
Leucine and Arginine Availability Modulate Mouse Embryonic Stem Cell Proliferation and Metabolism



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
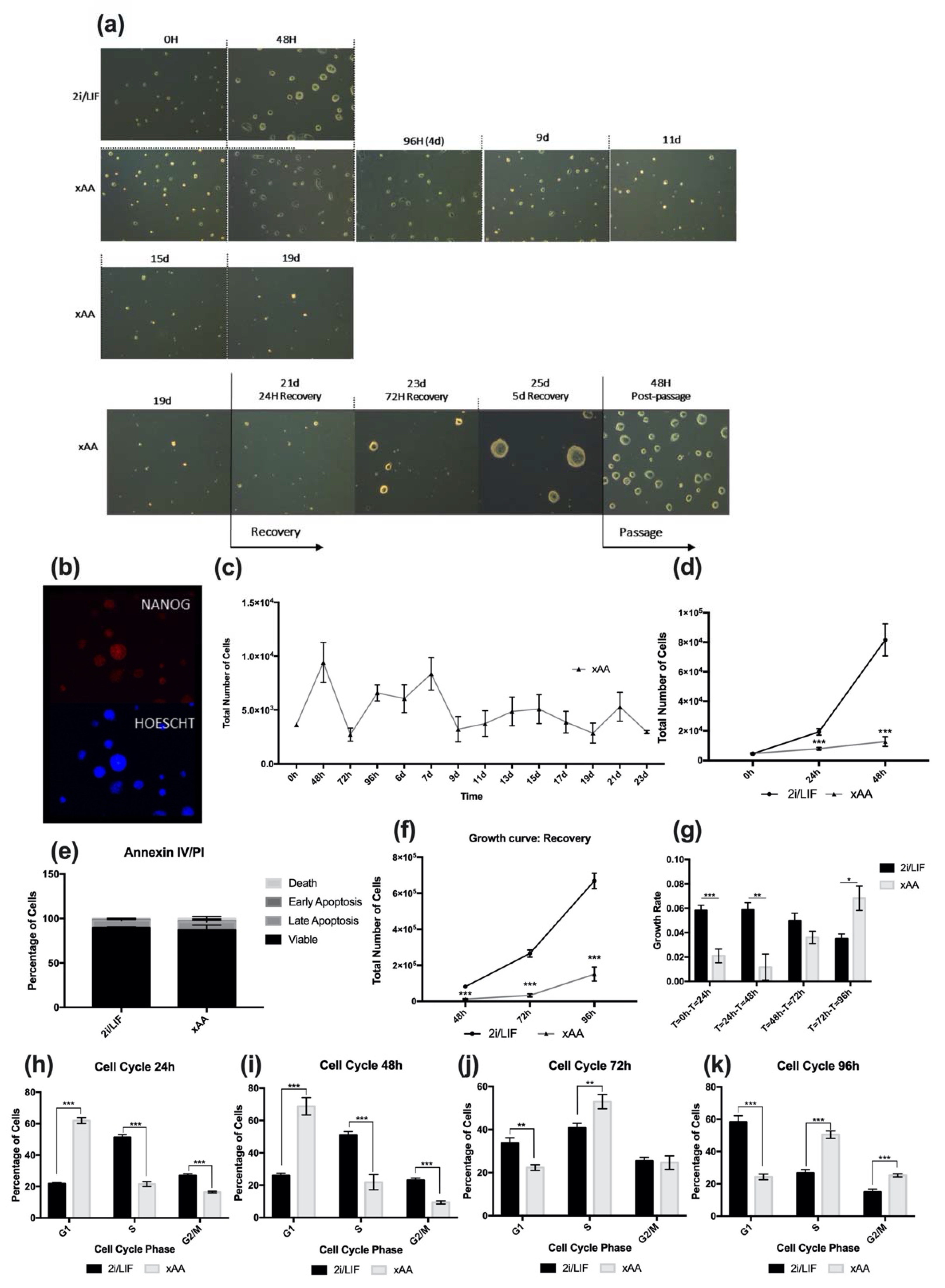
2.1. Leucine and Arginine Withdrawal Regulates mESC Self-Renewal and Viability
2.2. Effects of LIF Depletion along with Leucine and Arginine on mESC Self-Renewal, Cell Cycle, and Viability
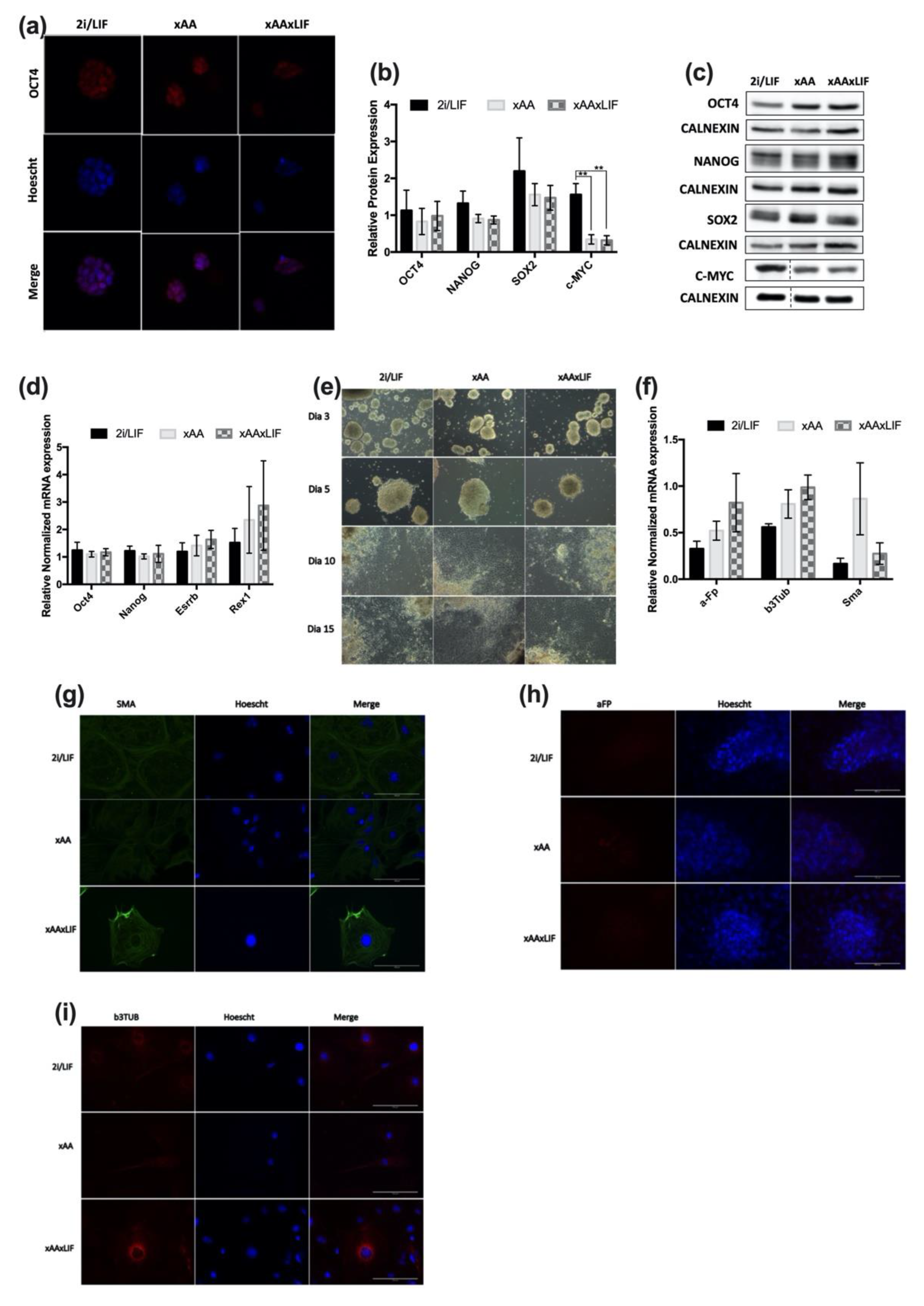
2.3. Leucine and Arginine Withdrawal Does Not Affect Pluripotency
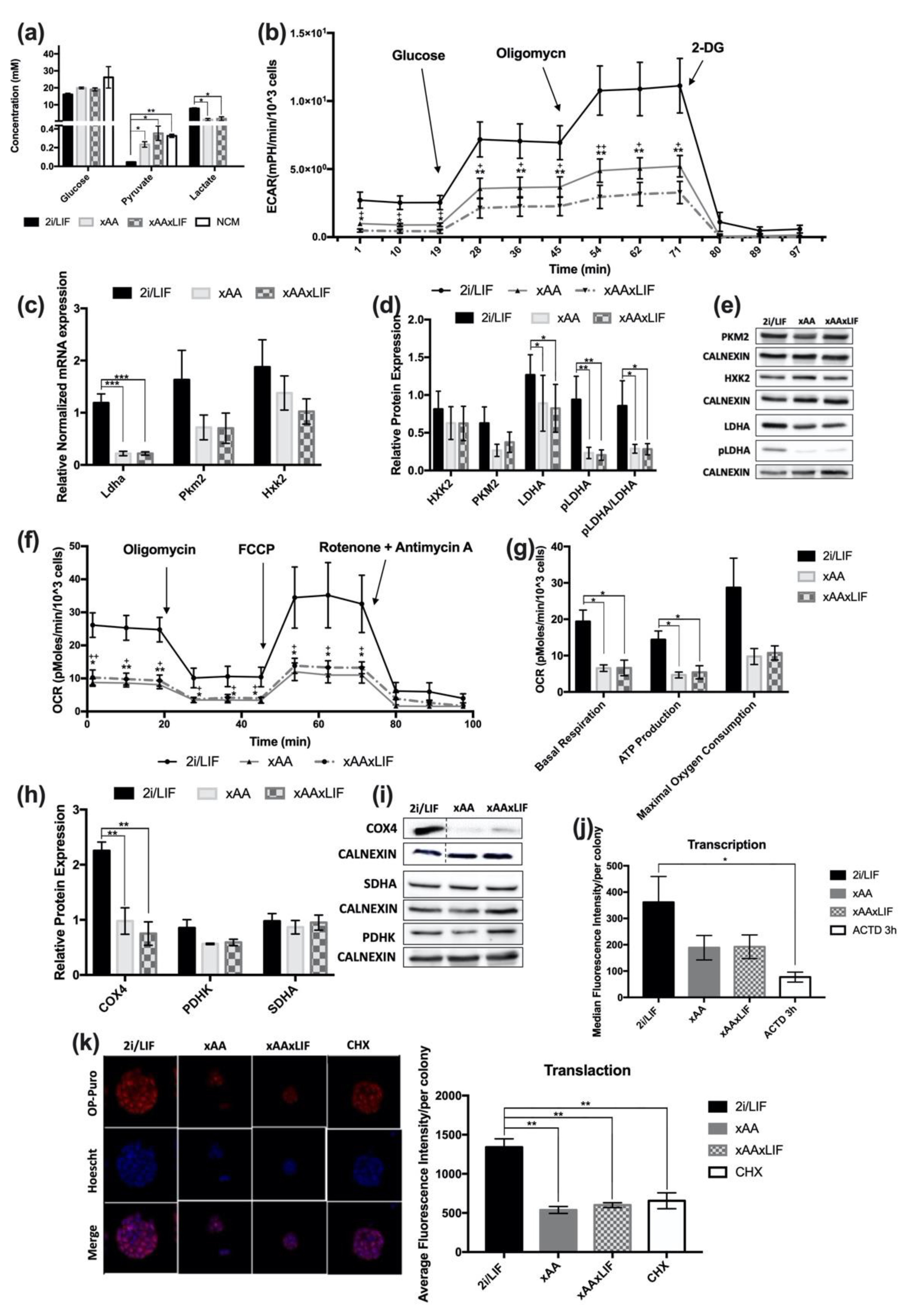
2.4. Lack of Leucine and Arginine Downregulates Glycolytic and Oxidative Function in mESCs as well as Overall Translation Initiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Maintenance
4.2. Cell Proliferation Assay
4.3. Flow Cytometry
4.4. Immunocytochemistry and Protein Translation Assay
4.5. RNA Isolation, DNA Cleanup, cDNA Synthesis, and Quantitative Real-Time PCR (qRT-PCR)
4.6. Protein Extraction and Quantification and Western Blotting
4.7. Quantification of Extracellular Metabolites in Media
4.8. Embryoid Body Assay
4.9. Live-Cell Metabolic Assays
5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chatot, C.L.; Lewis, J.L.; Torres, I.; Ziomek, C.A. Development of 1-cell embryos from different strains of mice in CZB medium. Biol. Reprod. 1990, 42, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Bazer, F.W.; Johnson, G.A.; Wu, G. Amino acids and conceptus development during the peri-implantation period of pregnancy. In Cell Signaling During Mammalian Early Embryo Development; Springer: New York, NY, USA, 2015; Volume 843, pp. 23–52. [Google Scholar] [CrossRef]
- Eckert, J.J.; Velazquez, M.A.; Fleming, T.P. Cell signalling during blastocyst morphogenesis. In Cell Signaling During Mammalian Early Embryo Development; Springer: New York, NY, USA, 2015; Volume 843, pp. 1–21. [Google Scholar] [CrossRef]
- Puscheck, E.E.; Awonuga, A.O.; Yang, Y.; Jiang, Z.; Rappolee, D.A. Molecular biology of the stress response in the early embryo and its stem cells. In Cell Signaling During Mammalian Early Embryo Development; Springer: New York, NY, USA, 2015; Volume 843, pp. 77–128. [Google Scholar] [CrossRef]
- Gardner, D.K.; Harvey, A.J. Blastocyst metabolism. Reprod. Fertil. Dev. 2015, 27, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, L.J. Amino Acid Transport and Metabolism Regulate Early Embryo Development: Species Differences, Clinical Significance, and Evolutionary Implications. Cells 2021, 10, 3154. [Google Scholar] [CrossRef] [PubMed]
- Wale, P.L.; Gardner, D.K. Oxygen regulates amino acid turnover and carbohydrate uptake during the preimplantation period of mouse embryo development. Biol. Reprod. 2012, 87, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Yu, Y. Metabolite availability as a window to view the early embryo microenvironment in vivo. Mol. Reprod. Dev. 2017, 84, 1027–1038. [Google Scholar] [CrossRef]
- Gwatkin, R.B.L. Amino acid requirements for attachment and outgrowth of the mouse blastocyst in vitro. J. Cell. Physiol. 1966, 68, 335–343. [Google Scholar] [CrossRef]
- Murakami, M.; Ichisaka, T.; Maeda, M.; Oshiro, N.; Hara, K.; Edenhofer, F.; Kiyama, H.; Yonezawa, K.; Yamanaka, S. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol. Cell. Biol. 2004, 24, 6710–6718. [Google Scholar] [CrossRef]
- Bazer, F.W.; Song, G.; Kim, J.; Erikson, D.W.; Johnson, G.A.; Burghardt, R.C.; Gao, H.; Satterfield, M.C.; Spencer, T.E.; Wu, G. Mechanistic mammalian target of rapamycin (MTOR) cell signaling: Effects of select nutrients and secreted phosphoprotein 1 on development of mammalian conceptuses. Mol. Cell. Endocrinol. 2012, 354, 22–33. [Google Scholar] [CrossRef]
- González, I.M.; Martin, P.M.; Burdsal, C.; Sloan, J.L.; Mager, S.; Harris, T.; Sutherland, A.E. Leucine and arginine regulate trophoblast motility through mTOR-dependent and independent pathways in the preimplantation mouse embryo. Dev. Biol. 2012, 361, 286–300. [Google Scholar] [CrossRef]
- Zeng, X.; Mao, X.; Huang, Z.; Wang, F.; Wu, G.; Qiao, S. Arginine enhances embryo implantation in rats through PI3K/PKB/mTOR/NO signaling pathway during early pregnancy. Reproduction 2013, 145, 1–7. [Google Scholar] [CrossRef]
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Wang, T. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Song, G.; Wu, G.; Gao, H.; Johnson, G.A.; Bazer, F.W. Arginine, Leucine, and Glutamine Stimulate Proliferation of Porcine Trophectoderm Cells Through the MTOR-RPS6K-RPS6-EIF4EBP1 Signal Transduction Pathway1. Biol. Reprod. 2013, 88, 113. [Google Scholar] [CrossRef] [PubMed]
- Naeslund, G. The effect of glucose, arginine and leucine deprivation on mouse blastocyst outgrowth in vitro. Upsala J. Med. Sci. 1979, 84, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Burghardt, R.C.; Wu, G.; Johnson, G.A.; Spencer, T.E.; Bazer, F.W. Select nutrients in the ovine uterine lumen. VII. Effects of arginine, leucine, glutamine, and glucose on trophectoderm cell signaling, proliferation, and migration. Biol. Reprod. 2011, 84, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Ogawa, K.; Shimosato, D.; Adachi, K. A parallel circuit of LIF signaling pathways maintains pluripotency of mouse ES cells. Nature 2009, 460, 118–122. [Google Scholar] [CrossRef]
- Martello, G.; Bertone, P.; Smith, A. Identification of the missing pluripotency mediator downstream of leukaemia inhibitory factor. EMBO J. 2013, 32, 2561–2574. [Google Scholar] [CrossRef]
- Stewart, C.L.; Kaspar, P.; Brunet, L.J.; Bhatt, H.; Gadi, I.; Abbondanzo, S.J. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature 1992, 359, 76. [Google Scholar] [CrossRef]
- Chen, J.R.; Cheng, J.G.; Shatzer, T.; Sewell, L.; Hernandez, L.; Stewart, C.L. Leukemia inhibitory factor can substitute for nidatory estrogen and is essential to inducing a receptive uterus for implantation but is not essential for subsequente embryogenesis. Endocrinology 2000, 141, 4365–4372. [Google Scholar] [CrossRef]
- Nichols, J.; Chambers, I.; Taga, T.; Smith, A. Physiological rationale for responsiveness of mouse embryonic stem cells to gp130 cytokines. Development 2001, 128, 2333–2339. [Google Scholar] [CrossRef]
- Fenelon, J.C.; Renfree, M.B. The history of the discovery of embryonic diapause in mammals. Biol. Reprod. 2018, 99, 241–251. [Google Scholar] [CrossRef]
- Zhou, W.; Choi, M.; Margineantu, D.; Margaretha, L.; Hesson, J.; Cavanaugh, C.; Blau, C.A.; Horwitz, M.S.; Hockenbery, D.; Ware, C.; et al. HIF1a induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J. 2012, 31, 2103–2116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ratanasirintrawoot, S.; Chandrasekaran, S.; Wu, Z.; Ficarro, S.B.; Yu, C.; Ross, C.A.; Cacchiarelli, D.; Xia, Q.; Seligson, M.; et al. LIN28 Regulates Stem Cell Metabolism and Conversion to Primed Pluripotency. Cell Stem Cell 2016, 19, 66–80. [Google Scholar] [CrossRef]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic control over mTOR-dependent diapause-like state. Dev. Cell 2020, 52, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Eckert, J.J.; Porter, R.; Watkins, A.J.; Burt, E.; Brooks, S.; Leese, H.J.; Humpherson, P.G.; Cameron, I.T.; Fleming, T.P. Metabolic induction and early responses of mouse blastocyst developmental programming following maternal low protein diet affecting life-long health. PLoS ONE 2012, 7, e52791. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Alexander, P.; Wu, L.; Hammer, R.; Cleaver, O.; McKnight, S.L. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009, 325, 435–439. [Google Scholar] [CrossRef] [PubMed]
- To, C.Y.; Freeman, M.; Van Winkle, L.J. Consumption of a Branched-Chain Amino Acid (BCAA) during Days 2–10 of Pregnancy Causes Abnormal Fetal and Placental Growth: Implications for BCAA Supplementation in Humans. Int. J. Environ. Res. Public Health 2020, 17, 2445. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, L.J.; Ryznar, R. One-carbon metabolism regulates embryonic stem cell fate through epigenetic DNA and histone modifications: Implications for transgenerational metabolic disorders in adults. Front. Cell Dev. Biol. 2019, 7, 300. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.B.; Ozsoy, S.; Zada, M.; Zada, M.; Zamfirescu, R.C.; Todorova, M.G.; Day, M.L. Selected amino acids promote mouse pre-implantation embryo development in a growth factor-like manner. Front. Physiol. 2020, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X.; et al. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2, e201900413. [Google Scholar] [CrossRef] [PubMed]
- Van Winkle, L.J.; Haghighat, N.; Campione, A.L. Glycine protects preimplantation mouse conceptuses from a detrimental effect on development of the inorganic ions in oviductal fluid. J. Exp. Zool. 1990, 253, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, T.; Loos, R.; Bertone, P.; Smith, A.; Nichols, J. The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat. Cell Biol. 2014, 16, 513–525. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, M.; Baumgärtner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Sutherland, A.E.; Van Winkle, L.J. Amino acid transport regulates blastocyst implantation. Biol. Reprod. 2003, 69, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Ying, Q.L.; Wray, J.; Nichols, J.; Batlle-Morera, L.; Doble, B.; Woodgett, J.; Cohen, F.; Smith, A. The ground state of embryonic stem cell self-renewal. Nature 2008, 453, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Wray, J.; Kalkan, T.; Smith, A.G. The ground state of pluripotency. Biochem. Soc. Trans. 2010, 38, 1027–1032. [Google Scholar] [CrossRef]
- Wray, J.; Kalkan, T.; Gomez-Lopez, S.; Eckardt, D.; Cook, A.; Kemler, R.; Smith, A. Inhibition of glycogen synthase kinase-3 alleviates Tcf3 repression of the pluripotency network and increases embryonic stem cell resistance to differentiation. Nat. Cell Biol. 2011, 13, 838–845. [Google Scholar] [CrossRef]
- Tai, C.I.; Ying, Q.L. Gbx2, a LIF/Stat3 target, promotes reprogramming to and retention of the pluripotent ground state. J. Cell Sci. 2013, 126, 1093–1098. [Google Scholar] [CrossRef]
- Ye, S.; Li, P.; Tong, C.; Ying, Q.L. Embryonic stem cell self-renewal pathways converge on the transcription factor Tfcp2l1. EMBO J. 2013, 32, 2548–2560. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, B.; Wang, S.; Wu, W.; Wang, Q.; Chen, Y.; Kong, S.; Lu, J.; Tang, Z.; Ran, H.; et al. Integral proteomic analysis of blastocysts reveals key molecular machinery governing embryonic diapause and reactivation for implantation in mice. Biol. Reprod. 2014, 90, 52. [Google Scholar] [CrossRef] [PubMed]
- Renfree, M.B.; Fenelon, J.C. The enigma of embryonic diapause. Development 2017, 144, 3199–3210. [Google Scholar] [CrossRef] [PubMed]
- Schiesari, L.; O’Connor, M.B. Diapause: Delaying the developmental clock in response to a changing environment. Curr. Top. Dev. Biol. 2013, 105, 213–246. [Google Scholar] [CrossRef] [PubMed]
- Hondo, E.; Stewart, C.L. Profiling gene expression in growth-arrested mouse embryos in diapause. Genome Biol. 2004, 6, 202. [Google Scholar] [CrossRef][Green Version]
- Fenelon, J.C.; Banerjee, A.; Murphy, B.D. Embryonic diapause: Development on hold. Int. J. Dev. Biol. 2014, 58, 163–174. [Google Scholar] [CrossRef]
- Hara, K.; Yonezawa, K.; Weng, Q.P.; Kozlowski, M.T.; Belham, C.; Avruch, J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J. Biol. Chem. 1998, 273, 14484–14494. [Google Scholar] [CrossRef]
- Schcolnik-Cabrera, A.; Juárez-López, D. Dual contribution of the mTOR pathway and of the metabolism of amino acids in prostate cancer. Cell. Oncol. 2022, 45, 831–859. [Google Scholar] [CrossRef]
- Desouki, M.M.; Geradts, J.; Milon, B.; Franklin, R.B.; Costello, L.C. hZip2 and hZip3 zinc transporters are down regulated in human prostate adenocarcinomatous glands. Mol. Cancer 2007, 6, 37. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Chung, T.Y.; Chu, C.Y.; Wang, H.J.; Hsiao, P.W.; Yeh, S.D.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef]
- Folmes, C.D.L.; Martinez-Fernandez, A.; Faustino, R.S.; Yamada, S.; Perez-Terzic, C.; Nelson, T.J.; Terzic, A. Nuclear reprogramming with c-Myc potentiates glycolytic capacity of derived induced pluripotent stem cells. J. Cardiovasc. Transl. Res 2013, 6, 10–21. [Google Scholar] [CrossRef]
- Cao, Y.; Guo, W.-T.; Tian, S.; He, X.; Wang, X.-W.; Liu, X.; Gu, K.-L.; Ma, X.; Huang, D.; Hu, L.; et al. miR-290/371-Mbd2-Myc circuit regulates glycolytic metabolism to promote pluripotency. EMBO J. 2015, 34, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Cliff, T.S.; Dalton, S. Metabolic switching and cell fate decisions: Implications for pluripotency, reprogramming and development. Curr. Opin. Genet. Dev. 2017, 46, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Böttinger, L.; Guiard, B.; Oeljeklaus, S.; Kulawiak, B.; Zufall, N.; Wiedemann, N.; Warscheid, B.; van der Laan, M.; Becker, T. A complex of Cox4 and mitochondrial Hsp70 plays an important role in the assembly of the cytochrome c oxidase. Mol. Biol. Cell 2013, 24, 2609–2619. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Momčilović, O.; Castro, C.; Ben-Yehudah, A.; Ramalho-Santos, J.; Navara, C. Enhancement of human embryonic stem cell pluripotency through inhibition of the mitochondrial respiratory chain. Stem Cell Res. 2009, 3, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Varum, S.; Rodrigues, A.S.; Moura, M.B.; Momcilovic, O.; Iv, C.A.E.; Ramalho-Santos, J.; Van Houten, B.; Schatten, G. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS ONE 2011, 6, e20914. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L.; Grãos, M.; Rodrigues, A.S.; Anjo, S.I.; Carvalho, R.A.; Oliveira, P.J.; Arenas, E.; Ramalho-Santos, J. Inhibition of mitochondrial complex III blocks neuronal differentiation and maintains embryonic stem cell pluripotency. PLoS ONE 2013, 8, e82095. [Google Scholar] [CrossRef]
- Rodrigues, A.S.; Correia, M.; Gomes, A.; Pereira, S.L.; Perestrelo, T.; Sousa, M.I.; Ramalho-Santos, J. Dichloroacetate, the pyruvate dehydrogenase complex and the modulation of mESC pluripotency. PLoS ONE 2015, 10, e0131663. [Google Scholar] [CrossRef]
- Rodrigues, A.S.; Pereira, S.L.; Correia, M.; Gomes, A.; Perestrelo, T.; Ramalho-Santos, J. Differentiate or die: 3-bromopyruvate and pluripotency in mouse embryonic stem cells. PLoS ONE 2015, 10, e0135617. [Google Scholar] [CrossRef]
- Sousa, M.I.; Correia, B.; Rodrigues, A.S.; Ramalho-Santos, J. Metabolic characterization of a paused-like pluripotent state. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129612. [Google Scholar] [CrossRef]
- Correia, M.; Rodrigues, A.S.; Perestrelo, T.; Pereira, S.L.; Ribeiro, M.F.; Sousa, M.I.; Ramalho-Santos, J. Different concentrations of kaempferol distinctly modulate murine embryonic stem cell function. Food Chem. Toxicol. 2016, 87, 148–156. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, B.; Sousa, M.I.; Branco, A.F.; Rodrigues, A.S.; Ramalho-Santos, J. Leucine and Arginine Availability Modulate Mouse Embryonic Stem Cell Proliferation and Metabolism. Int. J. Mol. Sci. 2022, 23, 14286. https://doi.org/10.3390/ijms232214286
Correia B, Sousa MI, Branco AF, Rodrigues AS, Ramalho-Santos J. Leucine and Arginine Availability Modulate Mouse Embryonic Stem Cell Proliferation and Metabolism. International Journal of Molecular Sciences. 2022; 23(22):14286. https://doi.org/10.3390/ijms232214286
Chicago/Turabian StyleCorreia, Bibiana, Maria Inês Sousa, Ana Filipa Branco, Ana Sofia Rodrigues, and João Ramalho-Santos. 2022. "Leucine and Arginine Availability Modulate Mouse Embryonic Stem Cell Proliferation and Metabolism" International Journal of Molecular Sciences 23, no. 22: 14286. https://doi.org/10.3390/ijms232214286
APA StyleCorreia, B., Sousa, M. I., Branco, A. F., Rodrigues, A. S., & Ramalho-Santos, J. (2022). Leucine and Arginine Availability Modulate Mouse Embryonic Stem Cell Proliferation and Metabolism. International Journal of Molecular Sciences, 23(22), 14286. https://doi.org/10.3390/ijms232214286