab—A novel therapy for non-organ-specific autoimmune diseases. Best Pract. Res. Clin. Rheumatol. 2012, 26, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Rizk, J.G.; Kalantar-Zadeh, K.; Mehra, M.R.; Lavie, C.J.; Rizk, Y.; Forthal, D.N. Pharmaco-Immunomodulatory Therapy in COVID-19. Drugs 2020, 80, 1267–1292. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Herold, S.; Hoegner, K.; Vadász, I.; Gessler, T.; Wilhelm, J.; Mayer, K.; Morty, R.E.; Walmrath, H.-D.; Seeger, W.; Lohmeyer, J. Inhaled Granulocyte/Macrophage Colony–Stimulating Factor as Treatment of Pneumonia-associated Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2014, 189, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, A.; Kurver, L.; Overheul, G.J.; Kooistra, E.J.; Abdo, W.F.; van Crevel, R.; Duivenvoorden, R.; Kox, M.; Oever, J.T.; Schouten, J.; et al. Interferon gamma immunotherapy in five critically ill COVID-19 patients with impaired cellular immunity: A case series. Med 2021, 2, 1163–1170.e2. [Google Scholar] [CrossRef]
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A.; et al. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276. [Google Scholar] [CrossRef]
- Chen, L.; Shi, M.; Deng, Q.; Liu, W.; Li, Q.; Ye, P.; Yu, X.; Zhang, B.; Xu, Y.; Li, X.; et al. A multi-center randomized prospective study on the treatment of infant bronchiolitis with interferon α1b nebulization. PLoS ONE 2020, 15, e0228391. [Google Scholar] [CrossRef]
- Dashti-Khavidaki, S.; Khalili, H. Considerations for Statin Therapy in Patients with COVID-19. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2020, 40, 484–486. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Wei, W. Angiotensin II in inflammation, immunity and rheumatoid arthritis. Clin. Exp. Immunol. 2015, 179, 137–145. [Google Scholar] [CrossRef]
- Cohen, J.I.; Burbelo, P.D. Reinfection With SARS-CoV-2: Implications for Vaccines. Clin. Infect. Dis. 2020, 73, e4223–e4228. [Google Scholar] [CrossRef]
- Galanti, M.; Shaman, J. Direct Observation of Repeated Infections with Endemic Coronaviruses. J. Infect. Dis. 2020, 223, 409–415. [Google Scholar] [CrossRef]
- Murchu, E.O.; Byrne, P.; Carty, P.G.; Gascun, D.C.; Keogan, M.; O’Neill, M.; Harrington, P.; Ryan, M. Quantifying the risk of SARS-CoV-2 reinfection over time. Rev. Med. Virol. 2021, 32, e2260. [Google Scholar] [CrossRef]
- Nguyen, Y.; Stirnemann, J.; Belmatoug, N. La maladie de Gaucher: Quand y penser? Revue Méd. Interne 2019, 40, 313–322. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Mistry, P.K. Therapies for lysosomal storage diseases: Principles, practice, and prospects for refinements based on evolving science. Mol. Genet. Metab. 2022, 137, 81–91. [Google Scholar] [CrossRef]
- Xu, Y.H.; Quinn, B.; Witte, D.; Grabowski, G.A. Viable mouse models of acid beta-glucosidase deficiency: The defect in Gaucher disease. Am. J. Pathol. 2003, 163, 2093–2101. [Google Scholar] [CrossRef]
- Pandey, M.K.; Grabowski, G.A. Advances in Gaucher Disease: Basic and Clinical Perspectives 78–93; Future Medicine Ltd.: London, UK, 2013. [Google Scholar]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and Classification of 304 Mutant Alleles in Patients with Type 1 and Type 3 Gaucher Disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.-H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Vitner, E.B.; Dekel, H.; Zigdon, H.; Shachar, T.; Farfel-Becker, T.; Eilam, R.; Karlsson, S.; Futerman, A.H. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum. Mol. Genet. 2010, 19, 3583–3590. [Google Scholar] [CrossRef]
- Dasgupta, N.; Xu, Y.-H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic Gaucher disease: Dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef]
- Orvisky, E.; Park, J.; Parker, A.; Walker, J.; Martin, B.; Stubblefield, B.; Uyama, E.; Tayebi, N.; Sidransky, E. The identification of eight novel glucocerebrosidase (GBA) mutations in patients with Gaucher disease. Hum. Mutat. 2002, 19, 458–459. [Google Scholar] [CrossRef]
- Conradi, N.G.; Sourander, P.; Nilsson, O.; Svennerholm, L.; Erikson, A. Neuropathology of the Norrbottnian type of Gaucher disease. Acta Neuropathol. 1984, 65, 99–109. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.R.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H.V. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137 Pt 3, 834–848. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Kelly, S.L.; Bame, J.R.; Duan, J.; Shinder, V.; Merrill, A.H., Jr.; Dobrenis, K.; Futerman, A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; Doummar, D.; Maire, I.; Villemeur, D.T.B. Type 2 Gaucher disease: 15 new cases and review of the literature. Brain Dev. 2006, 28, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Oppenheim, I.; Kauvar, E.; Tayebi, N.; Sidransky, E. Type 2 Gaucher disease: Phenotypic variation and genotypic heterogeneity. Blood Cells Mol. Dis. 2011, 46, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Taylor, D.; Vellodi, A. Ocular Motor Abnormalities in Gaucher Disease. Neuropediatrics 1999, 30, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Cogan, D.G.; Chu, F.C.; Reingold, D.; Barranger, J. Ocular Motor Signs in Some Metabolic Diseases. Arch. Ophthalmol. 1981, 99, 1802–1808. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Tsujl, S.; Stubblefield, B.K.; Gurrie, J.; FitzGibbon, E.J.; Glnns, E.I. Gaudier patients with oculomotor abnormalities do not have a unique genotype. Clin. Genet. 2008, 41, 1–5. [Google Scholar] [CrossRef]
- Patterson, M.C.; Horowitz, M.; Abel, R.B.; Currie, J.N.; Yu, K.-T.; Kaneski, C.; Higgins, J.J.; O’Neill, R.R.; Fedio, P.; Pikus, A.; et al. Isolated horizontal supranuclear gaze palsy as a marker of severe systemic involvement in Gaucher’s disease. Neurology 1993, 43, 1993. [Google Scholar] [CrossRef]
- King, J.O. Progressive myoclonic epilepsy due to Gaucher’s disease in an adult. J. Neurol. Neurosurg. Psychiatry 1975, 38, 849–854. [Google Scholar] [CrossRef]
- Winkelman, M.D.; Banker, B.Q.; Victor, M.; Moser, H.W. Non-infantile neuronopathic Gaucher’s disease: A clinicopathologic study. Neurology 1983, 33, 994. [Google Scholar] [CrossRef]
- Neil, J.F.; Glew, R.H.; Peters, S.P. Familial Psychosis and Diverse Neurologic Abnormalities in Adult-Onset Gaucher’s Disease. Arch. Neurol. 1979, 36, 95–99. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Fueki, N.; Sasaki, M.; Sakuragawa, N. Uncoupling of blood flow and oxygen metabolism in the cerebellum in type 3 Gaucher disease. Brain Dev. 1991, 13, 190–192. [Google Scholar] [CrossRef]
- Seeman, P.; Hoppner, J.; Lakner, V.; Liebisch, I.; Grau, G.; Rolfs, A.; Finckh, U. Two new missense mutations in a non-Jewish Caucasian family with type 3 Gaucher disease. Neurology 1996, 46, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Grover, W.D.; Tucker, S.H.; Wenger, D.A. Clinical variation in 2 related children with neuronopathic Gaucher disease. Ann. Neurol. 1978, 3, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Conradi, N.; Kyllerman, M.; Percy, A.K.; Svennerholm, L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: Neuropathological and neurochemical findings. Acta Neuropathol. 1991, 82, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Dobbelaere, D.; Sukno, S.; Defoort-Dhellemmes, S.; Lamblin, M.-D.; Largillière, C. Neurological outcome of a patient with Gaucher disease type III treated by enzymatic replacement therapy. J. Inherit. Metab. Dis. 1998, 21, 74–76. [Google Scholar] [CrossRef]
- Verghese, J.; Goldberg, R.F.; Desnick, R.J.; Grace, M.E.; Goldman, J.E.; Lee, S.C.; Dickson, D.W.; Rapin, I. Myoclonus from selective dentate nucleus degeneration in type 3 Gaucher disease. Arch. Neurol. 2000, 57, 389–395. [Google Scholar] [CrossRef][Green Version]
- Erikson, A. Gaucher disease-Norrbottnian type (III). Acta Paediatr. 1986, 75, 1–42. [Google Scholar] [CrossRef]
- Park, J.K.; Orvisky, E.; Tayebi, N.; Kaneski, C.; Lamarca, M.E.; Stubblefield, B.K.; Martin, B.M.; Schiffmann, R.; Sidransky, E. Myoclonic Epilepsy in Gaucher Disease: Genotype-Phenotype Insights from a Rare Patient Subgroup. Pediatr. Res. 2003, 53, 387–395. [Google Scholar] [CrossRef]
- Limgala, R.P.; Ioanou, C.; Plassmeyer, M.; Ryherd, M.; Kozhaya, L.; Austin, L.; Abidoglu, C.; Unutmaz, D.; Alpan, O.; Goker-Alpan, O. Time of Initiating Enzyme Replacement Therapy Affects Immune Abnormalities and Disease Severity in Patients with Gaucher Disease. PLoS ONE 2016, 11, e0168135. [Google Scholar] [CrossRef]
- Zahran, A.M.; Youssef, M.A.M.; Shafik, E.A.; Zahran, Z.A.M.; El-Badawy, O.; Elgheet, A.M.A.; Elsayh, K.I. Downregulation of B regulatory cells and upregulation of T helper 1 cells in children with Gaucher disease undergoing enzyme replacement therapy. Immunol. Res. 2020, 68, 73–80. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Dickson, P.I.; Muldowney, L.; Lee, J.J.; Rosenberg, A.; Abichandani, R.; Bluestone, J.A.; Burton, B.K.; Dewey, M.; Freitas, A.; et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol. Genet. Metab. 2016, 117, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T. Molecular diagnosis and gene therapy for Gaucher disease. Nihon Rinsho Jpn. J. Clin. Med. 1993, 51, 2300–2307. [Google Scholar]
- Bennett, L.L.; Fellner, C. Pharmacotherapy of Gaucher Disease: Current and Future Options. PTA Peer Rev. J. Formul. Manag. 2018, 43, 274–309. [Google Scholar]
- Silva, A.K.A.; Sagné, C.; Gazeau, F.; Abasolo, I. Enzyme replacement therapy: Current challenges and drug delivery prospects via extracellular vesicles. Rare Dis. Orphan Drugs J. 2022, 1, 13. [Google Scholar] [CrossRef]
- Veerhuis, R.; Nielsen, H.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef]
- Magdalon, J.; Mansur, F.; Silva, A.L.T.E.; Goes, D.V.A.; Reiner, O.; Sertié, A.L. Complement System in Brain Architecture and Neurodevelopmental Disorders. Front. Neurosci. 2020, 14, 23. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef]
- Yan, C.; Gao, H. New insights for C5a and C5a receptors in sepsis. Front. Immunol. 2012, 3, 368. [Google Scholar] [CrossRef]
- Law, S.K.; Levine, R.P. Interaction between the third complement protein and cell surface macromolecules. Proc. Natl. Acad. Sci. USA 1977, 74, 2701–2705. [Google Scholar] [CrossRef] [PubMed]
- Hein, E.; Garred, P. Immune Responses to Biosurfaces; Lambris, J.D., Ekdahl, K.N., Ricklin, D., Nilsson, B., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 77–92. [Google Scholar]
- Sørensen, R.; Thiel, S.; Jensenius, J.C. Mannan-binding-lectin-associated serine proteases, characteristics and disease associations. Springer Semin. Immunopathol. 2005, 27, 299–319. [Google Scholar] [CrossRef] [PubMed]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflamm. 2020, 17, 354. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Blasio, D.D.; Zangari, R.; Zanier, E.R.; de Simoni, M.G.; Orsini, F.; Blasio, D.D.; Zangari, R.; Zanier, E.R.; de Simoni, M.G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell Neurosci. 2014, 8, 380. [Google Scholar] [CrossRef]
- Kolev, M.; Friec, L.G.; Kemper, C. Complement—Tapping into new sites and effector systems. Nat. Rev. Immunol. 2014, 14, 811–820. [Google Scholar] [CrossRef]
- Cedzyński, M.; Thielens, N.M.; Mollnes, T.E.; Vorup-Jensen, T. Editorial: The Role of Complement in Health and Disease. Front. Immunol. 2019, 10, 1869. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2017, 14, 600–608. [Google Scholar] [CrossRef]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, K.; Safa, A.; Kotha, S.; Ratti, G.; Meri, S. Complement dysregulation in glomerulonephritis. Semin. Immunol. 2019, 45, 101331. [Google Scholar] [CrossRef]
- Köhl, J.; Baelder, R.; Lewkowich, I.P.; Pandey, M.K.; Hawlisch, H.; Wang, L.; Best, J.; Herman, N.S.; Sproles, A.A.; Zwirner, J.; et al. A regulatory role for the C5a anaphylatoxin in type 2 immunity in asthma. J. Clin. Investig. 2006, 116, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Lauer, N.; Hartmann, A.; Stippa, S.; Keilhauer, C.N.; Oppermann, M.; Pandey, M.K.; Köhl, J.; Zipfel, P.F.; Weber, B.H.; et al. An imbalance of human complement regulatory proteins CFHR1, CFHR3 and factor H influences risk for age-related macular degeneration (AMD). Hum. Mol. Genet. 2010, 19, 4694–4704. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, S.D.; Parikh, N.U.; Woodruff, T.M.; Jarvis, J.N.; Lopez, M.; Hennon, T.; Cunningham, P.; Quigg, R.J.; Schwartz, S.A.; Alexander, J.J. C5a alters blood-brain barrier integrity in a human in vitro model of systemic lupus erythematosus. Immunology 2015, 146, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Hack, B.; Chen, P.; Quigg, R.J.; Alexander, J.J. C5a/CD88 signaling alters blood-brain barrier integrity in lupus through nuclear factor-κB. J. Neurochem. 2011, 119, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Hack, B.; Chiang, E.; Garcia, J.G.N.; Quigg, R.J.; Alexander, J.J. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. 2010, 24, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Flierl, M.A.; Stahel, P.F.; Rittirsch, D.; Huber-Lang, M.; Niederbichler, A.D.; Hoesel, L.M.; Touban, B.M.; Morgan, S.J.; Smith, W.R.; Ward, P.A.; et al. Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit. Care 2009, 13, R12. [Google Scholar] [CrossRef]
- Landlinger, C.; Oberleitner, L.; Gruber, P.; Noiges, B.; Yatsyk, K.; Santic, R.; Mandler, M.; Staffler, G. Active immunization against complement factor C5a: A new therapeutic approach for Alzheimer’s disease. J. Neuroinflamm. 2015, 12, 150. [Google Scholar] [CrossRef]
- Woodruff, T.M.; Crane, J.W.; Proctor, L.M.; Buller, K.M.; Shek, A.B.; Vos, D.K.; Pollitt, S.; Williams, H.M.; Shiels, I.A.; Monk, P.N.; et al. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006, 20, 1407–1417. [Google Scholar] [CrossRef]
- Farkas, I.; Takahashi, M.; Fukuda, A.; Yamamoto, N.; Akatsu, H.; Baranyi, L.; Tateyama, H.; Yamamoto, T.; Okada, N.; Okada, H. Complement C5a Receptor-Mediated Signaling May Be Involved in Neurodegeneration in Alzheimer’s Disease. J. Immunol. 2003, 170, 5764–5771. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Fu, F.; Huang, S.; Lin, C.; Yang, G.; Ma, K.; Shi, H.; Yang, Z. C5a/C5aR Pathway Plays a Vital Role in Brain Inflammatory Injury via Initiating Fgl-2 in Intracerebral Hemorrhage. Mol. Neurobiol. 2016, 54, 6187–6197. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Mocco, J.; Hahn, D.K.; Kellner, C.P.; Komotar, R.J.; Ducruet, A.F.; Mack, W.J.; Connolly, E.S. Protective Effect of C5a Receptor Inhibition after Murine Reperfused Stroke. Neurosurgery 2008, 63, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.D.; Kumar, V.; Fung, J.N.T.; Ruitenberg, M.J.; Noakes, P.G.; Woodruff, T.M. Pharmacological inhibition of complement C5a-C5a1 receptor signalling ameliorates disease pathology in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Cereb. Blood Flow Metab. 2017, 174, 689–699. [Google Scholar] [CrossRef]
- Piatek, P.; Domowicz, M.; Lewkowicz, N.; Przygodzka, P.; Matysiak, M.; Dzitko, K.; Lewkowicz, P. C5a-Preactivated Neutrophils Are Critical for Autoimmune-Induced Astrocyte Dysregulation in Neuromyelitis Optica Spectrum Disorder. Front. Immunol. 2018, 9, 1694. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. The possible role of complement activation in Alzheimer disease. Trends Mol. Med. 2002, 8, 519–523. [Google Scholar] [CrossRef]
- Shen, Y.; Meri, S. Yin and Yang: Complement activation and regulation in Alzheimer’s disease. Prog. Neurobiol. 2003, 70, 463–472. [Google Scholar] [CrossRef]
- Yamada, T.; McGeer, P.L.; McGeer, E.G. Lewy bodies in Parkinson’s disease are recognized by antibodies to complement proteins. Acta Neuropathol. 1992, 84, 100–104. [Google Scholar] [CrossRef]
- Iseki, E.; Marui, W.; Akiyama, H.; Uéda, K.; Kosaka, K. Degeneration process of Lewy bodies in the brains of patients with dementia with Lewy bodies using α-synuclein-immunohistochemistry. Neurosci. Lett. 2000, 286, 69–73. [Google Scholar] [CrossRef]
- Tan, S.M.; Snelson, M.; Østergaard, J.A.; Coughlan, M.T. The Complement Pathway: New Insights into Immunometabolic Signaling in Diabetic Kidney Disease. Antioxid. Redox Signal 2022, 37, 781–801. [Google Scholar] [CrossRef]
- O’Brien, K.B.; Morrison, T.E.; Dundore, D.Y.; Heise, M.T.; Schultz-Cherry, S. A Protective Role for Complement C3 Protein during Pandemic 2009 H1N1 and H5N1 Influenza A Virus Infection. PLoS ONE 2011, 6, e17377. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, J.; Teng, Y.; Sun, H.; Tian, G.; He, L.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; et al. Complement Receptor C5aR1 Inhibition Reduces Pyroptosis in hDPP4-Transgenic Mice Infected with MERS-CoV. Viruses 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Posch, W.; Vosper, J.; Noureen, A.; Zaderer, V.; Witting, C.; Bertacchi, G.; Gstir, R.; Filipek, P.A.; Bonn, G.K.; Huber, L.A.; et al. C5aR inhibition of nonimmune cells suppresses inflammation and maintains epithelial integrity in SARS-CoV-2–infected primary human airway epithelia. J. Allergy Clin. Immunol. 2021, 147, 2083–2097.e6. [Google Scholar] [CrossRef]
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 thrombotic catastrophe complement-connected? J. Thromb. Haemost. 2020, 18, 2812–2822. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Bellander, B.-M. Is COVID-19 associated thrombosis caused by overactivation of the complement cascade? A literature review. Thromb. Res. 2020, 194, 36–41. [Google Scholar] [CrossRef]
- Boussier, J.; Yatim, N.; Marchal, A.; Hadjadj, J.; Charbit, B.; Sissy, E.C.; Carlier, N.; Pène, F.; Mouthon, L.; Tharaux, P.-L.; et al. Severe COVID-19 is associated with hyperactivation of the alternative complement pathway. J. Allergy Clin. Immunol. 2022, 149, 550–556.e2. [Google Scholar] [CrossRef]
- Santiesteban-Lores, L.E.; Amamura, T.A.; da Silva, T.F.; Midon, L.M.; Carneiro, M.C.; Isaac, L.; Bavia, L. A double edged-sword-The Complement System during SARS-CoV-2 infection. Life Sci. 2021, 272, 119245. [Google Scholar] [CrossRef]
- Droesch, C.; Do, M.H.; DeSancho, M.; Lee, E.-J.; Magro, C.; Harp, J. Livedoid and Purpuric Skin Eruptions Associated With Coagulopathy in Severe COVID-19. JAMA Dermatol. 2020, 156, 1022. [Google Scholar] [CrossRef]
- Yu, J.; Gerber, G.F.; Chen, H.; Yuan, X.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Complement dysregulation is associated with severe COVID-19 illness. Haematologica 2021, 107, 1095–1105. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 2020, 136, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Satyam, A.; Tsokos, M.G.; Brook, O.R.; Hecht, J.L.; Moulton, V.R.; Tsokos, G.C. Activation of classical and alternative complement pathways in the pathogenesis of lung injury in COVID-19. Clin. Immunol. 2021, 226, 108716. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K. The Role of Alpha-Synuclein Autoantibodies in the Induction of Brain Inflammation and Neurodegeneration in Aged Humans. Front. Aging Neurosci. 2022, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ Receptors: Old Friends and New Family Members. Immunity 2006, 24, 19–28. [Google Scholar] [CrossRef]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, C.; Wang, Z.; Lorenzi, J.C.C.; Muecksch, F.; Finkin, S.; Tokuyama, M.; Cho, A.; Jankovic, M.; Schaefer-Babajew, D.; Oliveira, T.Y.; et al. Evolution of antibody immunity to SARS-CoV-2. Nature 2021, 591, 639–644. [Google Scholar] [CrossRef]
- Turner, J.S.; Kim, W.; Kalaidina, E.; Goss, C.W.; Rauseo, A.M.; Schmitz, A.J.; Hansen, L.; Haile, A.; Klebert, M.K.; Pusic, I.; et al. SARS-CoV-2 infection induces long-lived bone marrow plasma cells in humans. Nature 2021, 595, 421–425. [Google Scholar] [CrossRef]
- Nguyen-Contant, P.; Embong, A.K.; Kanagaiah, P.; Chaves, F.A.; Yang, H.; Branche, A.R.; Topham, D.J.; Sangster, M.Y. S Protein-Reactive IgG and Memory B Cell Production after Human SARS-CoV-2 Infection Includes Broad Reactivity to the S2 Subunit. mBio 2020, 11, e01991-20. [Google Scholar] [CrossRef]
- Hartley, G.E.; Edwards, E.S.J.; Aui, P.M.; Varese, N.; Stojanovic, S.; McMahon, J.; Peleg, A.Y.; Boo, I.; Drummer, H.E.; Hogarth, P.M.; et al. Rapid Generation of Durable B Cell Memory to SARS-CoV-2 Spike and Nucleocapsid Proteins in COVID-19 and Convalescence. Sci. Immunol. 2020, 5, eabf8891. [Google Scholar] [CrossRef]
- Williams, J.W.; Tjota, M.Y.; Sperling, A.I. The Contribution of Allergen-Specific IgG to the Development of Th2-Mediated Airway Inflammation. J. Allergy 2012, 2012, 236075. [Google Scholar] [CrossRef]
- Karsten, C.M.; Köhl, J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology 2012, 217, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Bruhns, P.; Horiuchi, K.; Ravetch, J.V. FcγRIV: A Novel FcR with Distinct IgG Subclass Specificity. Immunity 2005, 23, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Seino, J.; Eveleigh, P.; Warnaar, S.; van Haarlem, L.J.M.; van Es, L.A.; Daha, M.R. Activation of human complement by mouse and mouse/human chimeric monoclonal antibodies. Clin. Exp. Immunol. 1993, 94, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.N.; Konrad, S.; Wiege, K.; Nieswandt, B.; Nimmerjahn, F.; Schmidt, R.E.; Gessner, J.E. Both FcγRIV and FcγRIII are essential receptors mediating type II and type III autoimmune responses via FcRγ-LAT-dependent generation of C5a. Eur. J. Immunol. 2009, 39, 3343–3356. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K. Molecular Basis for Downregulation of C5a-Mediated Inflammation by IgG1 Immune Complexes in Allergy and Asthma. Curr. Allergy Asthma Rep. 2013, 13, 596–606. [Google Scholar] [CrossRef]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef]
- Schäfer, A.; Muecksch, F.; Lorenzi, J.C.; Leist, S.R.; Cipolla, M.; Bournazos, S.; Schmidt, F.; Maison, R.M.; Gazumyan, A.; Martinez, D.R.; et al. Antibody potency, effector function, and combinations in protection and therapy for SARS-CoV-2 infection in vivo. J. Exp. Med. 2020, 218, 1–14. [Google Scholar] [CrossRef]
- Moncunill, G.; Mayor, A.; Santano, R.; Jiménez, A.; Vidal, M.; Tortajada, M.; Sanz, S.; Méndez, S.; Llupià, A.; Aguilar, R.; et al. SARS-CoV-2 Seroprevalence and Antibody Kinetics Among Health Care Workers in a Spanish Hospital After 3 Months of Follow-up. J. Infect. Dis. 2020, 223, 62–71. [Google Scholar] [CrossRef]
- Dasgupta, N.; Xu, Y.-H.; Oh, S.; Sun, Y.; Jia, L.; Keddache, M.; Grabowski, G.A. Gaucher Disease: Transcriptome Analyses Using Microarray or mRNA Sequencing in a Gba1 Mutant Mouse Model Treated with Velaglucerase alfa or Imiglucerase. PLoS ONE 2013, 8, e74912. [Google Scholar] [CrossRef]
- Xu, Y.-H.; Jia, L.; Quinn, B.; Zamzow, M.; Stringer, K.; Aronow, B.; Sun, Y.; Zhang, W.; Setchell, K.; Grabowski, G.A. Global gene expression profile progression in Gaucher disease mouse models. BMC Genom. 2011, 12, 20. [Google Scholar] [CrossRef]
- Guo, R.-F.; Ward, P.A. Role of C5a in Inflammatory Responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Köhl, J. A complex role for complement in allergic asthma. Expert Rev. Clin. Immunol. 2010, 6, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Farfel-Becker, T.; Ferreira, N.S.; Leshkowitz, D.; Sharma, P.; Lang, K.S.; Futerman, A.H. Induction of the type I interferon response in neurological forms of Gaucher disease. J. Neuroinflamm. 2016, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.-P.; Wang, G.; Qi, C.-F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B Cell Differentiation and Plasma Cell Generation by IL-21, a Novel Inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Boddupalli, C.S.; Verma, R.; Liu, J.; Yang, R.; Pastores, G.M.; Mistry, P.K.; Dhodapkar, M.V. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015, 125, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Ayeh, S.K.; Abbey, E.J.; Khalifa, B.A.A.; Nudotor, R.D.; Osei, A.D.; Chidambaram, V.; Osuji, N.; Khan, S.; Salia, E.L.; Oduwole, M.O.; et al. Statins use and COVID-19 outcomes in hospitalized patients. PLoS ONE 2021, 16, e0256899. [Google Scholar] [CrossRef]
- Lohia, P.; Kapur, S.; Benjaram, S.; Mir, T. Association between antecedent statin use and severe disease outcomes in COVID-19: A retrospective study with propensity score matching. J. Clin. Lipidol. 2021, 15, 451–459. [Google Scholar] [CrossRef]
- Gupta, A.; Madhavan, M.V.; Poterucha, T.J.; DeFilippis, E.M.; Hennessey, J.A.; Redfors, B.; Eckhardt, C.; Bikdeli, B.; Platt, J.; Nalbandian, A.; et al. Association between antecedent statin use and decreased mortality in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 1325. [Google Scholar] [CrossRef]
- Lohia, P.; Kapur, S.; Benjaram, S.; Cantor, Z.; Mahabadi, N.; Mir, T.; Badr, M.S. Statins and clinical outcomes in hospitalized COVID-19 patients with and without Diabetes Mellitus: A retrospective cohort study with propensity score matching. Cardiovasc. Diabetol. 2021, 20, 140. [Google Scholar] [CrossRef]
- Felice, C.; Nardin, C.; Tanna, D.G.L.; Grossi, U.; Bernardi, E.; Scaldaferri, L.; Romagnoli, M.; Tonon, L.; Cavasin, P.; Novello, S.; et al. Use of RAAS Inhibitors and Risk of Clinical Deterioration in COVID-19: Results From an Italian Cohort of 133 Hypertensives. Am. J. Hypertens. 2020, 33, 944–948. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, M.; Flannery, A.; Ferreira, J.P.; Cheng, M.P.; Kronfli, N.; Marelli, A.; Zannad, F.; Brophy, J.; Afillalo, J.; Huynh, T.; et al. Management of Renin-Angiotensin-Aldosterone System blockade in patients admitted to hospital with confirmed coronavirus disease (COVID-19) infection (The McGill RAAS-COVID-19): A structured summary of a study protocol for a randomized controlled trial. Trials 2021, 22, 115. [Google Scholar] [CrossRef] [PubMed]
- Hallaj, S.; Ghorbani, A.; Aghdas, S.A.M.; Mirza-Aghazadeh-Attari, M.; Sevbitov, A.; Hashemi, V.; Hallaj, T.; Jadidi-Niaragh, F. Angiotensin-converting enzyme as a new immunologic target for the new SARS-CoV-2. Immunol. Cell Biol. 2020, 99, 192–205. [Google Scholar] [CrossRef]
- Michon, A. ACE inhibitors—An effective treatment for hyaluronic acid soft tissue filler delayed inflammatory reaction following COVID-19 vaccination. J. Cosmet. Dermatol. 2022, 21, 1369–1370. [Google Scholar] [CrossRef] [PubMed]
- Kaps, L.; Labenz, C.; Grimm, D.; Schwarting, A.; Galle, P.R.; Schreiner, O. Treatment of cytokine storm syndrome with IL-1 receptor antagonist anakinra in a patient with ARDS caused by COVID-19 infection: A case report. Clin. Case Rep. 2020, 8, 2989–2993. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; Bentum-Puijk, V.W.; Berry, L.R.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar] [CrossRef]
- Amer, M.; Kamel, A.M.; Bawazeer, M.; Maghrabi, K.; Butt, A.; Dahhan, T.; Kseibi, E.; Khurshid, S.M.; Abujazar, M.; Alghunaim, R.; et al. Clinical characteristics and outcomes of critically ill mechanically ventilated COVID-19 patients receiving interleukin-6 receptor antagonists and corticosteroid therapy: A preliminary report from a multinational registry. Eur. J. Med. Res. 2021, 26, 117. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.-H.; Wang, C.-K.; Chang, S.-P.; Lu, L.-C.; Hung, S.-H.; Lin, W.-T. Janus kinase inhibitors for hospitalized patients with COVID-19: A meta-analysis of randomized controlled trials. Expert Rev. Anti Infect. Ther. 2021, 20, 773–779. [Google Scholar] [CrossRef]
- Lin, Z.; Niu, J.; Xu, Y.; Qin, L.; Ding, J.; Zhou, L. Clinical efficacy and adverse events of baricitinib treatment for coronavirus disease-2019 (COVID-19): A systematic review and meta-analysis. J. Med. Virol. 2021, 94, 1523–1534. [Google Scholar] [CrossRef]
- Chen, C.-X.; Wang, J.-J.; Li, H.; Yuan, L.-T.; Gale, R.P.; Liang, Y. JAK-inhibitors for coronavirus disease-2019 (COVID-19): A meta-analysis. Leukemia 2021, 35, 2616–2620. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chen, W.-C.; Hsu, C.-K.; Chao, C.-M.; Lai, C.-C. Clinical efficacy and safety of Janus kinase inhibitors for COVID-19: A systematic review and meta-analysis of randomized controlled trials. Int. Immunopharmacol. 2021, 99, 108027. [Google Scholar] [CrossRef]
- Fakharian, A.; Barati, S.; Mirenayat, M.; Rezaei, M.; Haseli, S.; Torkaman, P.; Yousefian, S.; Dastan, A.; Jamaati, H.; Dastan, F. Evaluation of adalimumab effects in managing severe cases of COVID-19: A randomized controlled trial. Int. Immunopharmacol. 2021, 99, 107961. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchié, A.; Wang, T.S.; Lee, E.; Cremer, P.C.; Carey, B.; Rajendram, P.; Hudock, K.M.; Korbee, L.; Van Tassell, B.W.; et al. Targeting GM-CSF in COVID-19 Pneumonia: Rationale and Strategies. Front Immunol. 2020, 11, 1625. [Google Scholar] [CrossRef]
- Criner, G.J.; Lang, F.M.; Gottlieb, R.L.; Mathews, K.S.; Wang, T.S.; Rice, T.W.; Madduri, D.; Bellam, S.; Jeanfreau, R.; Case, A.H.; et al. Anti-Granulocyte–Macrophage Colony–Stimulating Factor Monoclonal Antibody Gimsilumab for COVID-19 Pneumonia: A Randomized, Double-Blind, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2022, 205, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Zavattaro, E.; Cammarata, E.; Tarantino, V.; Soddu, D.; Gironi, L.C.; Savoia, P. Successful treatment of a bullous vasculitis with intravenous immunoglobulins in a COVID-19 patient. Dermatol. Ther. 2021, 34, e14853. [Google Scholar] [CrossRef]
- Huo, S.; Ferse, C.; Bösl, F.; Reincke, S.M.; Enghard, P.; Hinrichs, C.; Treskatsch, S.; Angermair, S.; Eckardt, K.-U.; Audebert, H.J.; et al. Intravenous immunoglobulins for treatment of severe COVID-19-related acute encephalopathy. J. Neurol. 2022, 269, 4013–4020. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, D.; Sasaki, K.; Karkar, A.; Sharif, S.; Lewis, K.; Mammen, M.J.; Alexander, P.; Ye, Z.; Lozano, L.E.C.; Munch, M.W.; et al. Corticosteroids in COVID-19 and non-COVID-19 ARDS: A systematic review and meta-analysis. Intensive Care Med. 2021, 47, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Kazachinskaia, E.; Chepurnov, A.; Shcherbakov, D.; Kononova, Y.; Saroyan, T.; Gulyaeva, M.; Shanshin, D.; Romanova, V.; Khripko, O.; Voevoda, M.; et al. IgG Study of Blood Sera of Patients with COVID-19. Pathogens 2021, 10, 1421. [Google Scholar] [CrossRef]
- Li, K.; Huang, B.; Wu, M.; Zhong, A.; Li, L.; Cai, Y.; Wang, Z.; Wu, L.; Zhu, M.; Li, J.; et al. Dynamic changes in anti-SARS-CoV-2 antibodies during SARS-CoV-2 infection and recovery from COVID-19. Nat. Commun. 2020, 11, 6044. [Google Scholar] [CrossRef]
- Ozturk, T.; Howell, C.; Benameur, K.; Ramonell, R.P.; Cashman, K.; Pirmohammed, S.; Bassit, L.; Roback, J.; Marconi, V.C.; Schinazi, R.F.; et al. Cross-sectional IgM and IgG profiles in SARS-CoV-2 infection. medRxiv 2020, 1–21. [Google Scholar] [CrossRef]
- Sun, B.; Feng, Y.; Mo, X.; Zheng, P.; Wang, Q.; Li, P.; Peng, P.; Liu, X.; Chen, Z.; Huang, H.; et al. Kinetics of SARS-CoV-2 specific IgM and IgG responses in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lai, D.-Y.; Lei, Q.; Xu, Z.-W.; Wang, F.; Hou, H.; Chen, L.; Wu, J.; Ren, Y.; Ma, M.-L.; et al. Systematic evaluation of IgG responses to SARS-CoV-2 spike protein-derived peptides for monitoring COVID-19 patients. Cell Mol. Immunol. 2021, 18, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Beavis, K.G.; Matushek, S.M.; Abeleda, A.P.F.; Bethel, C.; Hunt, C.; Gillen, S.; Moran, A.; Tesic, V. Evaluation of the EUROIMMUN Anti-SARS-CoV-2 ELISA Assay for detection of IgA and IgG antibodies. J. Clin. Virol. 2020, 129, 104468. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wiredja, D.; Yang, L.; Bulterys, P.L.; Costales, C.; Röltgen, K.; Manalac, J.; Yee, J.; Zehnder, J.; Shi, R.Z.; et al. Case-Control Study of Individuals with Discrepant Nucleocapsid and Spike Protein SARS-CoV-2 IgG Results. Clin. Chem. 2021, 67, 977–986. [Google Scholar] [CrossRef]
- Nakano, Y.; Kurano, M.; Morita, Y.; Shimura, T.; Yokoyama, R.; Qian, C.; Xia, F.; He, F.; Kishi, Y.; Okada, J.; et al. Time course of the sensitivity and specificity of anti-SARS-CoV-2 IgM and IgG antibodies for symptomatic COVID-19 in Japan. Sci. Rep. 2021, 11, 2776. [Google Scholar] [CrossRef]
- Liu, X.; Wang, J.; Xu, X.; Liao, G.; Chen, Y.; Hu, C.-H. Patterns of IgG and IgM antibody response in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1269–1274. [Google Scholar] [CrossRef]
- Meinberger, D.; Koch, M.; Roth, A.; Hermes, G.; Stemler, J.; Cornely, O.A.; Streichert, T.; Klatt, A.R. Analysis of IgM, IgA, and IgG isotype antibodies Directed against SARS-CoV-2 spike glycoprotein and ORF8 in the course of COVID-19. Sci. Rep. 2021, 11, 8920. [Google Scholar] [CrossRef]
- Zhou, C.; Bu, G.; Sun, Y.; Ren, C.; Qu, M.; Gao, Y.; Zhu, Y.; Wang, L.; Sun, L.; Liu, Y. Evaluation of serum IgM and IgG antibodies in COVID-19 patients by enzyme linked immunosorbent assay. J. Med. Virol. 2020, 93, 2857–2866. [Google Scholar] [CrossRef]
- Isho, B.; Abe, K.T.; Zuo, M.; Jamal, A.J.; Rathod, B.; Wang, J.H.; Li, Z.; Chao, G.; Rojas, O.L.; Bang, Y.M.; et al. Persistence of serum and saliva antibody responses to SARS-CoV-2 spike antigens in COVID-19 patients. Sci. Immunol. 2020, 5, eabe5511. [Google Scholar] [CrossRef]
- Sterlin, D.; Mathian, A.; Miyara, M.; Mohr, A.; Anna, F.; Claër, L.; Quentric, P.; Fadlallah, J.; Devilliers, H.; Ghillani, P.; et al. IgA dominates the early neutralizing antibody response to SARS-CoV-2. Sci. Transl. Med. 2021, 13, eabd2223. [Google Scholar] [CrossRef]
- Shah, J.; Liu, S.; Potula, H.-H.; Bhargava, P.; Cruz, I.; Force, D.; Bazerbashi, A.; Ramasamy, R. IgG and IgM antibody formation to spike and nucleocapsid proteins in COVID-19 characterized by multiplex immunoblot assays. BMC Infect. Dis. 2021, 21, 325. [Google Scholar] [CrossRef] [PubMed]
- Batra, M.; Tian, R.; Zhang, C.; Clarence, E.; Sacher, C.S.; Miranda, J.N.; Fuente, D.L.J.R.O.; Mathew, M.; Green, D.; Patel, S.; et al. Role of IgG against N-protein of SARS-CoV2 in COVID-19 clinical outcomes. Sci. Rep. 2021, 11, 3455. [Google Scholar] [CrossRef]
- Johnson, M.; Wagstaffe, H.R.; Gilmour, K.C.; Mai, A.L.; Lewis, J.; Hunt, A.; Sirr, J.; Bengt, C.; Grandjean, L.; Goldblatt, D. Evaluation of a novel multiplexed assay for determining IgG levels and functional activity to SARS-CoV-2. J. Clin. Virol. 2020, 130, 104572. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Wei, X.; Zheng, S.W.; Lenhart, B.J.; Xu, P.; Li, J.; Pan, J.; Albrecht, H.; Liu, C. Multiplex quantitative detection of SARS-CoV-2 specific IgG and IgM antibodies based on DNA-assisted nanopore sensing. Biosens. Bioelectron. 2021, 181, 113134. [Google Scholar] [CrossRef] [PubMed]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Platt, F.M. Sphingolipid lysosomal storage disorders. Nature 2014, 510, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Messner, M.C.; Cabot, M.C. Glucosylceramide in Humans. Adv. Exp. Med. Biol. 2010, 688, 156–164. [Google Scholar] [CrossRef]
- Melo, C.F.O.R.; de Oliveira, D.N.; Lima, E.D.O.; Guerreiro, T.M.; Esteves, C.Z.; Beck, R.M.; Padilla, M.A.; Milanez, G.P.; Arns, C.W.; Proenca-Modena, J.L.; et al. A Lipidomics Approach in the Characterization of Zika-Infected Mosquito Cells: Potential Targets for Breaking the Transmission Cycle. PLoS ONE 2016, 11, e0164377. [Google Scholar] [CrossRef]
- Khan, I.; Katikaneni, D.S.; Han, Q.; Sanchez-Felipe, L.; Hanada, K.; Ambrose, R.L.; Mackenzie, J.M.; Konan, K.V. Modulation of Hepatitis C Virus Genome Replication by Glycosphingolipids and Four-Phosphate Adaptor Protein. J. Virol. 2014, 88, 12276–12295. [Google Scholar] [CrossRef]
- Low, H.; Mukhamedova, N.; Cui, H.L.; McSharry, B.P.; Avdic, S.; Hoang, A.; Ditiatkovski, M.; Liu, Y.; Fu, Y.; Meikle, P.J.; et al. Cytomegalovirus Restructures Lipid Rafts via a US28/CDC42-Mediated Pathway, Enhancing Cholesterol Efflux from Host Cells. Cell Rep. 2016, 16, 186–200. [Google Scholar] [CrossRef]
- Chotiwan, N.; Andre, B.G.; Sanchez-Vargas, I.; Islam, M.N.; Grabowski, J.M.; Hopf-Jannasch, A.; Gough, E.; Nakayasu, E.; Blair, C.D.; Belisle, J.T.; et al. Dynamic remodeling of lipids coincides with dengue virus replication in the midgut of Aedes aegypti mosquitoes. PLOS Pathog. 2018, 14, e1006853. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.B.; Chng, C.; Guan, X.L.; Lei, Z.; Rozen, S.G.; Wenk, M.R. Lipidomics identifies a requirement for peroxisomal function during influenza virus replication. J. Lipid Res. 2014, 55, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Achdout, H.; Manaster, I.; Mandelboim, O. Influenza Virus Infection Augments NK Cell Inhibition through Reorganization of Major Histocompatibility Complex Class I Proteins. J. Virol. 2008, 82, 8030–8037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hidari, K.I.P.J.; Suzuki, Y.; Suzuki, T. Suppression of the Biosynthesis of Cellular Sphingolipids Results in the Inhibition of the Maturation of Influenza Virus Particles in MDCK Cells. Biol. Pharm. Bull. 2006, 29, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, 1–20. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Kochs, G.; Gripp, B.; Weigang, S.; Adams, C.; Carpinteiro, E.; Gulbins, A.; Keitsch, S.; et al. Pharmacological Inhibition of Acid Sphingomyelinase Prevents Uptake of SARS-CoV-2 by Epithelial Cells. Cell Rep. Med. 2020, 1, 100142. [Google Scholar] [CrossRef]
- Törnquist, K.; Asghar, M.Y.; Srinivasan, V.; Korhonen, L.; Lindholm, D. Sphingolipids as modulators of SARS-CoV-2 infection. Front. Cell Dev. Biol. 2021, 9, 1574. [Google Scholar] [CrossRef]
- Hong, B.Y.; Kim, E.Y.; Jung, S.-C. Upregulation of Proinflammatory Cytokines in the Fetal Brain of the Gaucher Mouse. J. Korean Med. Sci. 2006, 21, 733–738. [Google Scholar] [CrossRef]
- Kim, E.Y.; Hong, B.Y.; Go, S.H.; Lee, B.; Jung, S.-C. Downregulation of neurotrophic factors in the brain of a mouse model of Gaucher disease: Implications for neuronal loss in Gaucher disease. Exp. Mol. Med. 2006, 38, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Tybulewicz, V.L.J.; Tremblay, M.L.; Marca, L.; Willemsen, R.; Stubblefield, B.K.; Winfield, S.; Zablocka, B.; Sidransky, E.; Martin, B.M.; Huang, S.P.; et al. Animal model of Gaucher’s disease from targeted disruption of the mouse glucocerebrosidase gene. Nature 1992, 357, 407–410. [Google Scholar] [CrossRef]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Vardi, A.; Zigdon, H.; Meshcheriakova, A.; Klein, A.D.; Yaacobi, C.; Eilam, R.; Kenwood, B.M.; Rahim, A.A.; Massaro, G.; Merrill, A.H.; et al. Delineating pathological pathways in a chemically induced mouse model of Gaucher disease. J. Pathol. 2016, 239, 496–509. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, J.N.; Legler, G.; Sullivan, J.; Raghavan, S.S.; Mumford, R.A. The Gaucher mouse. Biochem. Biophys. Res. Commun. 1975, 67, 85–90. [Google Scholar] [CrossRef]
- Enquist, I.B.; Bianco, C.L.; Ooka, A.; Nilsson, E.; Månsson, J.-E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [PubMed]
- Lucisano, Y.M.; Mantovani, B. The role of complement in the stimulation of lysosomal enzyme release by polymorphonuclear leucocytes induced by immune complexes of IgG and of IgM. Immunology 1988, 65, 171–175. [Google Scholar]
- Cardella, C.J.; Davies, P.; Allison, A.C. Immune Complexes induce Selective Release of Lysosomal Hydrolases from Macrophages. Nature 1974, 247, 46–48. [Google Scholar] [CrossRef]
- Zurier, R.B.; Hoffstein, S.; Weissmann, G. Mechanisms of Lysosomal Enzyme Release from Human Leukocytes. J. Cell Biol. 1973, 58, 27–41. [Google Scholar] [CrossRef]
- Schorlemmer, H.-U.; Davies, P.; Allison, A.C. Ability of activated complement components to induce lysosomal enzyme release from macrophages. Nature 1976, 261, 48–49. [Google Scholar] [CrossRef]
- Schorlemmer, H.U.; Allison, A.C. Effects of activated complement components on enzyme secretion by macrophages. Immunology 1976, 31, 781–788. [Google Scholar]
- Knudsen, F.; Pedersen, J.O.; Juhl, O.; Nielsen, A.H.; Jersild, C. Complement and leukocytes during cardiopulmonary bypass: Effects on plasma C3d and C5a, leukocyte count, release of granulocyte elastase and granulocyte chemotaxis. J. Cardiothorac. Anesth. 1988, 2, 164–170. [Google Scholar] [CrossRef]
- Goldstein, I.M.; Hoffstein, S.T.; Weissmann, G. Influence of divalent cations upon complement-mediated enzyme release from human polymorphonuclear leukocytes. J. Immunol. 1975, 115, 665–670. [Google Scholar] [PubMed]
- Goldstein, I.; Hoffstein, S.; Gallin, J.; Weissmann, G. Mechanisms of Lysosomal Enzyme Release from Human Leukocytes: Microtubule Assembly and Membrane Fusion Induced by a Component of Complement. Proc. Natl. Acad. Sci. USA 1973, 70, 2916–2920. [Google Scholar] [CrossRef] [PubMed]
- Fierro, L.; Nesheiwat, N.; Naik, H.; Narayanan, P.; Mistry, P.K.; Balwani, M. Gaucher disease and SARS-CoV-2 infection: Experience from 181 patients in New York. Mol. Genet. Metab. 2020, 132, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Campos, M.; Escuder-Azuara, B.; de Frutos, L.L.; Gonzalo, I.S.; Giraldo, P. Direct and indirect effects of the SARS-CoV-2 pandemic on Gaucher Disease patients in Spain: Time to reconsider home-based therapies? Blood Cells Mol. Dis. 2020, 85, 102478. [Google Scholar] [CrossRef]
- Zimran, A.; Szer, J.; Revel-Vilk, S. Impact of Gaucher disease on COVID-19. Intern. Med. J. 2020, 50, 894–895. [Google Scholar] [CrossRef]
- Demirci, I.; Demir, T.; Dagdelen, S.; Haymana, C.; Tasci, I.; Atmaca, A.; Ertugrul, D.; Ata, N.; Sahin, M.; Salman, S.; et al. No association of Gaucher Disease with COVID-19-related outcomes: A nationwide cohort study. Intern. Med. J. 2021, 52, 379–385. [Google Scholar] [CrossRef]
- Khalili, M.; Baeis, M.G.; Saneifard, H.; Ghanaie, R.M.; Shamsian, B.S. Pediatric with Gaucher disease and COVID-19: Case report of uncommon manifestation of COVID-19 in chest Ct. Vis. J. Emerg. Med. 2021, 22, 100966. [Google Scholar] [CrossRef]
- Hamiel, U.; Kurolap, A.; Cohen, I.J.; Ruhrman-Shahar, N.; Hershkovitz, T.; Niederau, C.; Zimran, A.; Revel-Vilk, S.; Istaiti, M.; Cappellini, M.D.; et al. Experts’ views on COVID-19 vaccination and the impact of the pandemic on patients with Gaucher disease. Br. J. Haematol. 2021, 195, e135–e137. [Google Scholar] [CrossRef]
- Kristal, E.; Pode-Shakked, B.; Hazan, G.; Banne, E.; Ling, G.; David, O.; Shany, E.; Raas-Rothschild, A.; Anikster, Y.; Kneller, K.; et al. The effects of the COVID-19 pandemic on patients with lysosomal storage disorders in Israel. Orphanet J. Rare Dis. 2021, 16, 379. [Google Scholar] [CrossRef]
- Humbles, A.A.; Lu, B.; Nilsson, C.A.; Lilly, C.; Israel, E.; Fujiwara, Y.; Gerard, N.P.; Gerard, C. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature 2000, 406, 998–1001. [Google Scholar] [CrossRef]
- Krug, N.; Tschernig, T.; Erpenbeck, V.J.; Hohlfeld, J.M.; Köhl, J. Complement Factors C3a and C5a Are Increased in Bronchoalveolar Lavage Fluid after Segmental Allergen Provocation in Subjects with Asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 1841–1843. [Google Scholar] [CrossRef] [PubMed]
- Eglite, S.; Plüss, K.; Dahinden, C.A. Requirements for C5a Receptor-Mediated IL-4 and IL-13 Production and Leukotriene C4 Generation in Human Basophils. J. Immunol. 2000, 165, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Woolhiser, M. Activation of human mast cells by aggregated IgG through FcγRI: Additive effects of C3a. Clin. Immunol. 2004, 110, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Yang, K.; Foster, S.J.; Kondo, M.; Kelsoe, G. Inflammation Controls B Lymphopoiesis by Regulating Chemokine CXCL12 Expression. J. Exp. Med. 2004, 199, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Nagaoka, H.; Gonzalez-Aseguinolaza, G.; Tsuji, M.; Nussenzweig, M.C. Immunization and Infection Change the Number of Recombination Activating Gene (Rag)-Expressing B Cells in the Periphery by Altering Immature Lymphocyte Production. J. Exp. Med. 2000, 191, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Grabowski, G.A.; Köhl, J. An unexpected player in Gaucher disease: The multiple roles of complement in disease development. Semin. Immunol. 2018, 37, 30–42. [Google Scholar] [CrossRef]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Risitano, A.M.; Mastellos, D.C.; Huber-Lang, M.; Yancopoulou, D.; Garlanda, C.; Ciceri, F.; Lambris, J.D. Complement as a target in COVID-19? Nat. Rev. Immunol. 2020, 20, 343–344. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, e140711. [Google Scholar] [CrossRef]
- Lazo-Langner, A.; Chin-Yee, I.; Al-Ani, F. Eculizumab in the management of paroxysmal nocturnal hemoglobinuria: Patient selection and special considerations. Ther. Clin. Risk Manag. 2016, 12, 1161–1170. [Google Scholar] [CrossRef]
- Wong, E.K.; Kavanagh, D. Anticomplement C5 therapy with eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Transl. Res. 2015, 165, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Chemaitelly, H.; Nagelkerke, N.; Ayoub, H.H.; Coyle, P.; Tang, P.; Yassine, H.M.; Al-Khatib, H.A.; Smatti, M.K.; Hasan, M.R.; Al-Kanaani, Z.; et al. Duration of immune protection of SARS-CoV-2 natural infection against reinfection. J. Travel Med. 2022, 30, taac109. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, V.S.; Magnusen, A.F.; Rani, R.; Marsili, L.; Slavotinek, A.M.; Prows, D.R.; Hopkin, R.J.; McKay, M.A.; Pandey, M.K. Targeting the Complement–Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy. Int. J. Mol. Sci. 2022, 23, 14340. https://doi.org/10.3390/ijms232214340
Trivedi VS, Magnusen AF, Rani R, Marsili L, Slavotinek AM, Prows DR, Hopkin RJ, McKay MA, Pandey MK. Targeting the Complement–Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy. International Journal of Molecular Sciences. 2022; 23(22):14340. https://doi.org/10.3390/ijms232214340
Chicago/Turabian StyleTrivedi, Vyoma Snehal, Albert Frank Magnusen, Reena Rani, Luca Marsili, Anne Michele Slavotinek, Daniel Ray Prows, Robert James Hopkin, Mary Ashley McKay, and Manoj Kumar Pandey. 2022. "Targeting the Complement–Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy" International Journal of Molecular Sciences 23, no. 22: 14340. https://doi.org/10.3390/ijms232214340
APA StyleTrivedi, V. S., Magnusen, A. F., Rani, R., Marsili, L., Slavotinek, A. M., Prows, D. R., Hopkin, R. J., McKay, M. A., & Pandey, M. K. (2022). Targeting the Complement–Sphingolipid System in COVID-19 and Gaucher Diseases: Evidence for a New Treatment Strategy. International Journal of Molecular Sciences, 23(22), 14340. https://doi.org/10.3390/ijms232214340