
Macrophage-, Dendritic-, Smooth Muscle-, Endothelium-, and Stem Cells-Derived Foam Cells in Atherosclerosis



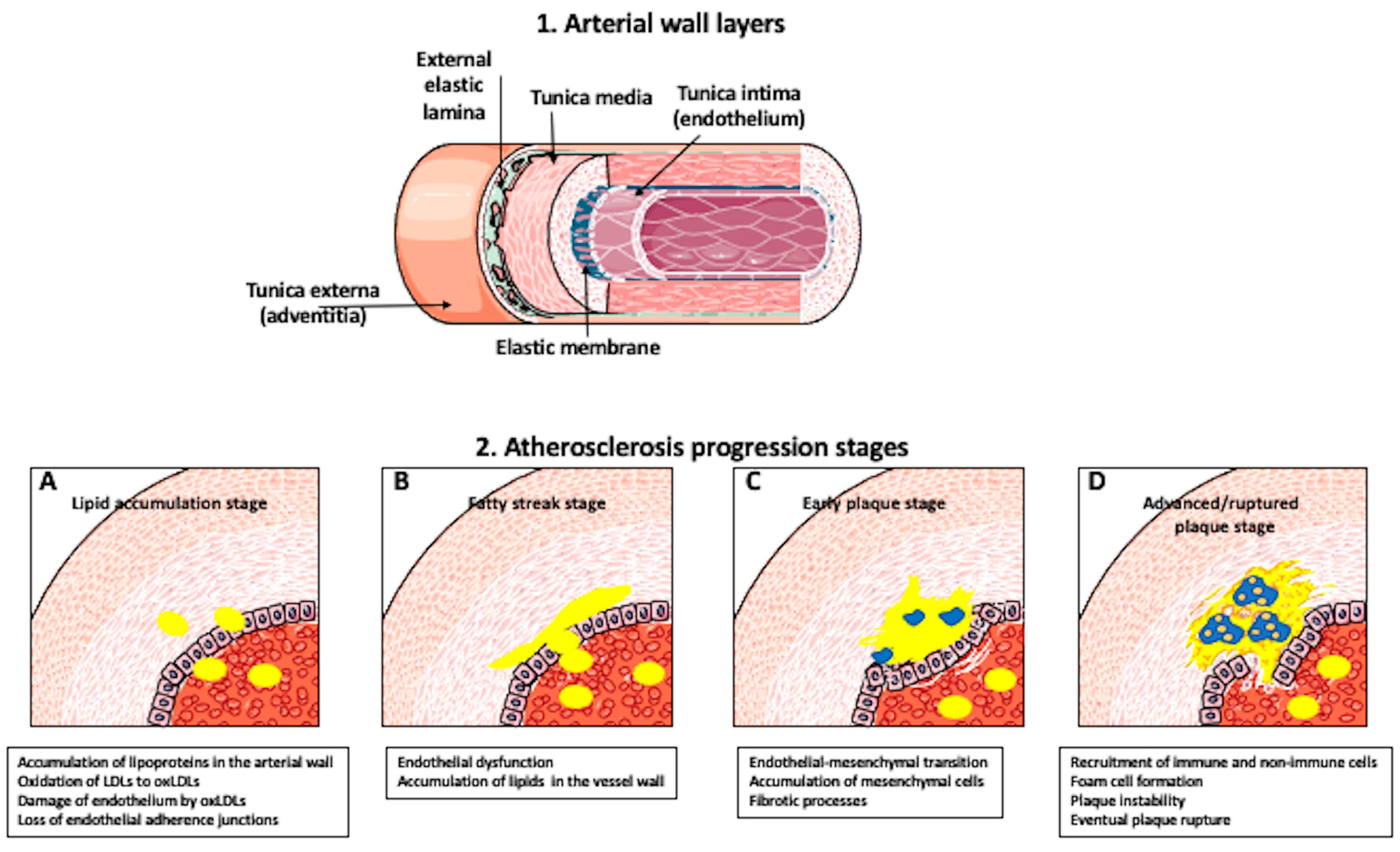{kind=link}
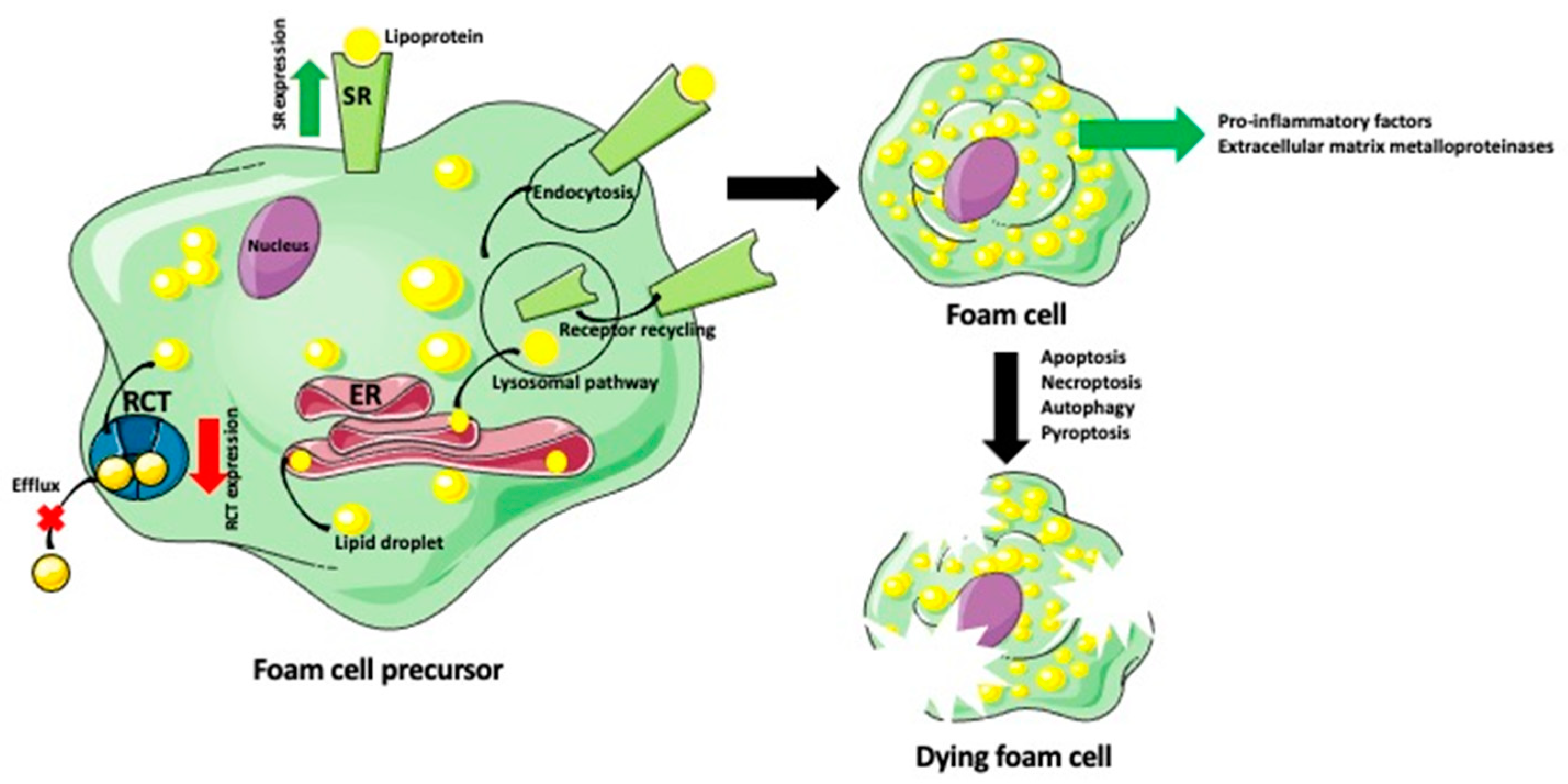{kind=link}
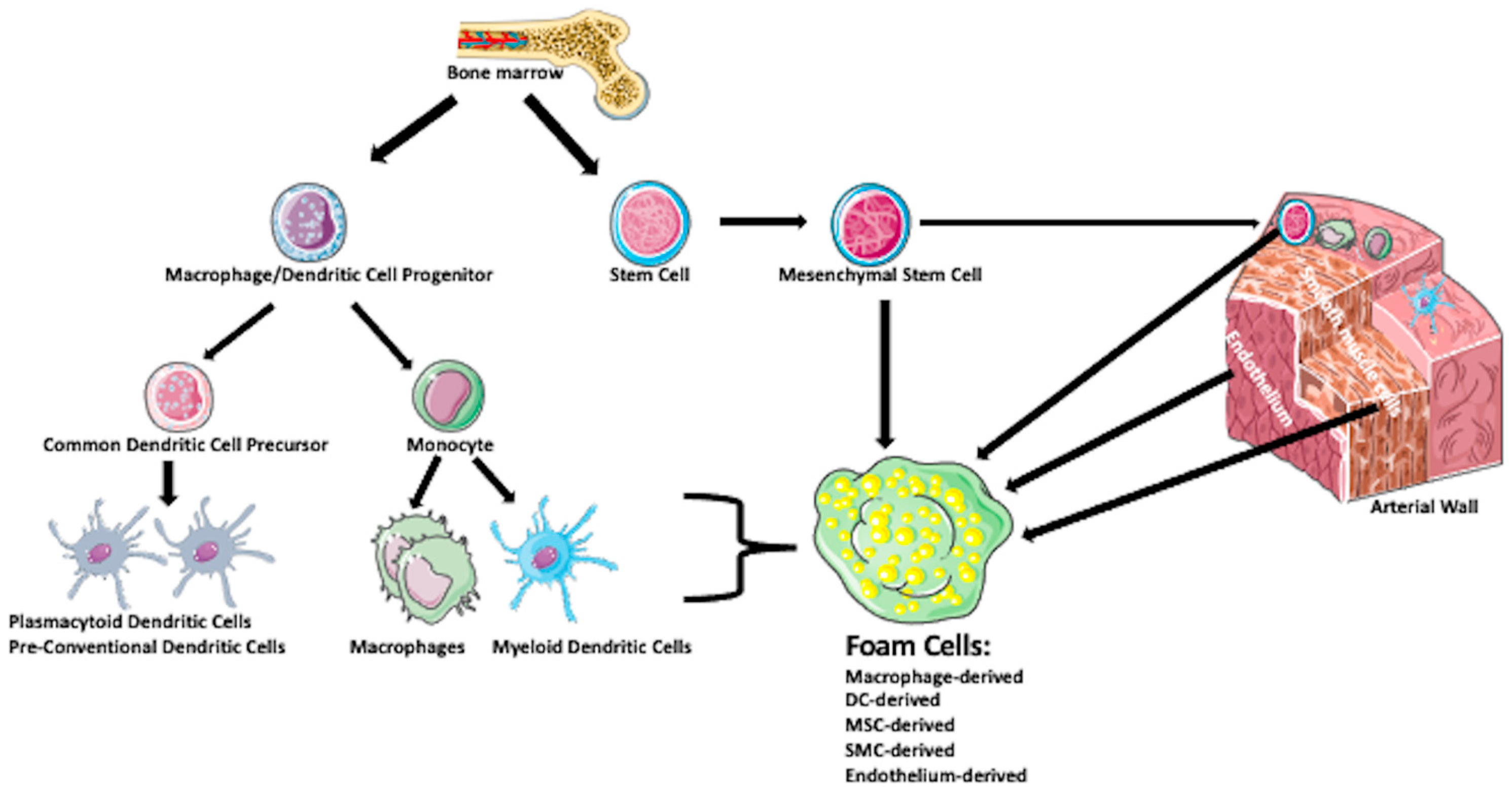{kind=link}
Abstract
1. Introduction
2. What Are the Origin and Mechanisms of Foam Cell Formation?
2.1. Macrophage-Derived Foam Cells
2.2. Dendritic-Derived Foam Cells
2.3. Smooth Muscle Cell-Derived Foam Cells
2.4. Endothelial Cell-Derived Foam Cells
2.5. Stem/Progenitor Cells-Derived Foam Cells
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AbCA1 | ATP-binding cassette A1 efflux transporter |
| AbCG1 | ATP-binding cassette G1 efflux transporter |
| ACAT1 | acyl-CoA: cholesterol acyltransferase |
| ActLDL | acetylated low-density lipoprotein |
| agLDL | aggregated low-density lipoprotein |
| apoA-I | apolipoprotein A-I |
| βVLDL | beta very low-density lipoprotein |
| CCL2 | C-C motif chemokine ligand 2 |
| CCL3 | C-C motif chemokine ligand 3 |
| CCL4 | C-C motif chemokine ligand 4 |
| CCL5 | C-C motif chemokine ligand 5 |
| CXCL16 | C-C motif chemokine ligand 16 |
| CCR2 | CC motif chemokine receptor 2 |
| CD34 | cluster of differentiation 34 protein |
| CD36 | scavenger receptor class B |
| CD68 | cluster of differentiation 68 protein |
| cDC | conventional DC |
| CDP | common DC precursor |
| CE | cholesteryl ester |
| CX3CL1 | C-X3-C motif chemokine ligand 1 |
| CX3CR1 | C-X3-C motif chemokine receptor1 |
| CXCR2 | C-X-C motif chemokine receptor 2 |
| DC | dendritic cell |
| EC | endothelial cell |
| ER | endoplasmic reticulum |
| FATP1 | fatty acid transport protein-1 |
| FC | free cholesterol |
| GM-CSF | granulocyte/macrophage colony-stimulating factor |
| Hsp | heat shock protein |
| IL-17A | interleukin-17A |
| IL-1β | interleukin-1β |
| IFN-y | interferon-gamma |
| KLF-4 | Kruppel-like factor 4 |
| LDL | low-density lipoprotein |
| LOX-1 | lectin-like oxLDL receptor-1 |
| LPC | phospholipid lysophosphatidylcholine |
| LPL | lysosomal acid lipase |
| LRP1 | low-density lipoprotein receptor-related protein 1 |
| LPR | low-density lipoprotein receptor |
| LXR | liver X receptor |
| M1 | pro-inflammatory macrophage subtype |
| M2 | ant-inflammatory macrophage subtype |
| Mac2 | galectin-3 protein |
| MCP-1 | monocyte chemoattractant protein-1 |
| M-CSF | macrophage colony-stimulating factor |
| MDP | macrophage and dendritic cell precursor |
| MHG | 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase |
| MMP | matrix metalloproteinase |
| MMP-9 | matrix metalloproteinase-9 |
| MSC | mesenchymal stem cell |
| MSRI | macrophage scavenger receptor type I |
| NLRP3 | NOD-, LRR- and pyrin domain-containing protein 3 |
| NR1H3 | nuclear receptor subfamily 1 group H member 3 |
| oxLDL | oxidized low-density lipoprotein |
| pDC | plasmacytoid DC |
| PPAR | peroxisome proliferator-activated receptor |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| RCT | reverse cholesterol transport system |
| PVAT | perivascular adipose tissue |
| SCa-1 | stem cell antigen-1 |
| SDF-1 | stromal cell-derived factor 1 |
| SMC | smooth muscle cell |
| SPC | stem/progenitor cell |
| SR | scavenger receptor |
| SR-A | scavenger receptor class A |
| SR-AI | scavenger receptor class A type I |
| SR-AII | scavenger receptor class A type II |
| SR-PSOX | scavenger receptor for phosphatidylserine and oxidized lipoprotein |
| TAM | tumor-specific macrophage |
| TB | tuberculosis |
| TNFα | tumor necrosis factor alpha |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VDC | vascular DC |
| VLA-4 | very late antigen-4 integrin |
References
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, R.; Silva, L.F.F.; Ribeiro, D.J.S.; Almeida, R.D.N.; Santos, I.O.; Corrêa, L.H.; de Sant’Ana, L.P.; Assunção, L.S.; Bozza, P.T.; Magalhães, K.G. Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front. Immunol. 2020, 10, 2927. [Google Scholar] [CrossRef] [PubMed]
- Stanek, A.; Fazeli, B.; Bartuś, S.; Sutkowska, E. The Role of Endothelium in Physiological and Pathological States: New Data. BioMed Res. Int. 2018, 2018, 1098039. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Phar-macotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Shi, H.; Winkler, M.A.; Lee, R.; Weintraub, N.L. Perivascular Adipose Tissue and Vascular Perturbation/Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Stanek, A.; Brożyna-Tkaczyk, K.; Myśliński, W. The Role of Obesity-Induced Perivascular Adipose Tissue (PVAT) Dysfunction in Vascular Homeostasis. Nutrients 2021, 13, 3843. [Google Scholar] [CrossRef]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Taki, A.; Kermani, A.; Ranjbarnavazi, S.M.; Pourmodheji, A. Overview of Different Medical Imaging Techniques for the Identification of Coronary Atherosclerotic Plaques. In Computing and Visualization for Intravascular Imaging and Computer-Assisted Stenting; Balocco, S., Zuluaga, M.A., Zahnd, G., Lee, S., Demirci, S., Eds.; The Elsevier and MICCAI Society Book Series; Academic Press: Cambridge, MA, USA, 2017; Chapter 4; pp. 79–106. ISBN 9780128110188. [Google Scholar] [CrossRef]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132, e157011. [Google Scholar] [CrossRef]
- Abedin, M.; Tintut, Y.; Demer, L.L. Mesenchymal stem cells and the artery wall. Circ. Res. 2004, 95, 671–676. [Google Scholar] [CrossRef]
- Bezsonov, E.E.; Sobenin, I.A.; Orekhov, A.N. Immunopathology of Atherosclerosis and Related Diseases: Focus on Molecular Biology. Int. J. Mol. Sci. 2021, 22, 4080. [Google Scholar] [CrossRef]
- Salnikova, D.; Orekhova, V.; Grechko, A.; Starodubova, A.; Bezsonov, E.; Popkova, T.; Orekhov, A. Mitochondrial Dysfunction in Vascular Wall Cells and Its Role in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8990. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, V.; Gennaro, M.L. Foam Cells: One Size Doesn’t Fit All. Trends Immunol. 2019, 40, 1163–1179. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Front. Cardiovasc. Med. 2022, 9, 845942. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Gordon, S.; Martinez, F.O. Foam Cell Macrophages in Tuberculosis. Front. Immunol. 2021, 12, 775326. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Yu, L.; Hu, J.; Xu, H.; Wang, J.; Shi, Y.; Luo, X.; Yan, W. Expression of PD-L1 in mononuclear cells, multinucleated cells, and foam cells in tenosynovial giant cell tumors. Int. J. Clin. Exp. Pathol. 2019, 12, 876–884. [Google Scholar] [PubMed]
- Murray, P.J. Cancer metastasis linked to macrophage size, shape, and metabolism. J. Exp. Med. 2020, 217, e20201259. [Google Scholar] [CrossRef]
- Kloc, M.; Li, X.C.; Ghobrial, R.M. Are Macrophages Responsible for Cancer Metastasis? J. Immunol. Biol. 2016, 1, 1. [Google Scholar] [CrossRef]
- Lee-Rueckert, M.; Lappalainen, J.; Kovanen, P.T.; Escola-Gil, J.C. Lipid-Laden Macrophages and Inflammation in Atherosclerosis and Cancer: An Integrative View. Front. Cardiovasc. Med. 2022, 9, 777822. [Google Scholar] [CrossRef]
- Jones, N.L.; Allen, N.S.; Willingham, M.C.; Lewis, J.C. Modified LDLs induce and bind to membrane ruffles on macrophages. Anat. Rec. 1999, 255, 44–56. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Kanuri, S.H.; Mehta, J.L. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr. Med. Chem. 2019, 26, 1693–1700. [Google Scholar] [CrossRef]
- Zhang, X.; Sessa, W.C.; Fernández-Hernando, C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fernández-Hernando, C. Transport of LDLs into the arterial wall: Impact in atherosclerosis. Curr. Opin. Lipidol. 2020, 31, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, F.; Crinelli, R.; Nasoni, M.G.; Benedetti, S.; Palma, F.; Fraternale, A.; Iuliano, L. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br. J. Pharmacol. 2021, 178, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, B.; Kallol, S.; Lüthi, M.; Albrecht, C.; Leiva, A. The polarized localization of lipoprotein receptors and cholesterol transporters in the syncytiotrophoblast of the placenta is reproducible in a monolayer of primary human trophoblasts. Placenta 2021, 105, 50–60. [Google Scholar] [CrossRef]
- Sukhorukov, V.N.; Khotina, V.A.; Chegodaev, Y.S.; Ivanova, E.; Sobenin, I.A.; Orekhov, A.N. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 2020, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kurihara, Y.; Takeya, M.; Kamada, N.; Kataoka, M.; Jishage, K.; Ueda, O.; Sakaguchi, H.; Higashi, T.; Suzuki, T.; et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 1997, 386, 292–296. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I. Introduction: Transporters, Porins, and Efflux Pumps. Chem. Rev. 2021, 121, 5095–5097. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Gu, H.M.; Zhang, D.W. Caveolin-1 and ATP binding cassette transporter A1 and G1-mediated cholesterol efflux. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Lippincott-Schwartz, J. Coated-pit dynamics. Nature 1999, 398, 753. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef]
- Meng, Y.; Heybrock, S.; Neculai, D.; Saftig, P. Cholesterol Handling in Lysosomes and Beyond. Trends Cell Biol. 2020, 30, 452–466. [Google Scholar] [CrossRef]
- Zhang, H. Lysosomal acid lipase and lipid metabolism: New mechanisms, new questions, and new therapies. Curr. Opin. Lipidol. 2018, 29, 218–223. [Google Scholar] [CrossRef]
- Kockx, M.; Dinnes, D.L.; Huang, K.Y.; Sharpe, L.J.; Jessup, W.; Brown, A.J.; Kritharides, L. Cholesterol accumulation inhibits ER to Golgi transport and protein secretion: Studies of apolipoprotein E and VSVGt. Biochem. J. 2012, 447, 51–60. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell 2017, 171, 1094–1109.e15. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Bobryshev, Y.V.; Orekhov, A.N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 2016, 20, 17–28. [Google Scholar] [CrossRef]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Westhorpe, C.L.; Dufour, E.M.; Maisa, A.; Jaworowski, A.; Crowe, S.M.; Muller, W.A. Endothelial cell activation promotes foam cell formation by monocytes following transendothelial migration in an in vitro model. Exp. Mol. Pathol. 2012, 93, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Uitz, E.; Bahadori, B.; McCarty, M.F.; Moghadasian, M.H. Practical strategies for modulating foam cell formation and behavior. World J. Clin. Cases 2014, 2, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Kakkar, V.; Lu, X. Impact of matrix metalloproteinases on atherosclerosis. Curr. Drug Targets 2014, 15, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, L.; Skepper, J.N.; Cary, N.R.; Mitchinson, M.J. Foam cell apoptosis and the development of the lipid core of human atherosclerosis. J. Pathol. 1996, 180, 423–429. [Google Scholar] [CrossRef]
- Lei, S.; Chen, J.; Song, C.; Li, J.; Zuo, A.; Xu, D.; Li, T.; Guo, Y. CTRP9 alleviates foam cells apoptosis by enhancing cholesterol efflux. Mol. Cell. Endocrinol. 2021, 522, 111138. [Google Scholar] [CrossRef]
- Chen, X.; Deng, Z.; Feng, J.; Chang, Q.; Lu, F.; Yuan, Y. Necroptosis in Macrophage Foam Cells Promotes Fat Graft Fibrosis in Mice. Front. Cell Dev. Biol. 2021, 9, 651360. [Google Scholar] [CrossRef]
- Robichaud, S.; Rasheed, A.; Pietrangelo, A.; Doyoung Kim, A.; Boucher, D.M.; Emerton, C.; Vijithakumar, V.; Gharibeh, L.; Fairman, G.; Mak, E.; et al. Autophagy Is Differentially Regulated in Leukocyte and Nonleukocyte Foam Cells During Atherosclerosis. Circ. Res. 2022, 130, 831–847. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Li, J.; Yu, Y.; Liu, Y.; Liu, H.; Peng, Q.; Guan, X. Autophagy blockage promotes the pyroptosis of ox-LDL-treated macrophages by modulating the p62/Nrf2/ARE axis. J. Physiol. Biochem. 2021, 77, 419–429. [Google Scholar] [CrossRef]
- Stöger, J.L.; Gijbels, M.J.; van der Velden, S.; Manca, M.; van der Loos, C.M.; Biessen, E.A.; Daemen, M.J.; Lutgens, E.; de Winther, M.P. Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef]
- Mushenkova, N.V.; Bezsonov, E.E.; Orekhova, V.A.; Popkova, T.V.; Starodubova, A.V.; Orekhov, A.N. Recognition of Oxidized Lipids by Macrophages and Its Role in Atherosclerosis Development. Biomedicines 2021, 9, 915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.J.; Li, T.J.; Yang, Y.; Guo, X.L.; Wu, M.C.; Rui, Y.C.; Wei, L.X. Lentiviral vector-mediated siRNA knockdown of SR-PSOX inhibits foam cell formation in vitro. Acta Pharmacol. Sin. 2008, 29, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- McLaren, J.E.; Michael, D.R.; Ashlin, T.G.; Ramji, D.P. Cytokines, macrophage lipid metabolism and foam cells: Implications for cardiovascular disease therapy. Prog. Lipid Res. 2011, 50, 331–347. [Google Scholar] [CrossRef]
- Walther, T.C.; Chung, J.; Farese, R.V., Jr. Lipid Droplet Biogenesis. Annu. Rev. Cell Dev. Biol. 2017, 33, 491–510. [Google Scholar] [CrossRef]
- Yu, X.H.; Zhang, D.W.; Zheng, X.L.; Tang, C.K. Cholesterol transport system: An integrated cholesterol transport model involved in atherosclerosis. Prog. Lipid Res. 2019, 73, 65–91. [Google Scholar] [CrossRef]
- Ben-Aicha, S.; Badimon, L.; Vilahur, G. Advances in HDL: Much More than Lipid Transporters. Int. J. Mol. Sci. 2020, 21, 732. [Google Scholar] [CrossRef]
- Matsuo, M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef]
- Subramaniam, K.; Babu, L.A.; Shah, N. A case of premature and recurrent myocardial infarction associated with ABCA.1 gene mutation. J. Postgrad. Med. 2021, 67, 29–32. [Google Scholar] [CrossRef]
- Wu, Y.T.; Li, J.B.; Lin, H.Q.; Zhang, G.X.; Hong, C.M.; Li, M.; Guo, Z.J.; Yang, Y.B. Inhibition of miR-200b-3p alleviates lipid accumulation and promotes cholesterol efflux by targeting ABCA1 in macrophage-derived foam cells. Exp. Ther. Med. 2021, 22, 831. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Baron, M.; Bouhlel, M.A.; Vanhoutte, J.; Copin, C.; Sebti, Y.; Derudas, B.; Mayi, T.; Bories, G.; Tailleux, A.; et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARγ and LXRα pathways. Circ. Res. 2011, 108, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Kidani, Y.; Bensinger, S.J. LXR and PPAR as integrators of lipid homeostasis and immunity. as integrators of lipid homeostasis and immunity. Immunol. Rev. 2012, 249, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A. Dendritic cells in atherosclerosis: Evidence in mice and humans. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Dendritic cells and their role in atherogenesis. Lab Investig. 2010, 90, 970–984. [Google Scholar] [CrossRef]
- Liu, P.; Yu, Y.R.; Spencer, J.A.; Johnson, A.E.; Vallanat, C.T.; Fong, A.M.; Patterson, C.; Patel, D.D. CX3CR1 deficiency impairs dendritic cell accumulation in arterial intima and reduces atherosclerotic burden. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 243–250. [Google Scholar] [CrossRef]
- Paulson, K.E.; Zhu, S.N.; Chen, M.; Nurmohamed, S.; Jongstra-Bilen, J.; Cybulsky, M.I. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ. Res. 2010, 106, 383–390. [Google Scholar] [CrossRef]
- Haka, A.S.; Grosheva, I.; Chiang, E.; Buxbaum, A.R.; Baird, B.A.; Pierini, L.M.; Maxfield, F.R. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. Mol. Biol. Cell 2009, 20, 4932–4940. [Google Scholar] [CrossRef]
- Haka, A.S.; Singh, R.K.; Grosheva, I.; Hoffner, H.; Capetillo-Zarate, E.; Chin, H.F.; Anandasabapathy, N.; Maxfield, F.R. Monocyte-Derived Dendritic Cells Upregulate Extracellular Catabolism of Aggregated Low-Density Lipoprotein on Maturation, Leading to Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2092–2103. [Google Scholar] [CrossRef]
- Salvatore, G.; Bernoud-Hubac, N.; Bissay, N.; Debard, C.; Daira, P.; Meugnier, E.; Proamer, F.; Hanau, D.; Vidal, H.; Aricò, M.; et al. Human monocyte-derived dendritic cells turn into foamy dendritic cells with IL-17A. J. Lipid Res. 2015, 56, 1110–1122. [Google Scholar] [CrossRef]
- Dubland, J.A.; Francis, G.A. So Much Cholesterol: The unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr. Opin. Lipidol. 2016, 27, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Nishi, H.; Long, X.; Ouimet, M.; Savji, N.; Martinez, F.O.; Cassella, C.P.; Moore, K.J.; Ramsey, S.A.; Miano, J.M.; et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Huff, M.W.; Pickering, J.G. Can a vascular smooth muscle-derived foam-cell really change its spots? Arterioscler. Thromb. Vasc. Biol. 2015, 35, 492–495. [Google Scholar] [CrossRef]
- Costales, P.; Fuentes-Prior, P.; Castellano, J.; Revuelta-Lopez, E.; Corral-Rodríguez, M.Á.; Nasarre, L.; Badimon, L.; Llorente-Cortes, V. K Domain CR9 of Low Density Lipoprotein (LDL) Receptor-related Protein 1 (LRP1) Is Critical for Aggregated LDL-induced Foam Cell Formation from Human Vascular Smooth Muscle Cells. J. Biol. Chem. 2015, 290, 14852–14865. [Google Scholar] [CrossRef]
- Wang, N.; Silver, D.L.; Thiele, C.; Tall, A.R. ATP-binding cassette transporter A1 (ABCA1) functions as a cholesterol efflux regulatory protein. J. Biol. Chem. 2001, 276, 23742–23747. [Google Scholar] [CrossRef]
- Ivan, L.; Antohe, F. Hyperlipidemia induces endothelial-derived foam cells in culture. J. Recept. Signal. Transduct. Res. 2010, 30, 106–114. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.C.; Shyu, W.C.; Lin, S.Z. Mesenchymal stem cells. Cell Transplant. 2011, 20, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Uosef, A.; Leśniak, M.; Kubiak, J.Z.; Ghobrial, R.M. Reciprocal interactions between mesenchymal stem cells and macrophages. Int. J. Dev. Biol. 2020, 64, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, T.; Sun, S.; Wang, R.; Deng, J.; Lyu, L.; Wu, H.; Yang, M.; Pu, X.; Du, L.; et al. Nonbone Marrow CD34+ Cells Are Crucial for Endothelial Repair of Injured Artery. Circ. Res. 2021, 129, e146–e165. [Google Scholar] [CrossRef] [PubMed]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schönberger, T.; Geisler, T.; Siegel-Axel, D.; et al. Platelets induce differentiation of human CD34+ progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef]
- Stellos, K.; Seizer, P.; Bigalke, B.; Daub, K.; Geisler, T.; Gawaz, M. Platelet aggregates-induced human CD34+ progenitor cell proliferation and differentiation to macrophages and foam cells is mediated by stromal cell derived factor 1 in vitro. Semin. Thromb. Hemost. 2010, 36, 139–145. [Google Scholar] [CrossRef]
- Chen, Y.; Wong, M.M.; Campagnolo, P.; Simpson, R.; Winkler, B.; Margariti, A.; Hu, Y.; Xu, Q. Adventitial stem cells in vein grafts display multilineage potential that contributes to neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1844–1851. [Google Scholar] [CrossRef][Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloc, M.; Kubiak, J.Z.; Ghobrial, R.M. Macrophage-, Dendritic-, Smooth Muscle-, Endothelium-, and Stem Cells-Derived Foam Cells in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 14154. https://doi.org/10.3390/ijms232214154
Kloc M, Kubiak JZ, Ghobrial RM. Macrophage-, Dendritic-, Smooth Muscle-, Endothelium-, and Stem Cells-Derived Foam Cells in Atherosclerosis. International Journal of Molecular Sciences. 2022; 23(22):14154. https://doi.org/10.3390/ijms232214154
Chicago/Turabian StyleKloc, Malgorzata, Jacek Z. Kubiak, and Rafik M. Ghobrial. 2022. "Macrophage-, Dendritic-, Smooth Muscle-, Endothelium-, and Stem Cells-Derived Foam Cells in Atherosclerosis" International Journal of Molecular Sciences 23, no. 22: 14154. https://doi.org/10.3390/ijms232214154
APA StyleKloc, M., Kubiak, J. Z., & Ghobrial, R. M. (2022). Macrophage-, Dendritic-, Smooth Muscle-, Endothelium-, and Stem Cells-Derived Foam Cells in Atherosclerosis. International Journal of Molecular Sciences, 23(22), 14154. https://doi.org/10.3390/ijms232214154