
De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species

 , and
, and
Abstract
1. Introduction
2. Results
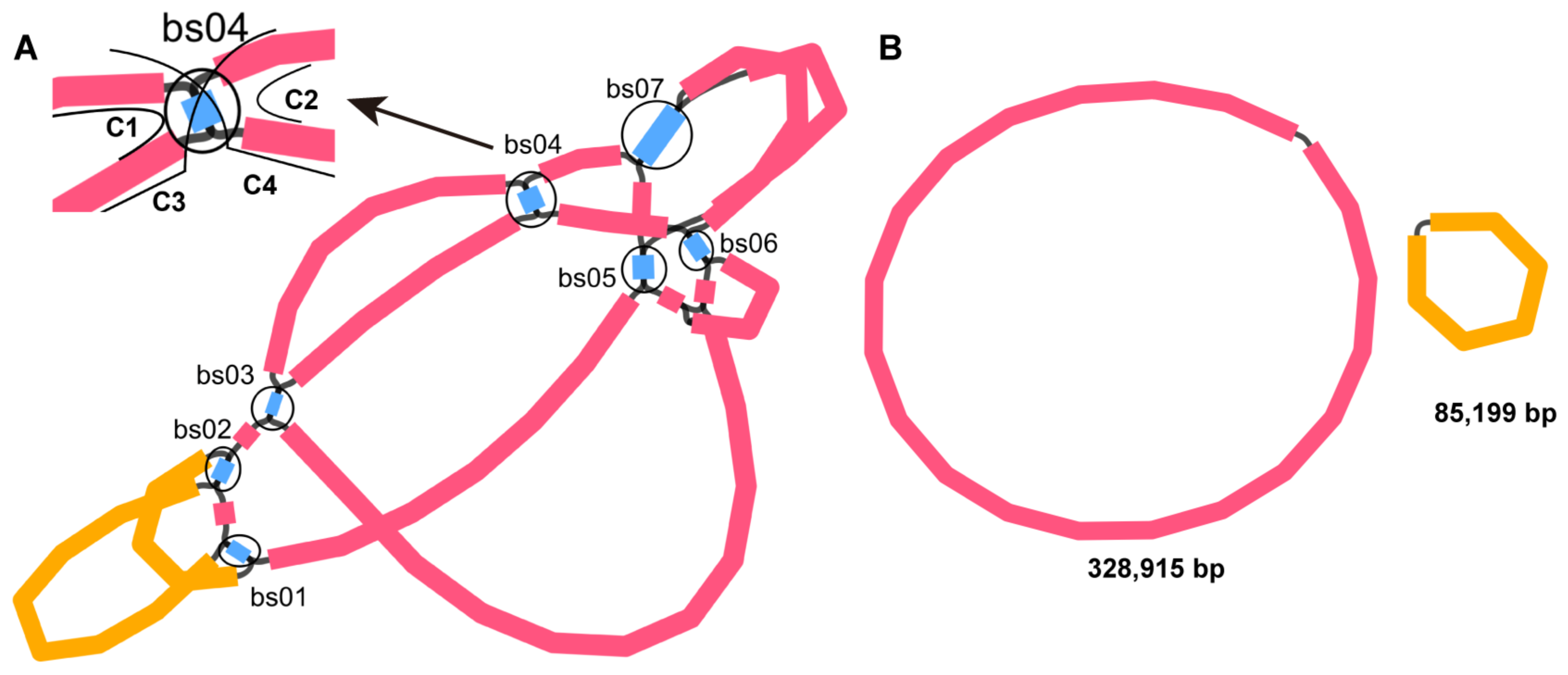
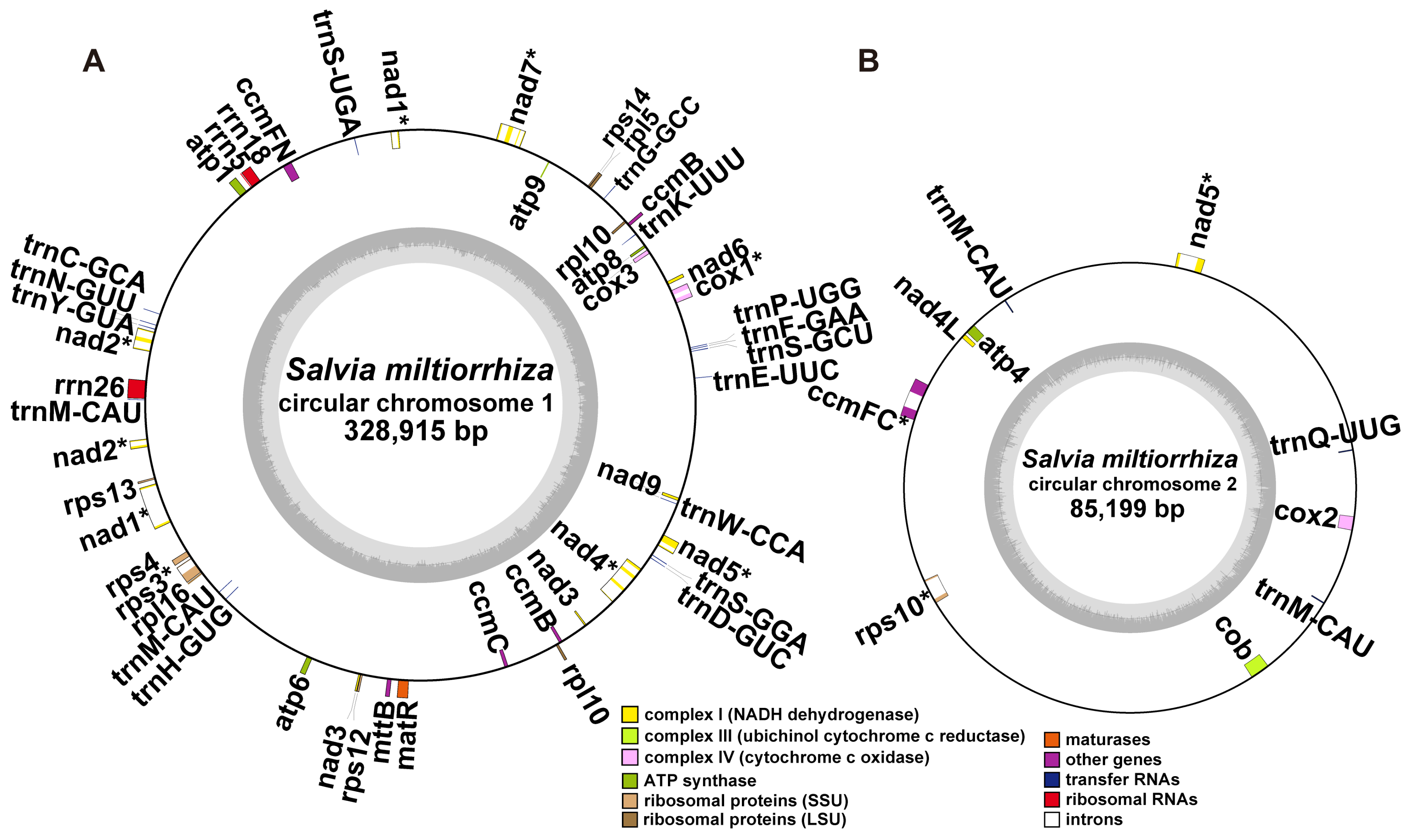
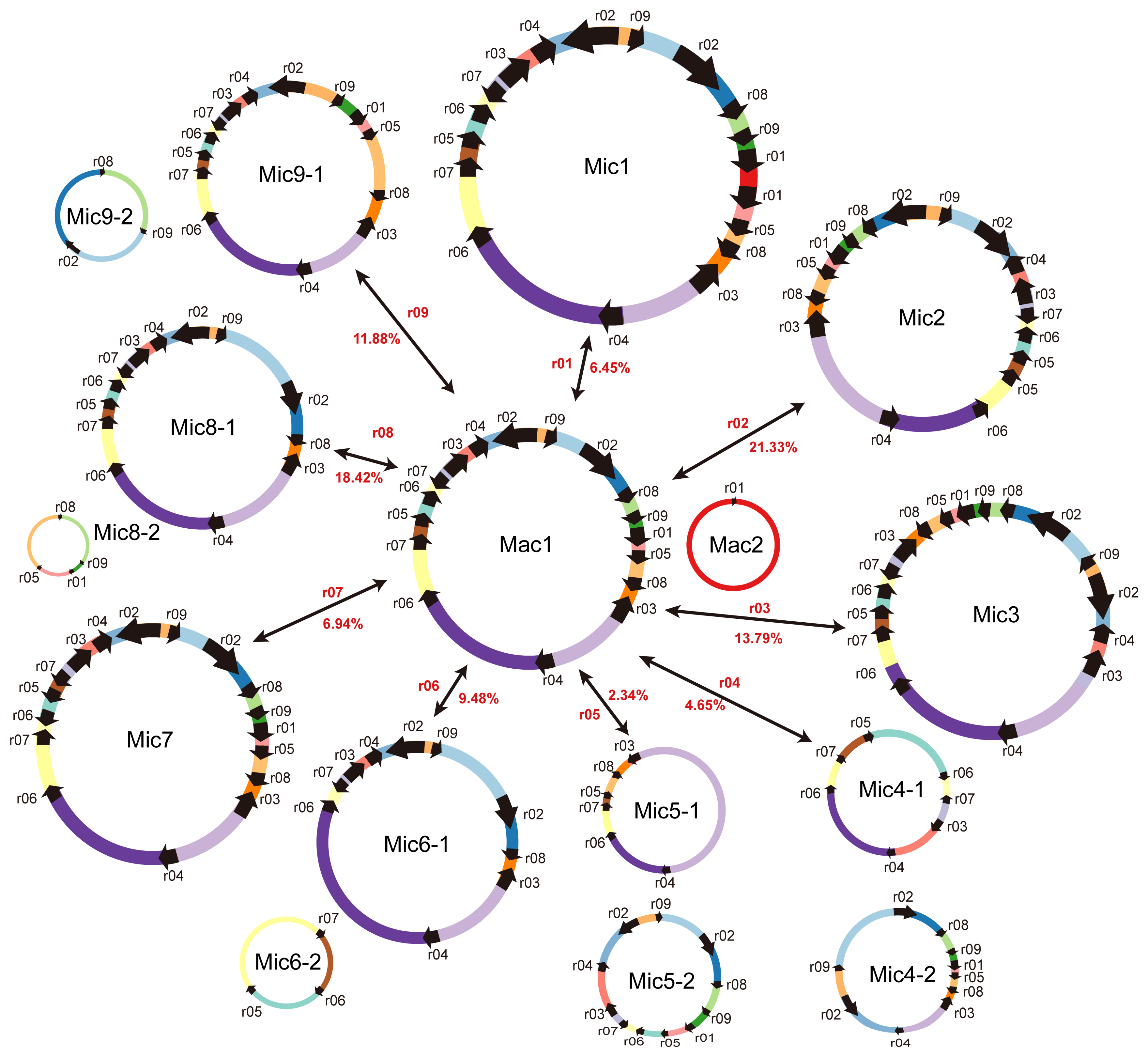
2.1. Structure Analysis of the S. miltiorrhiza Mitogenome
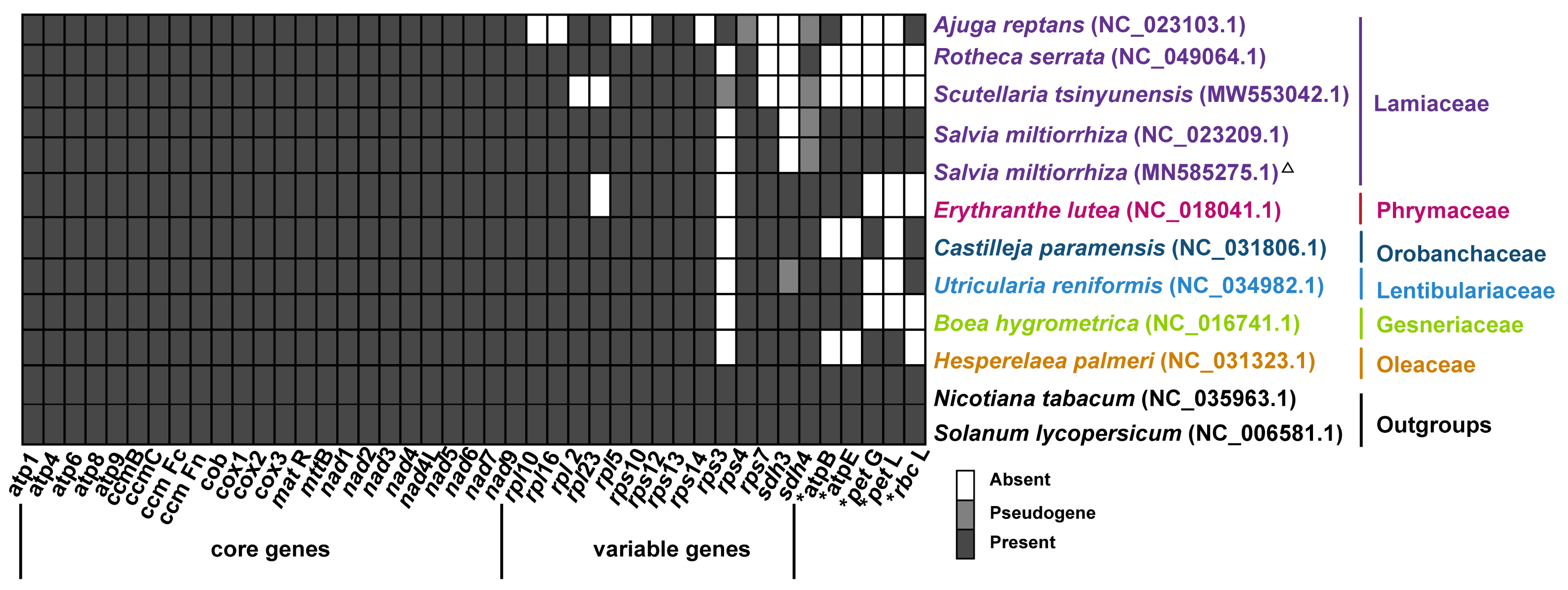
2.2. Gene Content of S. miltiorrhiza Mitogenome
2.3. Repeat Sequences in the Lamiales Mitogenomes
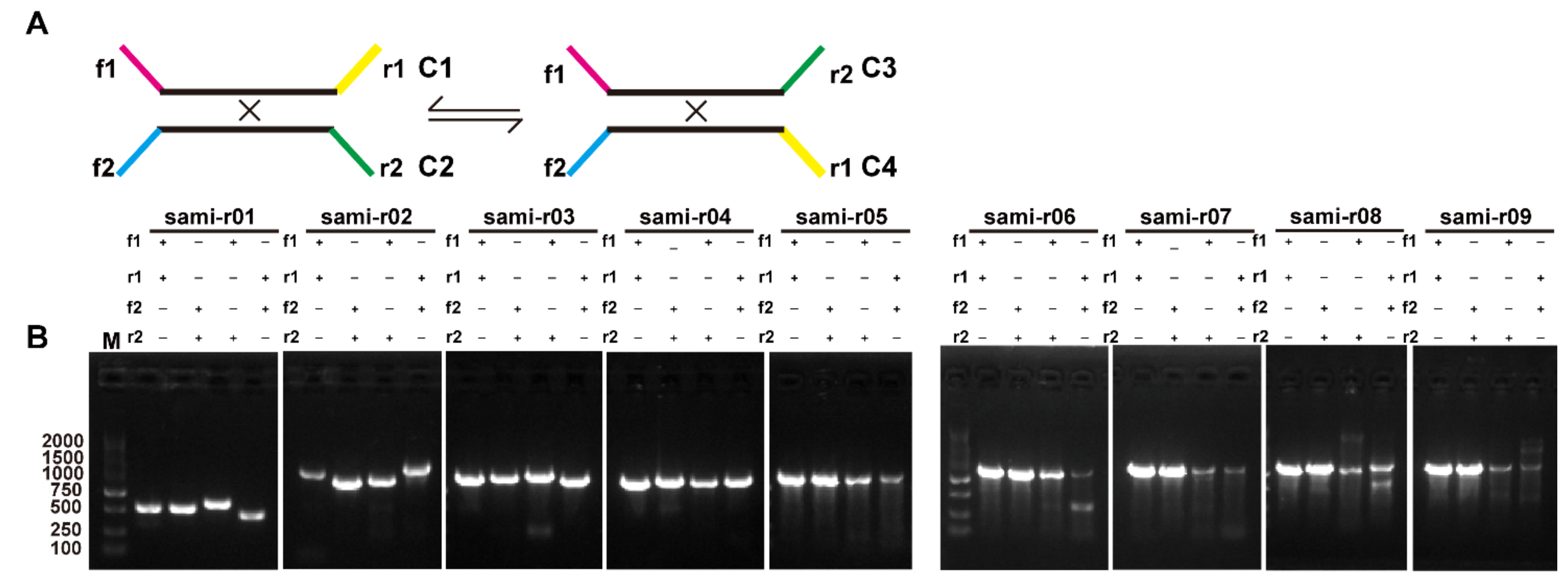
2.4. Homologous Recombination Mediated by Repeats
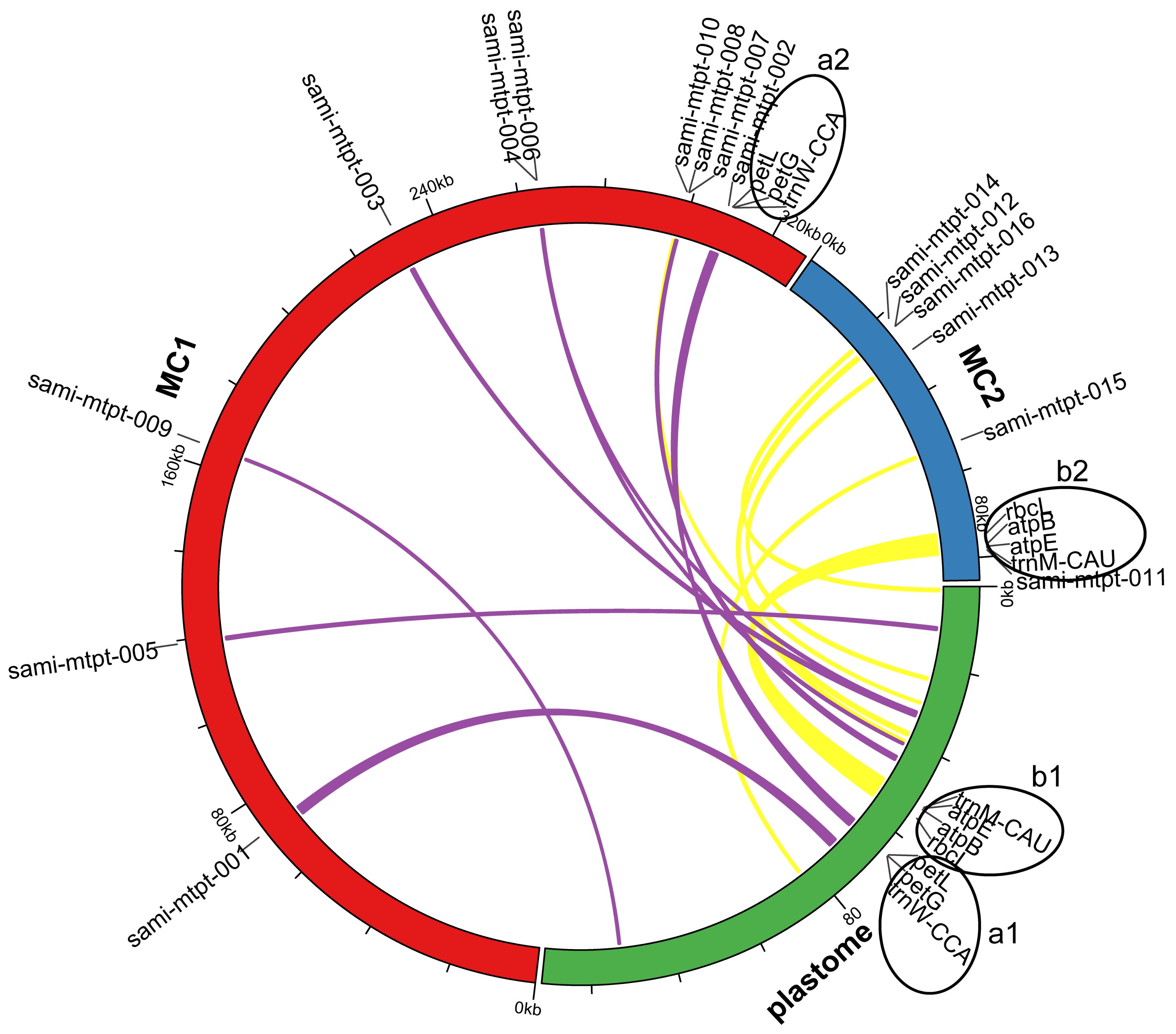
2.5. Identification of Mitochondrial Plastid Sequences (MTPTs)
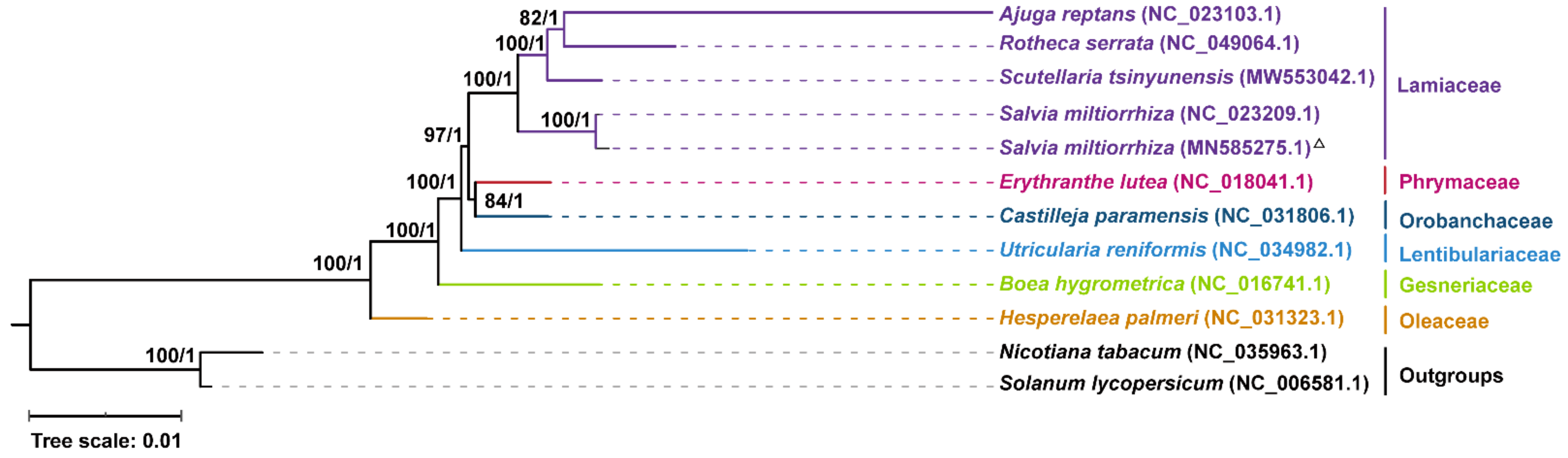
2.6. Phylogenetic Analysis
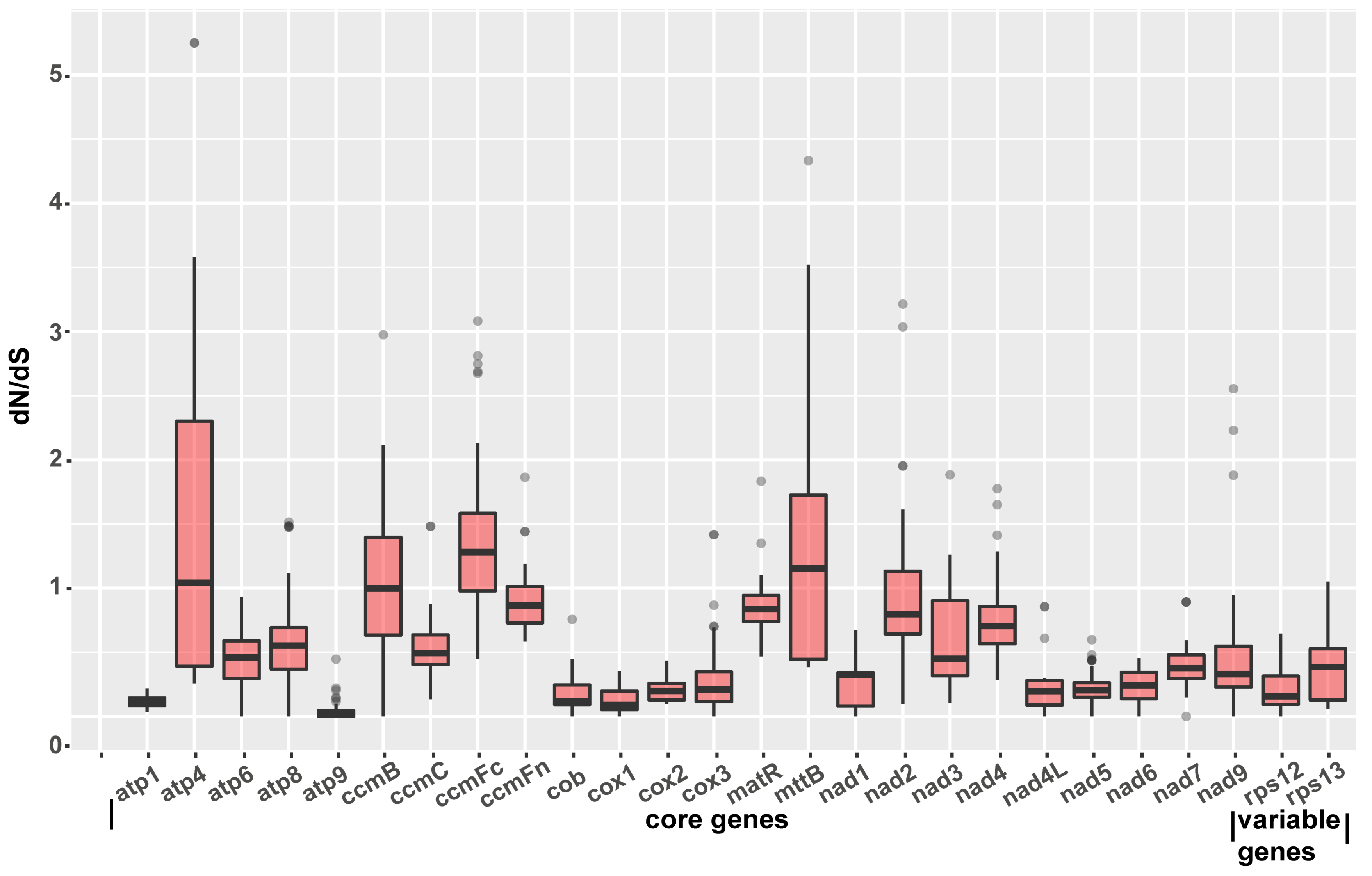
2.7. Identification of Genes under Selection
3. Discussion
3.1. Overview of the S. miltiorrhiza Mitogenome
3.2. Repeat Mediated Homologous Recombination in Lamiaceae
3.3. Current Limitation of the Plant Mitogenome Assembly Method
3.4. Future Studies of the S. miltiorrhiza Mitogenome
4. Materials and Methods
4.1. Plant Materials and DNA Extraction
4.2. DNA Sequencing and Mitogenome Assembly
4.3. Mitogenome Annotation
4.4. Repeat Sequence Analysis
4.5. Analysis of the Recombination Products
4.6. Identification of Mitochondrial Plastid Sequences (MTPTs)
4.7. Phylogenetic Analysis of the Nine Lamiales Species
4.8. Estimation of Nucleotide Substitution Rates
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mitogenome | Mitochondrial genome |
| NCBI | National Center for Biotechnology Information |
| PCG | Protein-coding genes |
| DNA | Deoxyribonucleic acid |
| RNA | Ribonucleic acid |
| C-to-U | Cytidine-to-Uridine |
| PacBio | Pacific Biosciences |
| Gb | Giga base pair |
| DBS | Double bifurcating structures |
| MC1/2 | Mitochondrial genome chromosomes |
| SSR | Simple sequence repeat |
| HSP | High-scoring pair |
| Mac | Major conformation |
| Mic | Minor conformation |
| MTPT | Mitochondrial plastid DNA |
| PCR | Polymerase chain reaction |
| sami | Salvia miltiorrhiza |
| NUMT | Nuclear mitochondrial sequence |
References
- Cheng, T.O. Danshen: A popular Chinese cardiac herbal drug. J. Am. Coll. Cardiol. 2006, 47, 1498. [Google Scholar] [CrossRef] [PubMed]
- Sui, C. Salvia miltiorrhiza Resources, Cultivation, and Breeding. In The Salvia miltiorrhiza Genome. Compendium of Plant Genomes; Lu, S., Ed.; Springer: Cham, Switzerland, 2019; pp. 17–32. [Google Scholar]
- Xu, H.-Y.; Zhang, Y.-Q.; Liu, Z.-M.; Chen, T.; Lv, C.-Y.; Tang, S.-H.; Zhang, X.-B.; Zhang, W.; Li, Z.-Y.; Zhou, R.-R. ETCM: An encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019, 47, D976–D982. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Song, J.; Luo, H.; Zhang, Y.; Li, Q.; Zhu, Y.; Xu, J.; Li, Y.; Song, C.; Wang, B.; et al. Analysis of the Genome Sequence of the Medicinal Plant Salvia miltiorrhiza. Mol. Plant 2016, 9, 949–952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Tian, Y.; Zhang, J.; Shu, L.; Yang, S.; Wang, W.; Sheng, J.; Dong, Y.; Chen, W. Hybrid de novo genome assembly of the Chinese herbal plant danshen (Salvia miltiorrhiza Bunge). Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Lin, C.; Xing, P.; Fen, Y.; Jin, H.; Zhou, C.; Gu, Y.Q.; Wang, J.; Li, X. A high-quality reference genome sequence of Salvia miltiorrhiza provides insights into tanshinone synthesis in its red rhizomes. Plant Genome 2020, 13, e20041. [Google Scholar] [CrossRef]
- Ma, Y.; Cui, G.; Chen, T.; Ma, X.; Wang, R.; Jin, B.; Yang, J.; Kang, L.; Tang, J.; Lai, C.; et al. Expansion within the CYP71D subfamily drives the heterocyclization of tanshinones synthesis in Salvia miltiorrhiza. Nat. Commun. 2021, 12, 685. [Google Scholar] [CrossRef]
- Chen, H.; Liu, C. The Chloroplast and Mitochondrial Genomes of Salvia miltiorrhiza. In The Salvia miltiorrhiza Genome; Springer: Cham, Switzerland, 2019; pp. 55–68. [Google Scholar]
- Wu, B.; Chen, H.; Shao, J.; Zhang, H.; Wu, K.; Liu, C. Identification of Symmetrical RNA Editing Events in the Mitochondria of Salvia miltiorrhiza by Strand-specific RNA Sequencing. Sci Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Rice, D.W.; Alverson, A.J.; Richardson, A.O.; Young, G.J.; Sanchez-Puerta, M.V.; Munzinger, J.; Barry, K.; Boore, J.L.; Zhang, Y.; DePamphilis, C.W. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm Amborella. Science 2013, 342, 1468–1473. [Google Scholar] [CrossRef]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes Reveal Extreme Contrasts in Gymnosperm Mitochondrial Evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Montier, L.L.C.; Deng, J.J.; Bai, Y. Number matters: Control of mammalian mitochondrial DNA copy number. J. Genet. Genom. 2009, 36, 125–131. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 1–17. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Štorchová, H.; Palmer, J.D.; Taylor, D.R. Extensive loss of translational genes in the structurally dynamic mitochondrial genome of the angiosperm Silene latifolia. BMC Evol. Biol. 2010, 10, 1–15. [Google Scholar] [CrossRef]
- Mower, J.P.; Case, A.L.; Floro, E.R.; Willis, J.H. Evidence against equimolarity of large repeat arrangements and a predominant master circle structure of the mitochondrial genome from a monkeyflower (Mimulus guttatus) lineage with cryptic CMS. Genome Biol. Evol. 2012, 4, 670–686. [Google Scholar] [CrossRef]
- Burger, G.; Gray, M.W.; Lang, B.F. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef]
- Handa, H. The complete nucleotide sequence and RNA editing content of the mitochondrial genome of rapeseed (Brassica napus L.): Comparative analysis of the mitochondrial genomes of rapeseed and Arabidopsis thaliana. Nucleic Acids Res. 2003, 31, 5907–5916. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhao, C.; Zhang, S.; Zhang, L.; Wu, H.; Liu, H.; Zhu, R.; Jia, Y.; Goffinet, B.; Liu, Y. Mitochondrial genomes of the early land plant lineage liverworts (Marchantiophyta): Conserved genome structure, and ongoing low frequency recombination. BMC Genom. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Wu, Z.Q.; Liao, X.Z.; Zhang, X.N.; Tembrock, L.R.; Broz, A. Genomic architectural variation of plant mitochondria—A review of multichromosomal structuring. J. Syst. Evol. 2020, 60, 160–168. [Google Scholar] [CrossRef]
- Sloan, D.B.; Wu, Z.; Sharbrough, J. Correction of persistent errors in Arabidopsis reference mitochondrial genomes. Plant. Cell 2018, 30, 525–527. [Google Scholar] [CrossRef]
- Tsujimura, M.; Kaneko, T.; Sakamoto, T.; Kimura, S.; Shigyo, M.; Yamagishi, H.; Terachi, T. Multichromosomal structure of the onion mitochondrial genome and a transcript analysis. Mitochondrion 2019, 46, 179–186. [Google Scholar] [CrossRef]
- Cossetin, L.F.; Santi, E.M.T.; Cossetin, J.F.; Dillmann, J.B.; Baldissera, M.D.; Garlet, Q.I.; de Souza, T.P.; Loebens, L.; Heinzmann, B.M.; Machado, M.M. In vitro safety and efficacy of lavender essential oil (Lamiales: Lamiaceae) as an insecticide against houseflies (Diptera: Muscidae) and blowflies (Diptera: Calliphoridae). J. Econ. Entomol. 2018, 111, 1974–1982. [Google Scholar] [CrossRef]
- Pathak, N.; Rai, A.; Kumari, R.; Bhat, K. Value addition in sesame: A perspective on bioactive components for enhancing utility and profitability. Pharmacogn. Rev. 2014, 8, 147. [Google Scholar] [PubMed]
- Zhu, A.; Guo, W.; Jain, K.; Mower, J.P. Unprecedented heterogeneity in the synonymous substitution rate within a plant genome. Mol. Biol. Evol. 2014, 31, 1228–1236. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Hu, S.; Yu, J. An efficient procedure for plant organellar genome assembly, based on whole genome data from the 454 GS FLX sequencing platform. Plant Methods 2011, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Van de Paer, C.; Hong-Wa, C.; Jeziorski, C.; Besnard, G. Mitogenomics of Hesperelaea, an extinct genus of Oleaceae. Gene 2016, 594, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.R.; Alvarenga, D.O.; Aranguren, Y.; Penha, H.A.; Fernandes, C.C.; Pinheiro, D.G.; Oliveira, M.T.; Michael, T.P.; Miranda, V.F.O.; Varani, A.M. The mitochondrial genome of the terrestrial carnivorous plant Utricularia reniformis (Lentibulariaceae): Structure, comparative analysis and evolutionary land.marks. PLoS ONE 2017, 12, e0180484. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 1–16. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Adams, K.L.; Qiu, Y.-L.; Stoutemyer, M.; Palmer, J.D. Punctuated evolution of mitochondrial gene content: High and variable rates of mitochondrial gene loss and transfer to the nucleus during angiosperm evolution. Proc. Natl. Acad. Sci. USA 2002, 99, 9905–9912. [Google Scholar] [CrossRef]
- Sperisen, C.; Büchler, U.; Gugerli, F.; Mátyás, G.; Geburek, T.; Vendramin, G. Tandem repeats in plant mitochondrial genomes: Application to the analysis of population differentiation in the conifer Norway spruce. Mol. Ecol 2001, 10, 257–263. [Google Scholar] [CrossRef]
- Nishikawa, T.; Vaughan, D.A.; Kadowaki, K.-I. Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theor. Appl. Genet. 2005, 110, 696–705. [Google Scholar] [CrossRef]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genomics 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Wang, X.-C.; Chen, H.; Yang, D.; Liu, C. Diversity of mitochondrial plastid DNAs (MTPTs) in seed plants. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2018, 29, 635–642. [Google Scholar] [CrossRef]
- Group, A.P.; Chase, M.W.; Christenhusz, M.J.; Fay, M.F.; Byng, J.; Judd, W.; Soltis, D.; Mabberley, D.; Sennikov, A.; Soltis, P. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot J. Linn. Soc. 2016, 181, 1–20. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ’master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Shearman, J.R.; Sonthirod, C.; Naktang, C.; Pootakham, W.; Yoocha, T.; Sangsrakru, D.; Jomchai, N.; Tragoonrung, S.; Tangphatsornruang, S. The two chromosomes of the mitochondrial genome of a sugarcane cultivar: Assembly and recombination analysis using long PacBio reads. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kazama, T.; Toriyama, K. Whole Mitochondrial Genome Sequencing and Re-Examination of a Cytoplasmic Male Sterility-Associated Gene in Boro-Taichung-Type Cytoplasmic Male Sterile Rice. PLoS ONE 2016, 11, e0159379. [Google Scholar] [CrossRef]
- Varré, J.-S.; d’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M. Complete sequence, multichromosomal architecture and transcriptome analysis of the Solanum tuberosum mitochondrial genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef] [PubMed]
- Emerman, A.B.; Bowman, S.K.; Barry, A.; Henig, N.; Patel, K.M.; Gardner, A.F.; Hendrickson, C.L. NEBNext Direct: A Novel, Rapid, Hybridization-Based Approach for the Capture and Library Conversion of Genomic Regions of Interest. Curr. Protoc. Mol. Biol. 2017, 119, 7–30. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Lee, E.; Harris, N.; Gibson, M.; Chetty, R.; Lewis, S. Apollo: A community resource for genome annotation editing. Bioinformatics 2009, 25, 1836–1837. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D. Basic local alignment search tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet--next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In Proceedings of the 2011 TeraGrid Conference: Extreme digital discovery, Salt Lake City, UT, USA 18–21 July 2011; Association for Computing Machinery: New York, NY, USA, 2011; pp. 1–8. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. Wiley Interdiscip Rev. Comput Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID of the HSP | ID of the DBS | Identity (%) | Alignment Length | Numbers of Mismatches | Numbers of Gap Openings | Positions of Repeat Copy 1 b | Positions of Repeat Copy 2 | E-Value | Type | Numbers of Long Reads Mapped to Each Conformation | Recombination Frequency (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | Mac1 | Mac2 | Mic1 | Mic2 | |||||||||
| r01 a | bs02 | 100 | 127 | 1 | 1 | 328,831 | 328,915 | 85,115 (MC2) | 85199 (MC2) | 3.51 × 10−37 | direct | 70 | 46 | 5 | 3 | 6.45% |
| r02 a | bs05 | 99.853 | 682 | 1 | 0 | 279,954 | 280,635 | 235,468 | 234,787 | 0 | inverted | 31 | 28 | 5 | 11 | 21.33% |
| r03 a | bs04 | 100 | 369 | 0 | 0 | 223,308 | 223,676 | 27,513 | 27,145 | 0 | inverted | 28 | 22 | 5 | 3 | 13.79% |
| r04 a | bs06 | 95.312 | 192 | 5 | 4 | 232,877 | 233,068 | 78,659 | 78,846 | 1.53 × 10−80 | direct | 24 | 17 | 1 | 1 | 4.65% |
| r05 a | bs03 | 100 | 87 | 0 | 0 | 185,367 | 185,453 | 1433 | 1519 | 2.72 × 10−38 | direct | 65 | 60 | 2 | 1 | 2.34% |
| r06 a | NA | 95.139 | 144 | 5 | 2 | 191,145 | 191,287 | 141,672 | 141,814 | 9.49 × 10−58 | direct | 46 | 59 | 6 | 5 | 9.48% |
| r07 a | NA | 91.176 | 136 | 11 | 1 | 175,216 | 175,351 | 209,605 | 209,471 | 5.80 × 10−45 | inverted | 30 | 37 | 3 | 2 | 6.94% |
| r08 a | NA | 98.649 | 74 | 1 | 0 | 300,772 | 300,845 | 19,641 | 19714 | 2.13 × 10−29 | direct | 33 | 29 | 11 | 3 | 18.42% |
| r09 a | NA | 98.305 | 59 | 0 | 1 | 311,788 | 311,846 | 252,783 | 252,840 | 1.67 × 10−20 | direct | 41 | 48 | 9 | 3 | 11.88% |
| r10 | bs07 | 99.966 | 5835 | 1 | 1 | 269,937 | 275,770 | 42,128 | 36,294 | 0 | inverted | 15 | 10 | 15 | 12 | 51.92% |
| r11 | bs01 | 94.828 | 116 | 5 | 1 | 27,842 | 27,957 | 326,567 (MC2) | 326,681 (MC2) | 1.54 × 10−44 | direct | 61 | 65 | 0 | 1 | 0.79% |
| Group of Genes | Name of Genes | |
|---|---|---|
| Core genes | ATP synthase | atp1, atp4, atp6, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFca, ccmFn | |
| Ubichinol cytochrome c reductase | cob | |
| Cytochrome c oxidase | cox1a, cox2a, cox3 | |
| Maturases | matR | |
| Transport membrane protein | mttB | |
| NADH dehydrogenase | nad1c, nad2c, nad3, nad4b, nad4L, nad5c, nad6, nad7b, nad9 | |
| Variable genes | Ribosomal protein large subunit | rpl5, rpl10, rpl16 |
| Ribosomal protein small subunit | rps3a, rps4, rps10a, rps12, rps13, rps14 | |
| rRNA genes | Ribosomal RNAs | rrn5, rrn18, rrn26 |
| tRNA genes | Transfer RNA | trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnH-GUG, trnK-UUU, trnM-CAU, trnM-CAU, trnN-GUU, trnP-UGG, trnS-GCU, trnS-GGA, trnS-UGA, trnW-CCA, trnY-GUA, trnQ-UUG, trnM-CAU, trnM-CAU |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Chen, H.; Ni, Y.; Li, J.; Cai, Y.; Ma, B.; Yu, J.; Wang, J.; Liu, C. De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. Int. J. Mol. Sci. 2022, 23, 14267. https://doi.org/10.3390/ijms232214267
Yang H, Chen H, Ni Y, Li J, Cai Y, Ma B, Yu J, Wang J, Liu C. De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. International Journal of Molecular Sciences. 2022; 23(22):14267. https://doi.org/10.3390/ijms232214267
Chicago/Turabian StyleYang, Heyu, Haimei Chen, Yang Ni, Jingling Li, Yisha Cai, Binxin Ma, Jing Yu, Jiehua Wang, and Chang Liu. 2022. "De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species" International Journal of Molecular Sciences 23, no. 22: 14267. https://doi.org/10.3390/ijms232214267
APA StyleYang, H., Chen, H., Ni, Y., Li, J., Cai, Y., Ma, B., Yu, J., Wang, J., & Liu, C. (2022). De Novo Hybrid Assembly of the Salvia miltiorrhiza Mitochondrial Genome Provides the First Evidence of the Multi-Chromosomal Mitochondrial DNA Structure of Salvia Species. International Journal of Molecular Sciences, 23(22), 14267. https://doi.org/10.3390/ijms232214267