
Delivery of Theranostic Nanoparticles to Various Cancers by Means of Integrin-Binding Peptides


Abstract

1. Introduction
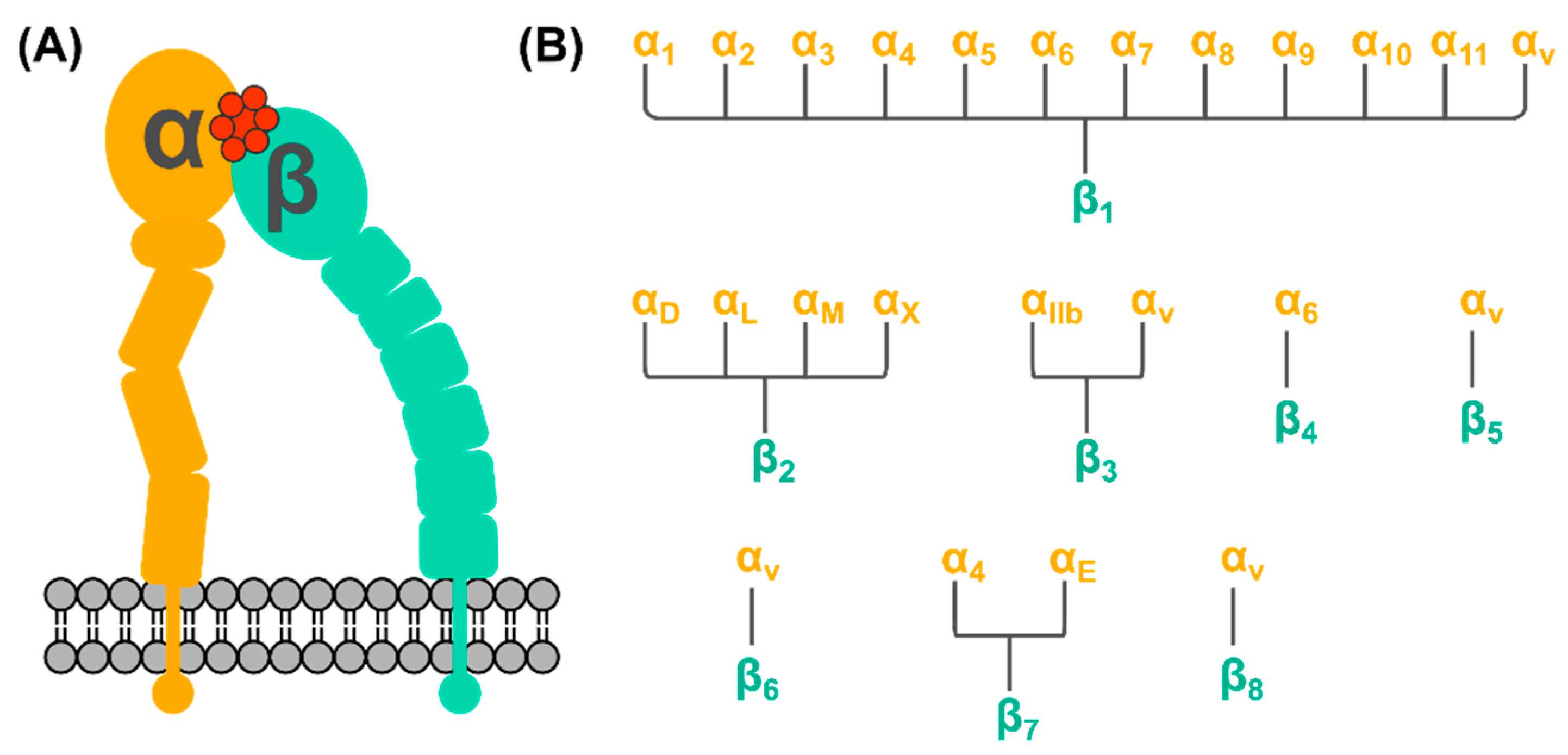
2. Integrin Receptors
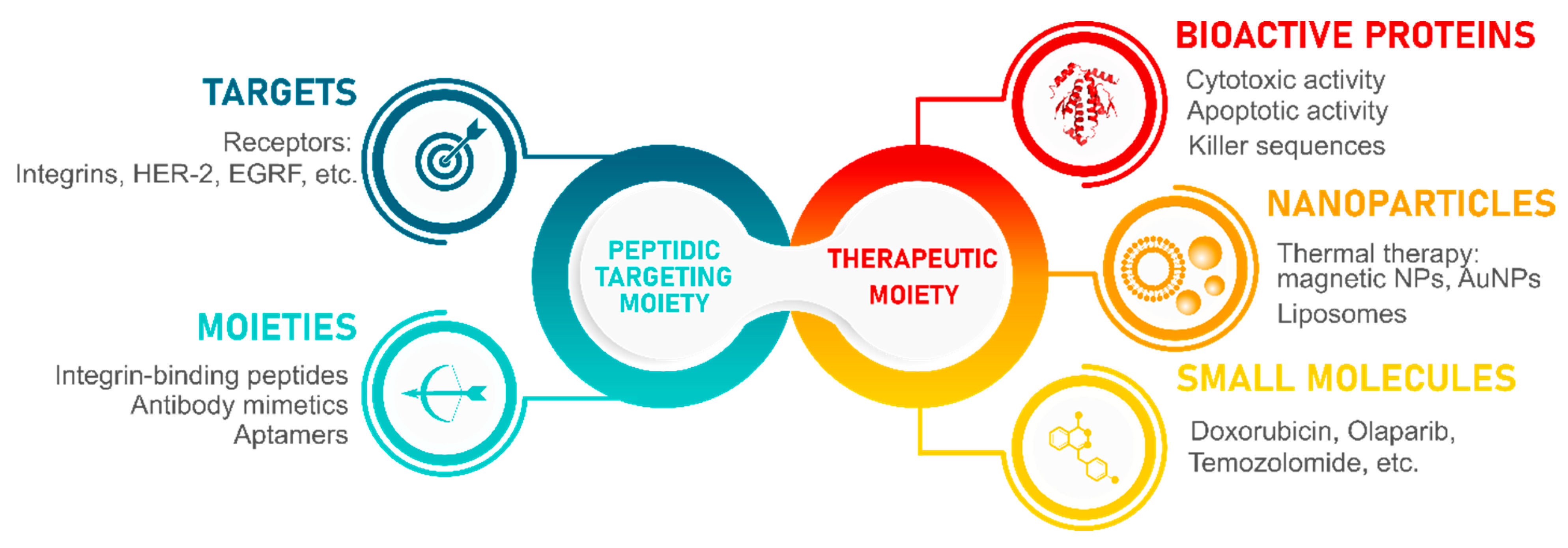
3. Integrin-Binding Peptides
3.1. General Overview of Integrin-Binding Peptides
3.2. Development of Integrin-Binding Peptides
4. Conjugation Strategies for Integrin-Binding Peptides
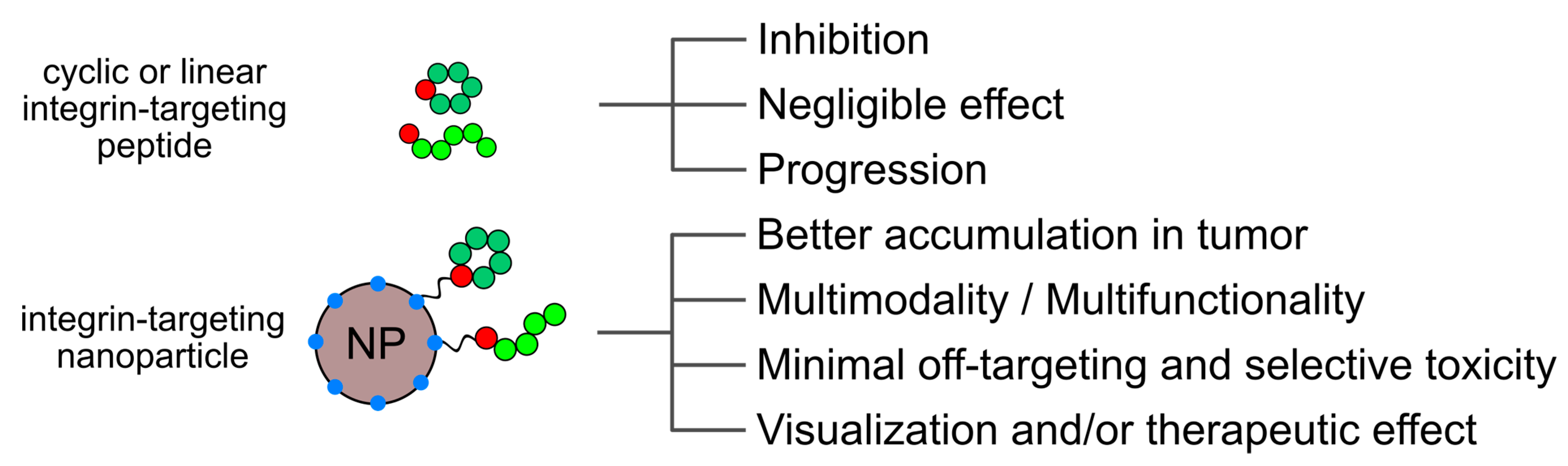
5. Integrin-Binding Peptides as Targeting Moieties in Nanoparticle Formulations
5.1. Lipid-Based Particles: Liposomes and Micelles
5.2. Metallic Nanoparticles
5.2.1. Quantum Dots
5.2.2. Gold Nanoparticles
5.2.3. Magnetic Nanoparticles
5.3. Selenium Nanoparticles
5.4. Other Types of Nanoparticles
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maeda, H.; Khatami, M. Analyses of Repeated Failures in Cancer Therapy for Solid Tumors: Poor Tumor-selective Drug Delivery, Low Therapeutic Efficacy and Unsustainable Costs. Clin. Transl. Med. 2018, 7, e11. [Google Scholar] [CrossRef]
- Yasuhiro, M.; Maeda, H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR Effect and beyond: Strategies to Improve Tumor Targeting and Cancer Nanomedicine Treatment Efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. The 35th Anniversary of the Discovery of EPR Effect: A New Wave of Nanomedicines for Tumor-Targeted Drug Delivery—Personal Remarks and Future Prospects. J. Pers. Med. 2021, 11, 229. [Google Scholar] [CrossRef]
- Zein, R.; Sharrouf, W.; Selting, K. Physical Properties of Nanoparticles That Result in Improved Cancer Targeting. J. Oncol. 2020, 2020, 5194780. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into Active Targeting of Nanoparticles in Drug Delivery: Advances in Clinical Studies and Design Considerations for Cancer Nanomedicine. Bioconjug. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef]
- Park, K. The Beginning of the End of the Nanomedicine Hype. J. Control. Release 2019, 305, 221–222. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Lammers, T. Macro-Nanomedicine: Targeting the Big Picture. J. Control. Release 2019, 294, 372–375. [Google Scholar] [CrossRef]
- Bazak, R.; Houri, M.; El Achy, S.; Kamel, S.; Refaat, T. Cancer Active Targeting by Nanoparticles: A Comprehensive Review of Literature. J. Cancer Res. Clin. Oncol. 2015, 141, 769–784. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1033. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. A Reevaluation of Integrins as Regulators of Angiogenesis. Nat. Med. 2002, 8, 918–921. [Google Scholar] [CrossRef] [PubMed]
- Sökeland, G.; Schumacher, U. The Functional Role of Integrins during Intra- and Extravasation within the Metastatic Cascade. Mol. Cancer 2019, 18, 12. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Kikkawa, Y.; Hozumi, K.; Katagiri, F.; Nomizu, M.; Kleinman, H.K.; Koblinski, J.E. Laminin-111-Derived Peptides and Cancer. Cell Adh. Migr. 2013, 7, 150–159. [Google Scholar] [CrossRef]
- Zhang, W.-M.; Käpylä, J.; Puranen, J.S.; Knight, C.G.; Tiger, C.-F.; Pentikäinen, O.T.; Johnson, M.S.; Farndale, R.W.; Heino, J.; Gullberg, D. α11β1 Integrin Recognizes the GFOGER Sequence in Interstitial Collagens. J. Biol. Chem. 2003, 278, 7270–7277. [Google Scholar] [CrossRef]
- Mardilovich, A.; Craig, J.A.; McCammon, M.Q.; Garg, A.; Kokkoli, E. Design of a Novel Fibronectin-Mimetic Peptide−Amphiphile for Functionalized Biomaterials. Langmuir 2006, 22, 3259–3264. [Google Scholar] [CrossRef]
- Clark, A.Y.; Martin, K.E.; García, J.R.; Johnson, C.T.; Theriault, H.S.; Han, W.M.; Zhou, D.W.; Botchwey, E.A.; García, A.J. Integrin-Specific Hydrogels Modulate Transplanted Human Bone Marrow-Derived Mesenchymal Stem Cell Survival, Engraftment, and Reparative Activities. Nat. Commun. 2020, 11, 114. [Google Scholar] [CrossRef]
- Ponce, M.L.; Hibino, S.; Lebioda, A.M.; Mochizuki, M.; Nomizu, M.; Kleinman, H.K. Identification of a potent peptide antagonist to an active laminin-1 sequence that blocks angiogenesis and tumor growth. Cancer Res. 2003, 63, 5060–5064. [Google Scholar] [PubMed]
- Liu, R.; Li, X.; Xiao, W.; Lam, K.S. Tumor-Targeting Peptides from Combinatorial Libraries. Adv. Drug Deliv. Rev. 2017, 110–111, 13–37. [Google Scholar] [CrossRef]
- Yacobovich, S.; Tuchinsky, L.; Kirby, M.; Kardash, T.; Agranyoni, O.; Nesher, E.; Redko, B.; Gellerman, G.; Tobi, D.; Gurova, K.; et al. Novel Synthetic Cyclic Integrin αvβ3 Binding Peptide ALOS4: Antitumor Activity in Mouse Melanoma Models. Oncotarget 2016, 7, 63549–63560. [Google Scholar] [CrossRef] [PubMed]
- Clemons, T.D.; Singh, R.; Sorolla, A.; Chaudhari, N.; Hubbard, A.; Iyer, K.S. Distinction Between Active and Passive Targeting of Nanoparticles Dictate Their Overall Therapeutic Efficacy. Langmuir 2018, 34, 15343–15349. [Google Scholar] [CrossRef]
- Ryppa, C.; Mann-Steinberg, H.; Fichtner, I.; Weber, H.; Satchi-Fainaro, R.; Biniossek, M.L.; Kratz, F. In vitro and in vivo evaluation of doxorubicin conjugates with the divalent peptide e-[c(RGDFK)2] that targets integrin αvβ3. Bioconjug. Chem. 2008, 19, 1414–1422. [Google Scholar] [CrossRef] [PubMed]
- Redko, B.; Tuchinsky, H.; Segal, T.; Tobi, D.; Luboshits, G.; Ashur-Fabian, O.; Pinhasov, A.; Gerlitz, G.; Gellerman, G. Toward the Development of a Novel Non-RGD Cyclic Peptide Drug Conjugate for Treatment of Human Metastatic Melanoma. Oncotarget 2017, 8, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gray, B.P.; McGuire, M.J.; Brown, K.C. Synthesis and Biological Evaluation of a Peptide–Paclitaxel Conjugate Which Targets the Integrin αvβ6. Bioorg. Med. Chem. 2011, 19, 5480–5489. [Google Scholar] [CrossRef] [PubMed]
- Keat, N.; Kenny, J.; Chen, K.; Onega, M.; Garman, N.; Slack, R.J.; Parker, C.A.; Lumbers, R.T.; Hallett, W.; Saleem, A.; et al. A Microdose PET Study of the Safety, Immunogenicity, Biodistribution, and Radiation Dosimetry of 18F-FB-A20FMDV2 for Imaging the Integrin αvβ6. J. Nucl. Med. Technol. 2018, 46, 136–143. [Google Scholar] [CrossRef]
- Shah, B.P.; Pasquale, N.; De, G.; Tan, T.; Ma, J.; Lee, K.-B. Core–Shell Nanoparticle-Based Peptide Therapeutics and Combined Hyperthermia for Enhanced Cancer Cell Apoptosis. ACS Nano 2014, 8, 9379–9387. [Google Scholar] [CrossRef]
- Li, Y.; Hu, P.; Wang, X.; Hou, X.; Liu, F.; Jiang, X. Integrin αvβ3-Targeted Polydopamine-Coated Gold Nanostars for Photothermal Ablation Therapy of Hepatocellular Carcinoma. Regen. Biomater. 2021, 8, rbab046. [Google Scholar] [CrossRef]
- Gal, O.; Betzer, O.; Rousso-Noori, L.; Sadan, T.; Motiei, M.; Nikitin, M.; Friedmann-Morvinski, D.; Popovtzer, R.; Popovtzer, A. Antibody Delivery into the Brain by Radiosensitizer Nanoparticles for Targeted Glioblastoma Therapy. J. Nanotheranostics 2022, 3, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.A.; Learmonth, D.A.; Sousa, R.A.; Salgado, A.J. αvβ3 and α5β1 Integrin-Specific Ligands: From Tumor Angiogenesis Inhibitors to Vascularization Promoters in Regenerative Medicine? Biotechnol. Adv. 2018, 36, 208–227. [Google Scholar] [CrossRef]
- Weber, M.R.; Zuka, M.; Lorger, M.; Tschan, M.; Torbett, B.E.; Zijlstra, A.; Quigley, J.P.; Staflin, K.; Eliceiri, B.P.; Krueger, J.S.; et al. Activated Tumor Cell Integrin αvβ3 Cooperates with Platelets to Promote Extravasation and Metastasis from the Blood Stream. Thromb. Res. 2016, 140, S27–S36. [Google Scholar] [CrossRef]
- Erdreich-Epstein, A.; Shimada, H.; Groshen, S.; Liu, M.; Metelitsa, L.S.; Kim, K.S.; Stins, M.F.; Seeger, R.C.; Durden, D.L. Integrins αvβ3 and αvβ5 Are Expressed by Endothelium of High-Risk Neuroblastoma and Their Inhibition Is Associated with Increased Endogenous Ceramide. Cancer Res. 2000, 60, 712–721. [Google Scholar]
- Arun, A.S.; Tepper, C.G.; Lam, K.S. Identification of integrin drug targets for 17 solid tumor types. Oncotarget 2018, 9, 30146–30162. [Google Scholar] [CrossRef]
- Saraf, P.; Li, X.; Jasti, B. Integrin Targeting Using RGD-Based Peptide Amphiphiles; Springer: Berlin/Heidelberg, Germany, 2015; pp. 135–155. [Google Scholar] [CrossRef]
- Marelli, U.K.; Rechenmacher, F.; Sobahi, T.R.A.; Mas-Moruno, C.; Kessler, H. Tumor Targeting via Integrin Ligands. Front. Oncol. 2013, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-Methylated Cyclic RGD Peptides as Highly Active and Selective αvβ3 Integrin Antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-Penetrating Delivery of Compounds and Nanoparticles into Tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Braun, G.B.; de Mendoza, T.H.; Kotamraju, V.R.; French, R.P.; Lowy, A.M.; Teesalu, T.; Ruoslahti, E. Tumor-Penetrating iRGD Peptide Inhibits Metastasis. Mol. Cancer Ther. 2015, 14, 120–128. [Google Scholar] [CrossRef]
- Oba, M.; Fukushima, S.; Kanayama, N.; Aoyagi, K.; Nishiyama, N.; Koyama, H.; Kataoka, K. Cyclic RGD peptide-conjugated polyplex micelles as a targetable gene delivery system directed to cells possessing alphavbeta3 and alphavbeta5 integrins. Bioconjug. Chem. 2007, 18, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Takenaka, T.; Toh, K.; Wu, S.; Nishihara, H.; Kano, M.R.; Ino, Y.; Nomoto, T.; Matsumoto, Y.; Koyama, H.; et al. Cyclic RGD-linked polymeric micelles for targeted delivery of platinum anticancer drugs to glioblastoma through the blood-brain tumor barrier. ACS Nano 2013, 7, 8583–8592. [Google Scholar] [PubMed]
- Ge, Z.; Chen, Q.; Osada, K.; Liu, X.; Tockary, T.A.; Uchida, S.; Dirisala, A.; Ishii, T.; Nomoto, T.; Toh, K.; et al. Targeted gene delivery by polyplex micelles with crowded PEG palisade and cRGD moiety for systemic treatment of pancreatic tumors. Biomaterials 2014, 35, 3416–3426. [Google Scholar] [CrossRef] [PubMed]
- Dirisala, A.; Osada, K.; Chen, Q.; Tockary, T.A.; Machitani, K.; Osawa, S.; Liu, X.; Ishii, T.; Miyata, K.; Oba, M.; et al. Optimized rod length of polyplex micelles for maximizing transfection efficiency and their performance in systemic gene therapy against stroma-rich pancreatic tumors. Biomaterials 2014, 35, 5359–5368. [Google Scholar] [CrossRef]
- Chen, Q.; Osada, K.; Ge, Z.; Uchida, S.; Tockary, T.A.; Dirisala, A.; Matsui, A.; Toh, K.; Takeda, K.M.; Liu, X.; et al. Polyplex micelle installing intracellular self-processing functionalities without free catiomers for safe and efficient systemic gene therapy through tumor vasculature targeting. Biomaterials 2017, 113, 253–265. [Google Scholar] [CrossRef]
- Maltsev, O.V.; Marelli, U.K.; Kapp, T.G.; di Leva, F.S.; di Maro, S.; Nieberler, M.; Reuning, U.; Schwaiger, M.; Novellino, E.; Marinelli, L.; et al. Stable Peptides Instead of Stapled Peptides: Highly Potent αvβ6-Selective Integrin Ligands. Angew. Chem. Int. Ed. 2016, 55, 1535–1539. [Google Scholar] [CrossRef]
- Reichart, F.; Maltsev, O.V.; Kapp, T.G.; Räder, A.F.B.; Weinmüller, M.; Marelli, U.K.; Notni, J.; Wurzer, A.; Beck, R.; Wester, H.-J.; et al. Selective Targeting of Integrin αvβ8 by a Highly Active Cyclic Peptide. J. Med. Chem. 2019, 62, 2024–2037. [Google Scholar] [CrossRef]
- Kaneda, Y.; Yamamoto, Y.; Okada, N.; Tsutsumi, Y.; Nakagawa, S.; Kakiuch, M.; Maeda, M.; Kawasaki, K.; Mayumi, T. Antimetastatic Effect of Synthetic Glu-Lle-Leu-Asp-Val Peptide Derivatives Containing D-Amino Acids. Anticancer Drugs 1997, 8, 702–707. [Google Scholar] [CrossRef]
- Cringoli, M.C.; Romano, C.; Parisi, E.; Waddington, L.J.; Melchionna, M.; Semeraro, S.; De Zorzi, R.; Grönholm, M.; Marchesan, S. Bioadhesive Supramolecular Hydrogel from Unprotected, Short D,L-Peptides with Phe-Phe and Leu-Asp-Val Motifs. Chem. Commun. 2020, 56, 3015–3018. [Google Scholar] [CrossRef]
- Vanderslice, P.; Ren, K.; Revelle, J.K.; Kim, D.C.; Scott, D.; Bjercke, R.J.; Yeh, E.T.; Beck, P.J.; Kogan, T.P. A cyclic hexapeptide is a potent antagonist of alpha 4 integrins. J. Immunol. 1997, 158, 1710–1718. [Google Scholar]
- Viles, J.H.; Mitchell, J.B.O.; Gough, S.L.; Doyle, P.M.; Harris, C.J.; Sadler, P.J.; Thornton, J.M. Multiple Solution Conformations of the Integrin-Binding Cyclic Pentapeptide Cyclo(-Ser-d-Leu-Asp-Val-Pro-). Analysis of the (Phi,Psi) Space Available to Cyclic Pentapeptides. Eur. J. Biochem. 1996, 242, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Doyle, P.M.; Harris, J.C.; Moody, C.M.; Sadler, P.J.; Sims, M.; Thornton, J.M.; Uppenbrink, J.; Viles, J.H. Solution Structure of a Biologically Active Cyclic LDV Peptide Analogue Containing a Type II′β-Turn Mimetic. Int. J. Pept. Protein Res. 2009, 47, 427–436. [Google Scholar] [CrossRef]
- Meena, C.L.; Singh, D.; Weinmüller, M.; Reichart, F.; Dangi, A.; Marelli, U.K.; Zahler, S.; Sanjayan, G.J. Novel Cilengitide-Based Cyclic RGD Peptides as αvβ Integrin Inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127039. [Google Scholar] [CrossRef]
- Wang, W.; Wang, F.; Lu, F.; Xu, S.; Hu, W.; Huang, J.; Gu, Q.; Sun, X. The Antiangiogenic Effects of Integrin α5β1 Inhibitor (ATN-161) In Vitro and In Vivo. Investig. Opthalmology Vis. Sci. 2011, 52, 7213. [Google Scholar] [CrossRef]
- van Golen, K.L.; Bao, L.; Brewert, G.J.; Pienta, K.J.; Kamradt, J.M.; Livant, D.L.; Merajver, S.D. Suppression of Tumor Recurrence and Metastasis by a Combination of the PHSCN Sequence and the Antiangiogenic Compound Tetrathiomolybdate in Prostate Carcinoma. Neoplasia 2002, 4, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Veine, D.M.; Livant, D.L. Therapeutic Inhibition of Breast Cancer Bone Metastasis Progression and Lung Colonization: Breaking the Vicious Cycle by Targeting α5β1 Integrin. Breast Cancer Res. Treat. 2016, 157, 489–501. [Google Scholar] [CrossRef]
- Doñate, F.; Parry, G.C.; Shaked, Y.; Hensley, H.; Guan, X.; Beck, I.; Tel-Tsur, Z.; Plunkett, M.L.; Manuia, M.; Shaw, D.E.; et al. Pharmacology of the Novel Antiangiogenic Peptide ATN-161 (Ac-PHSCN-NH2): Observation of a U-Shaped Dose-Response Curve in Several Preclinical Models of Angiogenesis and Tumor Growth. Clin. Cancer Res. 2008, 14, 2137–2144. [Google Scholar] [CrossRef]
- Cianfrocca, M.E.; Kimmel, K.A.; Gallo, J.; Cardoso, T.; Brown, M.M.; Hudes, G.; Lewis, N.; Weiner, L.; Lam, G.N.; Brown, S.C.; et al. Phase 1 Trial of the Antiangiogenic Peptide ATN-161 (Ac-PHSCN-NH2), a Beta Integrin Antagonist, in Patients with Solid Tumours. Br. J. Cancer 2006, 94, 1621–1626. [Google Scholar] [CrossRef]
- Levi, B.; Yacobovich, S.; Kirby, M.; Becker, M.; Agranyoni, O.; Redko, B.; Gellerman, G.; Pinhasov, A.; Koman, I.; Nesher, E. Anti-Cancer Effects of Cyclic Peptide ALOS4 in a Human Melanoma Mouse Model. Int. J. Mol. Sci. 2021, 22, 9579. [Google Scholar] [CrossRef]
- Thevenard, J.; Floquet, N.; Ramont, L.; Prost, E.; Nuzillard, J.-M.; Dauchez, M.; Yezid, H.; Alix, A.J.P.; Maquart, F.-X.; Monboisse, J.-C.; et al. Structural and Antitumor Properties of the YSNSG Cyclopeptide Derived from Tumstatin. Chem. Biol. 2006, 13, 1307–1315. [Google Scholar] [CrossRef][Green Version]
- Thevenard, J.; Ramont, L.; Devy, J.; Brassart, B.; Dupont-Deshorgue, A.; Floquet, N.; Schneider, L.; Ouchani, F.; Terryn, C.; Maquart, F.-X.; et al. The YSNSG Cyclopeptide Derived from Tumstatin Inhibits Tumor Angiogenesis by Down-Regulating Endothelial Cell Migration. Int. J. Cancer 2010, 126, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Slimano, F.; Djerada, Z.; Guerin, J.; Bellouch, M.I.; Brassart-Pasco, S.; Dukic, S. Intratumoral Distribution of YSNSG Cyclopeptide in a Mouse Melanoma Model Using Microdialysis. Eur. J. Pharm. Sci. 2020, 143, 105201. [Google Scholar] [CrossRef]
- Jackson, D.Y.; Quan, C.; Artis, D.R.; Rawson, T.; Blackburn, B.; Struble, M.; Fitzgerald, G.; Chan, K.; Mullins, S.; Burnier, J.P.; et al. Potent α4β1 Peptide Antagonists as Potential Anti-Inflammatory Agents. J. Med. Chem. 1997, 40, 3359–3368. [Google Scholar] [CrossRef]
- Tilley, J.; Chen, L.; Sidduri, A.; Fotouhi, N. The Discovery of VLA-4 Antagonists. Curr. Top. Med. Chem. 2004, 4, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Sawicki, L.A.; Ovadia, E.M.; Pradhan, L.; Cowart, J.E.; Ross, K.E.; Wu, C.H.; Kloxin, A.M. Tunable Synthetic Extracellular Matrices to Investigate Breast Cancer Response to Biophysical and Biochemical Cues. APL Bioeng. 2019, 3, 016101. [Google Scholar] [CrossRef] [PubMed]
- Sephel, G.C.; Tashiro, K.-I.; Sasaki, M.; Greatorex, D.; Martin, G.R.; Yamada, Y.; Kleinman, H.K. Laminin a Chain Synthetic Peptide Which Supports Neurite Outgrowth. Biochem. Biophys. Res. Commun. 1989, 162, 821–829. [Google Scholar] [CrossRef]
- Sieni, E.; Bazzolo, B.; Pieretti, F.; Zamuner, A.; Tasso, A.; Dettin, M.; Conconi, M.T. Breast Cancer Cells Grown on Hyaluronic Acid-Based Scaffolds as 3D in Vitro Model for Electroporation. Bioelectrochemistry 2020, 136, 107626. [Google Scholar] [CrossRef]
- Hozumi, K.; Akizuki, T.; Yamada, Y.; Hara, T.; Urushibata, S.; Katagiri, F.; Kikkawa, Y.; Nomizu, M. Cell Adhesive Peptide Screening of the Mouse Laminin α1 Chain G Domain. Arch. Biochem. Biophys. 2010, 503, 213–222. [Google Scholar] [CrossRef]
- Mardilovich, A.; Kokkoli, E. Biomimetic Peptide−Amphiphiles for Functional Biomaterials: The Role of GRGDSP and PHSRN. Biomacromolecules 2004, 5, 950–957. [Google Scholar] [CrossRef]
- Logan, D.; Abu-Ghazaleh, R.; Blakemore, W.; Curry, S.; Jackson, T.; King, A.; Lea, S.; Lewis, R.; Newman, J.; Parry, N.; et al. Structure of a Major Immunogenic Site on Foot-and-Mouth Disease Virus. Nature 1993, 362, 566–568. [Google Scholar] [CrossRef]
- Hausner, S.H.; DiCara, D.; Marik, J.; Marshall, J.F.; Sutcliffe, J.L. Use of a Peptide Derived from Foot-and-Mouth Disease Virus for the Noninvasive Imaging of Human Cancer: Generation and Evaluation of 4-[18F]Fluorobenzoyl A20FMDV2 for In Vivo Imaging of Integrin αvβ6 Expression with Positron Emission Tomography. Cancer Res. 2007, 67, 7833–7840. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowich-Knipp, S.J.; Chakrabarti, S.; Siahaan, T.J.; Williams, T.D.; Dillman, R.K. Solution Stability of Linear vs. Cyclic RGD Peptides. J. Pept. Res. 1999, 53, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowich-Knipp, S.J.; Jois, D.S.S.; Siahaan, T.J. The Effect of Conformation on the Solution Stability of Linear vs. Cyclic RGD Peptides. J. Pept. Res. 1999, 53, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Hölzemann, G.; Sulyok, G.A.G.; Kessler, H. Nanomolar Small Molecule Inhibitors for αvβ6, αvβ5, and αvβ3 Integrins. J. Med. Chem. 2002, 45, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Haworth, D.; Rees, A.; Alcock, P.J.; Wood, L.J.; Dutta, A.S.; Gormley, J.J.; Jones, H.B.; Jamieson, A.; Reilly, C.F. Anti-Inflammatory Activity of c(ILDV-NH(CH2)5CO), a Novel, Selective, Cyclic Peptide Inhibitor of VLA-4-Mediated Cell Adhesion. Br. J. Pharmacol. 1999, 126, 1751–1760. [Google Scholar] [CrossRef]
- Ding, J.; Tasker, C.; Lespinasse, P.; Dai, J.; Fitzgerald-Bocarsly, P.; Lu, W.; Heller, D.; Chang, T.L.-Y. Integrin α4β7 Expression Increases HIV Susceptibility in Activated Cervical CD4+ T Cells by an HIV Attachment-Independent Mechanism. J. Acquir. Immune Defic. Syndr. 2015, 69, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Magnuson, A.; Benoist, C.; Pittet, M.J.; Weissleder, R. Age-Related Tumor Growth in Mice Is Related to Integrin α 4 in CD8+ T Cells. JCI Insight 2018, 3, e122961. [Google Scholar] [CrossRef]
- Nair-Gupta, P.; Rudnick, S.I.; Luistro, L.; Smith, M.; McDaid, R.; Li, Y.; Pillarisetti, K.; Joseph, J.; Heidrich, B.; Packman, K.; et al. Blockade of VLA4 Sensitizes Leukemic and Myeloma Tumor Cells to CD3 Redirection in the Bone Marrow Microenvironment. Blood Cancer J. 2020, 10, 65. [Google Scholar] [CrossRef]
- Bagati, A.; Kumar, S.; Jiang, P.; Pyrdol, J.; Zou, A.E.; Godicelj, A.; Mathewson, N.D.; Cartwright, A.N.R.; Cejas, P.; Brown, M.; et al. Integrin αvβ6–TGFβ–SOX4 Pathway Drives Immune Evasion in Triple-Negative Breast Cancer. Cancer Cell 2021, 39, 54–67.e9. [Google Scholar] [CrossRef]
- Whilding, L.M.; Vallath, S.; Maher, J. The Integrin αvβ6: A Novel Target for CAR T-Cell Immunotherapy? Biochem. Soc. Trans. 2016, 44, 349–355. [Google Scholar] [CrossRef]
- Niu, J.; Li, Z. The Roles of Integrin αvβ6 in Cancer. Cancer Lett. 2017, 403, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Tchaicha, J.H.; Reyes, S.B.; Shin, J.; Hossain, M.G.; Lang, F.F.; McCarty, J.H. Glioblastoma Angiogenesis and Tumor Cell Invasiveness Are Differentially Regulated by β8 Integrin. Cancer Res. 2011, 71, 6371–6381. [Google Scholar] [CrossRef]
- Guerrero, P.A.; Tchaicha, J.H.; Chen, Z.; Morales, J.E.; McCarty, N.; Wang, Q.; Sulman, E.P.; Fuller, G.; Lang, F.F.; Rao, G.; et al. Glioblastoma Stem Cells Exploit the αvβ8 Integrin-TGFβ1 Signaling Axis to Drive Tumor Initiation and Progression. Oncogene 2017, 36, 6568–6580. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Wu, F.; Tian, D.; Wang, T.; Lu, T.; Huang, X.; Zhang, P.; Qin, L. MiR-199a-3p Enhances Cisplatin Sensitivity of Ovarian Cancer Cells by Targeting ITGB8. Oncol. Rep. 2018, 39, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Zuo, H. IRGD: A Promising Peptide for Cancer Imaging and a Potential Therapeutic Agent for Various Cancers. J. Oncol. 2019, 2019, 9367845. [Google Scholar] [CrossRef]
- Mamluk, R.; Gechtman, Z.; Kutcher, M.E.; Gasiunas, N.; Gallagher, J.; Klagsbrun, M. Neuropilin-1 Binds Vascular Endothelial Growth Factor 165, Placenta Growth Factor-2, and Heparin via Its B1b2 Domain. J. Biol. Chem. 2002, 277, 24818–24825. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Hurtado de Mendoza, T.; Mose, E.S.; Botta, G.P.; Braun, G.B.; Kotamraju, V.R.; French, R.P.; Suzuki, K.; Miyamura, N.; Teesalu, T.; Ruoslahti, E.; et al. Tumor-Penetrating Therapy for β5 Integrin-Rich Pancreas Cancer. Nat. Commun. 2021, 12, 1541. [Google Scholar] [CrossRef]
- Atchison, N.A.; Fan, W.; Papas, K.K.; Hering, B.J.; Tsapatsis, M.; Kokkoli, E. Binding of the Fibronectin-Mimetic Peptide, PR_b, to α5β1 on Pig Islet Cells Increases Fibronectin Production and Facilitates Internalization of PR_b Functionalized Liposomes. Langmuir 2010, 26, 14081–14088. [Google Scholar] [CrossRef]
- Levine, R.M.; Dinh, C.V.; Harris, M.A.; Kokkoli, E. Targeting HPV-infected Cervical Cancer Cells with PEGylated Liposomes Encapsulating siRNA and the Role of siRNA Complexation with Polyethylenimine. Bioeng. Transl. Med. 2016, 1, 168–180. [Google Scholar] [CrossRef]
- Levine, R.M.; Kokkoli, E. Dual-Ligand α5β1 and α6β4 Integrin Targeting Enhances Gene Delivery and Selectivity to Cancer Cells. J. Control. Release 2017, 251, 24–36. [Google Scholar] [CrossRef] [PubMed]
- DiCara, D.; Rapisarda, C.; Sutcliffe, J.L.; Violette, S.M.; Weinreb, P.H.; Hart, I.R.; Howard, M.J.; Marshall, J.F. Structure-Function Analysis of Arg-Gly-Asp Helix Motifs in αvβ6 Integrin Ligands. J. Biol. Chem. 2007, 282, 9657–9665. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Hafeji, M.; Rogers, R.; Ludbrook, S.B.; Marshall, J.F.; Flint, D.J.; Pyne, S.; Denyer, J.C. Pharmacological Characterization of the αvβ6 Integrin Binding and Internalization Kinetics of the Foot-and-Mouth Disease Virus Derived Peptide A20FMDV2. Pharmacology 2016, 97, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Hausner, S.H.; Bauer, N.; Hu, L.Y.; Knight, L.M.; Sutcliffe, J.L. The Effect of Bi-Terminal PEGylation of an Integrin αvβ6 –Targeted 18F Peptide on Pharmacokinetics and Tumor Uptake. J. Nucl. Med. 2015, 56, 784–790. [Google Scholar] [CrossRef]
- Cardle, I.I.; Jensen, M.C.; Pun, S.H.; Sellers, D.L. Optimized Serum Stability and Specificity of an αvβ6 Integrin-Binding Peptide for Tumor Targeting. J. Biol. Chem. 2021, 296, 100657. [Google Scholar] [CrossRef]
- Hung, K.; Harris, P.W.R.; Desai, A.; Marshall, J.F.; Brimble, M.A. Structure-Activity Relationship Study of the Tumour-Targeting Peptide A20FMDV2 via Modification of Lys16, Leu13, and N- and/or C-Terminal Functionality. Eur. J. Med. Chem. 2017, 136, 154–164. [Google Scholar] [CrossRef]
- Nakamura, S.; Matsuno, A.; Ueda, M. Improvement of Biodistribution Profile of a Radiogallium-Labeled, αvβ6 Integrin-Targeting Peptide Probe by Incorporation of Negatively Charged Amino Acids. Ann. Nucl. Med. 2020, 34, 575–582. [Google Scholar] [CrossRef]
- Ui, T.; Ueda, M.; Higaki, Y.; Kamino, S.; Sano, K.; Kimura, H.; Saji, H.; Enomoto, S. Development and Characterization of a 68Ga-Labeled A20FMDV2 Peptide Probe for the PET Imaging of αvβ6 Integrin-Positive Pancreatic Ductal Adenocarcinoma. Bioorg. Med. Chem. 2020, 28, 115189. [Google Scholar] [CrossRef]
- Ganguly, T.; Tang, S.Y.; Bauer, N.; Sutcliffe, J.L. Correction to: Evaluation of Two Optical Probes for Imaging the Integrin αvβ6− In Vitro and In Vivo in Tumor-Bearing Mice. Mol. Imaging Biol. 2020, 22, 1182–1183. [Google Scholar] [CrossRef]
- Saleem, A.; Helo, Y.; Win, Z.; Dale, R.; Cook, J.; Searle, G.E.; Wells, P. Integrin αvβ6 Positron Emission Tomography Imaging in Lung Cancer Patients Treated With Pulmonary Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2020, 107, 370–376. [Google Scholar] [CrossRef]
- Livant, D.L.; Brabec, R.K.; Pienta, K.J.; Allen, D.L.; Kurachi, K.; Markwart, S.; Upadhyaya, A. Anti-invasive, antitumorigenic, and antimetastatic activities of the PHSCN sequence in prostate carcinoma. Cancer Res. 2000, 60, 309–320. [Google Scholar] [PubMed]
- Beddingfield, B.J.; Iwanaga, N.; Chapagain, P.P.; Zheng, W.; Roy, C.J.; Hu, T.Y.; Kolls, J.K.; Bix, G.J. The Integrin Binding Peptide, ATN-161, as a Novel Therapy for SARS-CoV-2 Infection. JACC Basic Transl. Sci. 2021, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Amruta, N.; Chastain, W.H.; Paz, M.; Solch, R.J.; Murray-Brown, I.C.; Befeler, J.B.; Gressett, T.E.; Longo, M.T.; Engler-Chiurazzi, E.B.; Bix, G. SARS-CoV-2 Mediated Neuroinflammation and the Impact of COVID-19 in Neurological Disorders. Cytokine Growth Factor Rev. 2021, 58, 1–15. [Google Scholar] [CrossRef]
- Yoshida, Y. The Potent Peptide Antagonist to Angiogenesis, C16Y, and Cisplatin Act Synergistically in the down-Regulation of the Bcl-2/Bax Ratio and the Induction of Apoptosis in Human Ovarian Cancer Cells. Int. J. Oncol. 2011, 39, 1359–1364. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ji, T.; Zhao, Y.; Zhang, Y.; Zhao, X.; Zhao, R.; Lang, J.; Zhao, X.; Shi, J.; Sukumar, S.; et al. Improvement of Stability and Efficacy of C16Y Therapeutic Peptide via Molecular Self-Assembly into Tumor-Responsive Nanoformulation. Mol. Cancer Ther. 2015, 14, 2390–2400. [Google Scholar] [CrossRef]
- Hamano, N.; Negishi, Y.; Fujisawa, A.; Manandhar, M.; Sato, H.; Katagiri, F.; Nomizu, M.; Aramaki, Y. Modification of the C16Y Peptide on Nanoparticles Is an Effective Approach to Target Endothelial and Cancer Cells via the Integrin Receptor. Int. J. Pharm. 2012, 428, 114–117. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Ashley, J.D.; Stefanick, J.F.; Qi, Y.M.; Alves, N.J.; Handlogten, M.W.; Suckow, M.A.; Navari, R.M.; Bilgicer, B. Rationally Engineered Nanoparticles Target Multiple Myeloma Cells, Overcome Cell-Adhesion-Mediated Drug Resistance, and Show Enhanced Efficacy in vivo. Blood Cancer J. 2012, 2, e64. [Google Scholar] [CrossRef]
- Stefanick, J.F.; Omstead, D.T.; Ashley, J.D.; Deak, P.E.; Mustafaoglu, N.; Kiziltepe, T.; Bilgicer, B. Optimizing Design Parameters of a Peptide Targeted Liposomal Nanoparticle in an in Vivo Multiple Myeloma Disease Model after Initial Evaluation in vitro. J. Control. Release 2019, 311–312, 190–200. [Google Scholar] [CrossRef]
- Shroff, K.; Pearce, T.R.; Kokkoli, E. Enhanced Integrin Mediated Signaling and Cell Cycle Progression on Fibronectin Mimetic Peptide Amphiphile Monolayers. Langmuir 2012, 28, 1858–1865. [Google Scholar] [CrossRef]
- Hosoyama, K.; Lazurko, C.; Muñoz, M.; McTiernan, C.D.; Alarcon, E.I. Peptide-Based Functional Biomaterials for Soft-Tissue Repair. Front. Bioeng. Biotechnol. 2019, 7, 205. [Google Scholar] [CrossRef]
- Tysseling, V.M.; Sahni, V.; Pashuck, E.T.; Birch, D.; Hebert, A.; Czeisler, C.; Stupp, S.I.; Kessler, J.A. Self-Assembling Peptide Amphiphile Promotes Plasticity of Serotonergic Fibers Following Spinal Cord Injury. J. Neurosci. Res. 2010, 88, 3161–3170. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; den Berk, L.; Wondergem, J.A.J.; Tong, C.; Kwakernaak, M.C.; ter Braak, B.; Heinrich, D.; Water, B.; Kieltyka, R.E. Squaramide-Based Supramolecular Materials Drive HepG2 Spheroid Differentiation. Adv. Healthc. Mater. 2021, 10, 2001903. [Google Scholar] [CrossRef] [PubMed]
- Balion, Z.; Sipailaite, E.; Stasyte, G.; Vailionyte, A.; Mazetyte-Godiene, A.; Seskeviciute, I.; Bernotiene, R.; Phopase, J.; Jekabsone, A. Investigation of Cancer Cell Migration and Proliferation on Synthetic Extracellular Matrix Peptide Hydrogels. Front. Bioeng. Biotechnol. 2020, 8, 773. [Google Scholar] [CrossRef]
- Hedegaard, C.L.; Redondo-Gómez, C.; Tan, B.Y.; Ng, K.W.; Loessner, D.; Mata, A. Peptide-Protein Coassembling Matrices as a Biomimetic 3D Model of Ovarian Cancer. Sci. Adv. 2020, 6, eabb3298. [Google Scholar] [CrossRef]
- Srikanth, M.; Das, S.; Berns, E.J.; Kim, J.; Stupp, S.I.; Kessler, J.A. Nanofiber-Mediated Inhibition of Focal Adhesion Kinase Sensitizes Glioma Stemlike Cells to Epidermal Growth Factor Receptor Inhibition. Neuro Oncol. 2013, 15, 319–329. [Google Scholar] [CrossRef]
- Zununi Vahed, S.; Salehi, R.; Davaran, S.; Sharifi, S. Liposome-Based Drug Co-Delivery Systems in Cancer Cells. Mater. Sci. Eng. C 2017, 71, 1327–1341. [Google Scholar] [CrossRef] [PubMed]
- Rabe, M.; Zope, H.R.; Kros, A. Interplay between Lipid Interaction and Homo-Coiling of Membrane-Tethered Coiled-Coil Peptides. Langmuir 2015, 31, 9953–9964. [Google Scholar] [CrossRef]
- Shi, P.; Chen, H. Peptide-Directed Binding of Quantum Dots to Integrins in Human Fibroblast. IEEE Trans. Nanobioscience 2006, 5, 15–19. [Google Scholar] [CrossRef]
- Knudsen, N.Ø.; Schiffelers, R.M.; Jorgensen, L.; Hansen, J.; Frokjaer, S.; Foged, C. Design of Cyclic RKKH Peptide-Conjugated PEG Liposomes Targeting the Integrin α2β1 Receptor. Int. J. Pharm. 2012, 428, 171–177. [Google Scholar] [CrossRef]
- Dai, W.; Yang, T.; Wang, X.; Wang, J.; Zhang, X.; Zhang, Q. PHSCNK-Modified and Doxorubicin-Loaded Liposomes as a Dual Targeting System to Integrin-Overexpressing Tumor Neovasculature and Tumor Cells. J. Drug Target. 2010, 18, 254–263. [Google Scholar] [CrossRef]
- Chen, X.; Yu, Q.; Liu, Y.; Sheng, Q.; Shi, K.; Wang, Y.; Li, M.; Zhang, Z.; He, Q. Synergistic Cytotoxicity and Co-Autophagy Inhibition in Pancreatic Tumor Cells and Cancer-Associated Fibroblasts by Dual Functional Peptide-Modified Liposomes. Acta Biomater. 2019, 99, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhang, H.; Peng, Y.; Fu, Q.; Yue, Q.; Zhao, Y.; Guo, L.; Wu, Y. Dual-Targeting Liposomes with Active Recognition of GLUT5 and αvβ3 for Triple-Negative Breast Cancer. Eur. J. Med. Chem. 2019, 183, 111720. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Wang, H.; Tang, G.; Huang, T.; Tang, X.; Liang, X.; Yao, S.; Nie, D. In Vivo Cancer Dual-Targeting and Dual-Modality Imaging with Functionalized Quantum Dots. J. Nucl. Med. 2015, 56, 1278–1284. [Google Scholar] [CrossRef]
- Arosio, D.; Manzoni, L.; Araldi, E.M.V.; Scolastico, C. Cyclic RGD Functionalized Gold Nanoparticles for Tumor Targeting. Bioconjug. Chem. 2011, 22, 664–672. [Google Scholar] [CrossRef]
- Shipunova, V.O.; Sogomonyan, A.S.; Zelepukin, I.V.; Nikitin, M.P.; Deyev, S.M. PLGA Nanoparticles Decorated with Anti-HER2 Affibody for Targeted Delivery and Photoinduced Cell Death. Molecules 2021, 26, 3955. [Google Scholar] [CrossRef] [PubMed]
- Shipunova, V.O.; Kolesnikova, O.A.; Kotelnikova, P.A.; Soloviev, V.D.; Popov, A.A.; Proshkina, G.M.; Nikitin, M.P.; Deyev, S.M. Comparative Evaluation of Engineered Polypeptide Scaffolds in HER2-Targeting Magnetic Nanocarrier Delivery. ACS Omega 2021, 6, 16000–16008. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, A.; Gasparri, A.M.; Monieri, M.; Anderluzzi, G.; Colombo, B.; Gori, A.; Corti, A.; Curnis, F. Nanogold Functionalized With Lipoamide-IsoDGR: A Simple, Robust and Versatile Nanosystem for αvβ3-Integrin Targeting. Front. Chem. 2021, 9, 690357. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-H.; Jeon, J.; Hong, S.H.; Rhim, W.-K.; Lee, Y.-S.; Youn, H.; Chung, J.-K.; Lee, M.C.; Lee, D.S.; Kang, K.W.; et al. Tumor Targeting and Imaging Using Cyclic RGD-PEGylated Gold Nanoparticle Probes with Directly Conjugated Iodine-125. Small 2011, 7, 2052–2060. [Google Scholar] [CrossRef]
- Liang, G.; Jin, X.; Zhang, S.; Xing, D. RGD Peptide-Modified Fluorescent Gold Nanoclusters as Highly Efficient Tumor-Targeted Radiotherapy Sensitizers. Biomaterials 2017, 144, 95–104. [Google Scholar] [CrossRef]
- Ali, M.R.K.; Wu, Y.; Tang, Y.; Xiao, H.; Chen, K.; Han, T.; Fang, N.; Wu, R.; El-Sayed, M.A. Targeting Cancer Cell Integrins Using Gold Nanorods in Photothermal Therapy Inhibits Migration through Affecting Cytoskeletal Proteins. Proc. Natl. Acad. Sci. USA 2017, 114, E5655–E5663. [Google Scholar] [CrossRef]
- Egorova, E.A.; van Rijt, M.M.J.; Sommerdijk, N.; Gooris, G.S.; Bouwstra, J.A.; Boyle, A.L.; Kros, A. One Peptide for Them All: Gold Nanoparticles of Different Sizes Are Stabilized by a Common Peptide Amphiphile. ACS Nano 2020, 14, 5874–5886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-X.; Zhao, W.-Y.; Liu, L.; Ju, R.-J.; Mu, L.-M.; Zhao, Y.; Zeng, F.; Xie, H.-J.; Yan, Y.; Lu, W.-L. A Nanostructure of Functional Targeting Epirubicin Liposomes Dually Modified with Aminophenyl Glucose and Cyclic Pentapeptide Used for Brain Glioblastoma Treatment. Oncotarget 2015, 6, 32681–32700. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, Y.; Wang, L.; Liu, W.; Xiao, L.; Lin, Q.; Gong, T.; Sun, X.; He, Q.; Zhang, Z.; et al. Improved Melanoma Suppression with Target-Delivered TRAIL and Paclitaxel by a Multifunctional Nanocarrier. J. Control. Release 2020, 325, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Song, S.; Duan, N.; Wang, F.; Wang, Y.; Yang, Y.; Peng, C.; Li, J.; Nie, D.; Zhang, X.; et al. MT1-MMP-Activated Liposomes to Improve Tumor Blood Perfusion and Drug Delivery for Enhanced Pancreatic Cancer Therapy. Adv. Sci. 2020, 7, 1902746. [Google Scholar] [CrossRef]
- Yakavets, I.; Francois, A.; Guiot, M.; Lequeux, N.; Fragola, A.; Pons, T.; Bezdetnaya, L.; Marchal, F. NIR Imaging of the Integrin-Rich Head and Neck Squamous Cell Carcinoma Using Ternary Copper Indium Selenide/Zinc Sulfide-Based Quantum Dots. Cancers 2020, 12, 3727. [Google Scholar] [CrossRef]
- Arriortua, O.K.; Insausti, M.; Lezama, L.; Gil de Muro, I.; Garaio, E.; de la Fuente, J.M.; Fratila, R.M.; Morales, M.P.; Costa, R.; Eceiza, M.; et al. RGD-Functionalized Fe3O4 Nanoparticles for Magnetic Hyperthermia. Colloids Surf. B Biointerfaces 2018, 165, 315–324. [Google Scholar] [CrossRef]
- Kato, N.; Sato, T.; Fuchigami, Y.; Suga, T.; Geng, L.; Tsurumaru, M.; Hagimori, M.; Mukai, H.; Kawakami, S. Synthesis and Evaluation of a Novel Adapter Lipid Derivative for Preparation of Cyclic Peptide-Modified PEGylated Liposomes: Application of Cyclic RGD Peptide. Eur. J. Pharm. Sci. 2022, 176, 106239. [Google Scholar] [CrossRef]
- Ringaci, A.; Shevchenko, K.G.; Zelepukin, I.V.; Popova, A.V.; Nikitin, M.P. Phage-mimicking nanoagents for rapid depolymerase specificity screening against multidrug resistant bacteria. Biosens. Bioelectron. 2022, 213, 114444. [Google Scholar] [CrossRef]
- Zhang, M.-Z.; Li, C.; Fang, B.-Y.; Yao, M.-H.; Ren, Q.-Q.; Zhang, L.; Zhao, Y.-D. High Transfection Efficiency of Quantum Dot-Antisense Oligonucleotide Nanoparticles in Cancer Cells through Dual-Receptor Synergistic Targeting. Nanotechnology 2014, 25, 255102. [Google Scholar] [CrossRef]
- Wilder, L.M.; Fies, W.A.; Rabin, C.; Webb, L.J.; Crooks, R.M. Conjugation of an α-Helical Peptide to the Surface of Gold Nanoparticles. Langmuir 2019, 35, 3363–3371. [Google Scholar] [CrossRef]
- Gasparri, A.M.; Sacchi, A.; Basso, V.; Cortesi, F.; Freschi, M.; Rrapaj, E.; Bellone, M.; Casorati, G.; Dellabona, P.; Mondino, A.; et al. Boosting Interleukin-12 Antitumor Activity and Synergism with Immunotherapy by Targeted Delivery with isoDGR-Tagged Nanogold. Small 2019, 15, 1903462. [Google Scholar] [CrossRef]
- Corti, A.; Sacchi, A.; Gasparri, A.M.; Monieri, M.; Anderluzzi, G.; Colombo, B.; Gori, A.; Mondino, A.; Curnis, F. Enhancement of Doxorubicin Anti-Cancer Activity by Vascular Targeting Using isoDGR/Cytokine-Coated Nanogold. J. Nanobiotechnology 2021, 19, 128. [Google Scholar] [CrossRef]
- Proshkina, G.M.; Shramova, E.I.; Shilova, M.V.; Zelepukin, I.V.; Shipunova, V.O.; Ryabova, A.V.; Deyev, S.M.; Kotlyar, A.B. DARPin_9-29-Targeted Gold Nanorods Selectively Suppress HER2-Positive Tumor Growth in Mice. Cancers 2021, 13, 5235. [Google Scholar] [CrossRef]
- Nikitin, M.P.; Zelepukin, I.V.; Shipunova, V.O.; Sokolov, I.L.; Deyev, S.M.; Nikitin, P.I. Enhancement of the blood-circulation time and performance of nanomedicines via the forced clearance of erythrocytes. Nat. Biomed. Eng. 2020, 4, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ji, Y. RGD-Modified Polymer and Liposome Nanovehicles: Recent Research Progress for Drug Delivery in Cancer Therapeutics. Eur. J. Pharm. Sci. 2019, 128, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Xia, Y.; Zou, Y.; Yang, W.; Zhang, J.; Zhong, Z.; Meng, F. ATN-161 Peptide Functionalized Reversibly Cross-Linked Polymersomes Mediate Targeted Doxorubicin Delivery into Melanoma-Bearing C57BL/6 Mice. Mol. Pharm. 2017, 14, 2538–2547. [Google Scholar] [CrossRef]
- Yan, Y.; Gong, J.; Chen, J.; Zeng, Z.; Huang, W.; Pu, K.; Liu, J.; Chen, P. Recent Advances on Graphene Quantum Dots: From Chemistry and Physics to Applications. Adv. Mater. 2019, 31, 1808283. [Google Scholar] [CrossRef]
- Tang, R.; Xue, J.; Xu, B.; Shen, D.; Sudlow, G.P.; Achilefu, S. Tunable Ultrasmall Visible-to-Extended Near-Infrared Emitting Silver Sulfide Quantum Dots for Integrin-Targeted Cancer Imaging. ACS Nano 2015, 9, 220–230. [Google Scholar] [CrossRef]
- Li, M.-M.; Cao, J.; Yang, J.-C.; Shen, Y.-J.; Cai, X.-L.; Chen, Y.-W.; Qu, C.-Y.; Zhang, Y.; Shen, F.; Zhou, M.; et al. Biodistribution and Toxicity Assessment of Intratumorally Injected Arginine–Glycine–Aspartic Acid Peptide Conjugated to CdSe/ZnS Quantum Dots in Mice Bearing Pancreatic Neoplasm. Chem. Biol. Interact. 2018, 291, 103–110. [Google Scholar] [CrossRef]
- Yang, K.; Wang, Y.-W.; Tang, H.; Chen, D.; Bai, Y.-L. Toxicity Assessment of Repeated Intravenous Injections of Arginine-Glycine-Aspartic Acid Peptide Conjugated CdSeTe/ZnS Quantum Dots in Mice. Int. J. Nanomed. 2014, 4809. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiu, W.; Sun, Y.; Zhu, D.; Zhang, Q.; Yuwen, L.; Weng, L.; Teng, Z.; Wang, L. RGD-QD-MoS2 Nanosheets for Targeted Fluorescent Imaging and Photothermal Therapy of Cancer. Nanoscale 2017, 9, 15835–15845. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Crawford, B.M.; Vo-Dinh, T. Gold Nanoparticles-Mediated Photothermal Therapy and Immunotherapy. Immunotherapy 2018, 10, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Shipunova, V.O.; Belova, M.M.; Kotelnikova, P.A.; Shilova, O.N.; Mirkasymov, A.B.; Danilova, N.V.; Komedchikova, E.N.; Popovtzer, R.; Deyev, S.M.; Nikitin, M.P. Photothermal Therapy with HER2-Targeted Silver Nanoparticles Leading to Cancer Remission. Pharmaceutics 2022, 14, 1013. [Google Scholar] [CrossRef]
- Jose, J.; Kumar, R.; Harilal, S.; Mathew, G.E.; Parambi, D.G.T.; Prabhu, A.; Uddin, M.S.; Aleya, L.; Kim, H.; Mathew, B. Magnetic Nanoparticles for Hyperthermia in Cancer Treatment: An Emerging Tool. Environ. Sci. Pollut. Res. 2020, 27, 19214–19225. [Google Scholar] [CrossRef]
- Vilas-Boas, V.; Carvalho, F.; Espiña, B. Magnetic Hyperthermia for Cancer Treatment: Main Parameters Affecting the Outcome of In Vitro and In Vivo Studies. Molecules 2020, 25, 2874. [Google Scholar] [CrossRef]
- Nam, J.-M.; Ahmed, K.M.; Costes, S.; Zhang, H.; Onodera, Y.; Olshen, A.B.; Hatanaka, K.C.; Kinoshita, R.; Ishikawa, M.; Sabe, H.; et al. Β1-Integrin via NF-ΚB Signaling Is Essential for Acquisition of Invasiveness in a Model of Radiation Treated in Situ Breast Cancer. Breast Cancer Res. 2013, 15, R60. [Google Scholar] [CrossRef]
- Peiris, P.M.; Deb, P.; Doolittle, E.; Doron, G.; Goldberg, A.; Govender, P.; Shah, S.; Rao, S.; Carbone, S.; Cotey, T.; et al. Vascular Targeting of a Gold Nanoparticle to Breast Cancer Metastasis. J. Pharm. Sci. 2015, 104, 2600–2610. [Google Scholar] [CrossRef]
- Poon, W.; Zhang, X.; Bekah, D.; Teodoro, J.G.; Nadeau, J.L. Targeting B16 Tumors in Vivo with Peptide-Conjugated Gold Nanoparticles. Nanotechnology 2015, 26, 285101. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Cai, J.; Li, X.; Cheng, D.; Su, H.; Zhang, J.; Liu, S.; Shi, H.; Zhang, Y.; et al. Tumor Angiogenesis Targeted Radiosensitization Therapy Using Gold Nanoprobes Guided by MRI/SPECT Imaging. ACS Appl. Mater. Interfaces 2016, 8, 1718–1732. [Google Scholar] [CrossRef]
- Yang, C.; Bromma, K.; Chithrani, D. Peptide Mediated In Vivo Tumor Targeting of Nanoparticles through Optimization in Single and Multilayer In Vitro Cell Models. Cancers 2018, 10, 84. [Google Scholar] [CrossRef]
- Huang, X.; Kang, B.; Qian, W.; Mackey, M.A.; Chen, P.C.; Oyelere, A.K.; El-Sayed, I.H.; El-Sayed, M.A. Comparative Study of Photothermolysis of Cancer Cells with Nuclear-Targeted or Cytoplasm-Targeted Gold Nanospheres: Continuous Wave or Pulsed Lasers. J. Biomed. Opt. 2010, 15, 058002. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Selim, S.A.; Aili, D. Effects of Macrophage Polarization on Gold Nanoparticle-Assisted Plasmonic Photothermal Therapy. RSC Adv. 2021, 11, 25047–25056. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, C.; Cheng, R.; Cheng, L.; Meng, F.; Liu, Z.; Zhong, Z. CRGD-Directed, NIR-Responsive and Robust AuNR/PEG–PCL Hybrid Nanoparticles for Targeted Chemotherapy of Glioblastoma in vivo. J. Control. Release 2014, 195, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Tan, G.; Zhong, Y.; Jiang, Y.; Cai, L.; Yu, Z.; Liu, S.; Ren, F. Smart Nanoplatform for Sequential Drug Release and Enhanced Chemo-Thermal Effect of Dual Drug Loaded Gold Nanorod Vesicles for Cancer Therapy. J. Nanobiotechnology 2019, 17, 44. [Google Scholar] [CrossRef]
- Wei, P.; Chen, J.; Hu, Y.; Li, X.; Wang, H.; Shen, M.; Shi, X. Dendrimer-Stabilized Gold Nanostars as a Multifunctional Theranostic Nanoplatform for CT Imaging, Photothermal Therapy, and Gene Silencing of Tumors. Adv. Healthc. Mater. 2016, 5, 3203–3213. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.-H.; Onodera, Y.; Ichikawa, Y.; Rankin, E.; Giaccia, A.; Watanabe, Y.; Qian, W.; Hashimoto, T.; Shirato, H.; Nam, J.-M. Targeting Integrins with RGD-Conjugated Gold Nanoparticles in Radiotherapy Decreases the Invasive Activity of Breast Cancer Cells. Int. J. Nanomed. 2017, 12, 5069–5085. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wang, S.; Wang, X.; Rong, P.; Ma, Y.; Liu, G.; Huang, P.; Lu, G.; Chen, X. In Vivo Volumetric Photoacoustic Molecular Angiography and Therapeutic Monitoring with Targeted Plasmonic Nanostars. Small 2014, 10, 1585–1593. [Google Scholar] [CrossRef]
- Nicolson, F.; Andreiuk, B.; Andreou, C.; Hsu, H.-T.; Rudder, S.; Kircher, M.F. Non-Invasive In Vivo Imaging of Cancer Using Surface-Enhanced Spatially Offset Raman Spectroscopy (SESORS). Theranostics 2019, 9, 5899–5913. [Google Scholar] [CrossRef]
- Shipunova, V.O.; Kotelnikova, P.A.; Aghayeva, U.F.; Stremovskiy, O.A.; Novikov, I.A.; Schulga, A.A.; Nikitin, M.P.; Deyev, S.M. Self-assembling nanoparticles biofunctionalized with magnetite-binding protein for the targeted delivery to HER2/neu overexpressing cancer cells. J. Magn. Magn. Mater. 2019, 469, 450–455. [Google Scholar] [CrossRef]
- Ringaci, A.; Yaremenko, A.V.; Shevchenko, K.G.; Zvereva, S.D.; Nikitin, M.P. Metal-organic frameworks for simultaneous gene and small molecule delivery in vitro and in vivo. Chem. Eng. J. 2021, 418, 129386. [Google Scholar] [CrossRef]
- Bragina, V.A.; Khomyakova, E.; Orlov, A.V.; Znoyko, S.L.; Mochalova, E.N.; Paniushkina, L.; Shender, V.O.; Erbes, T.; Evtushenko, E.G.; Bagrov, D.V.; et al. Highly Sensitive Nanomagnetic Quantification of Extracellular Vesicles by Immunochromatographic Strips: A Tool for Liquid Biopsy. Nanomaterials 2022, 12, 1579. [Google Scholar] [CrossRef] [PubMed]
- Znoyko, S.L.; Orlov, A.V.; Bragina, V.A.; Nikitin, M.P.; Nikitin, P.I. Nanomagnetic lateral flow assay for high-precision quantification of diagnostically relevant concentrations of serum TSH. Talanta 2020, 216, 120961. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.D.; Ringaci, A.; Alenichev, M.K.; Drozhzhennikova, E.B.; Shevchenko, K.G.; Cherkasov, V.R.; Nikitin, M.P.; Nikitin, P.I. Dynamic light scattering biosensing based on analyte-induced inhibition of nanoparticle aggregation. Anal. Bioanal. Chem. 2020, 412, 3423–3431. [Google Scholar] [CrossRef] [PubMed]
- Mochalova, E.N.; Kotov, I.V.; Rozenberg, J.M.; Nikitin, M.P. Precise Quantitative Analysis of Cell Targeting by Particle-Based Agents Using Imaging Flow Cytometry and Convolutional Neural Network. Cytometry A 2020, 97, 279–287. [Google Scholar] [CrossRef]
- Wust, P.; Hildebrandt, B.; Sreenivasa, G.; Rau, B.; Gellermann, J.; Riess, H.; Felix, R.; Schlag, P. Hyperthermia in Combined Treatment of Cancer. Lancet Oncol. 2002, 3, 487–497. [Google Scholar] [CrossRef]
- Shakil, M.S.; Hasan, M.A.; Sarker, S.R. Iron Oxide Nanoparticles for Breast Cancer Theranostics. Curr. Drug Metab. 2019, 20, 446–456. [Google Scholar] [CrossRef]
- Wu, T.; Ding, X.; Su, B.; Soodeen-Lalloo, A.K.; Zhang, L.; Shi, J.-Y. Magnetic Resonance Imaging of Tumor Angiogenesis Using Dual-Targeting RGD10–NGR9 Ultrasmall Superparamagnetic Iron Oxide Nanoparticles. Clin. Transl. Oncol. 2018, 20, 599–606. [Google Scholar] [CrossRef]
- Cho, H.-Y.; Mavi, A.; Chueng, S.-T.D.; Pongkulapa, T.; Pasquale, N.; Rabie, H.; Han, J.; Kim, J.H.; Kim, T.-H.; Choi, J.-W.; et al. Tumor Homing Reactive Oxygen Species Nanoparticle for Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 23909–23918. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Turovsky, E.A.; Blinova, E.V. Therapeutic Potential and Main Methods of Obtaining Selenium Nanoparticles. Int. J. Mol. Sci. 2021, 22, 10808. [Google Scholar] [CrossRef]
- Varlamova, E.G.; Goltyaev, M.V.; Mal’tseva, V.N.; Turovsky, E.A.; Sarimov, R.M.; Simakin, A.V.; Gudkov, S.V. Mechanisms of the Cytotoxic Effect of Selenium Nanoparticles in Different Human Cancer Cell Lines. Int. J. Mol. Sci. 2021, 22, 7798. [Google Scholar] [CrossRef]
- Xia, Y.; Tang, G.; Wang, C.; Zhong, J.; Chen, Y.; Hua, L.; Li, Y.; Liu, H.; Zhu, B. Functionalized selenium nanoparticles for targeted siRNA delivery silence Derlin1 and promote antitumor efficacy against cervical cancer. Drug Deliv. 2020, 27, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Tang, G.; Guo, M.; Xu, T.; Chen, H.; Lin, Z.; Li, Y.; Chen, Y.; Zhu, B.; Liu, H.; et al. Silencing KLK12 expression via RGDfC-decorated selenium nanoparticles for the treatment of colorectal cancer in vitro and in vivo. Mater. Sci. Eng. C 2020, 110, 110594. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Tang, G.; Chen, Y.; Wang, C.; Guo, M.; Xu, T.; Zhao, M.; Zhou, Y. Tumor-targeted delivery of siRNA to silence Sox2 gene expression enhances therapeutic response in hepatocellular carcinoma. Bioact. Mater. 2021, 6, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Kolesanova, E.F.; Melnikova, M.V.; Bolshakova, T.N.; Rybalkina, E.Y.; Sivov, I.G. Bacteriophage MS2 As a Tool for Targeted Delivery in Solid Tumor Chemotherapy. Acta Nat. 2019, 11, 98–101. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, P.; Xi, Y.; Chen, X.; He, Z.; Meng, T.; Chen, J.; Li, L.; Zhu, J.-J. Hierarchical Nanocarriers for Precisely Regulating the Therapeutic Process via Dual-Mode Controlled Drug Release in Target Tumor Cells. ACS Appl. Mater. Interfaces 2017, 9, 36655–36664. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, H.; Li, X.; Lv, Y.; Ma, T.; Guo, S.; Huang, Z.; Wang, X.; Xu, P. Dual Targeting Hyaluronic Acid—RGD Mesoporous Silica Coated Gold Nanorods for Chemo-Photothermal Cancer Therapy. Mater. Sci. Eng. C 2017, 81, 261–270. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, M.; Zhang, W.; Huang, L.; Miao, D.; Zhu, H.; Su, G. Rattle-Type Gold Nanorods/Porous-SiO2 Nanocomposites as Near-Infrared Light-Activated Drug Delivery Systems for Cancer Combined Chemo–Photothermal Therapy. Mol. Pharm. 2019, 16, 1929–1938. [Google Scholar] [CrossRef]
- Chen, G.; Xie, Y.; Peltier, R.; Lei, H.; Wang, P.; Chen, J.; Hu, Y.; Wang, F.; Yao, X.; Sun, H. Peptide-Decorated Gold Nanoparticles as Functional Nano-Capping Agent of Mesoporous Silica Container for Targeting Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 11204–11209. [Google Scholar] [CrossRef]
- Kobatake, E.; Ikeda, Y.; Mie, M. Construction of Protein Nanoparticles for Targeted Delivery of Drugs to Cancer Cells. Mater. Adv. 2022, 3, 6262–6269. [Google Scholar] [CrossRef]
- Song, Y.; Li, W.; Meng, S.; Zhou, W.; Su, B.; Tang, L.; Zhao, Y.; Wu, X.; Yin, D.; Fan, M.; et al. Dual Integrin αvβ3 and NRP-1-Targeting Paramagnetic Liposome for Tumor Early Detection in Magnetic Resonance Imaging. Nanoscale Res. Lett. 2018, 13, 380. [Google Scholar] [CrossRef]
- Garcia Ribeiro, R.S.; Belderbos, S.; Danhier, P.; Gallo, J.; Manshian, B.; Gallez, B.; Bañobre, M.; de Cuyper, M.; Soenen, S.; Gsell, W.; et al. Targeting Tumor Cells and Neovascularization Using RGD-Functionalized Magnetoliposomes. Int. J. Nanomed. 2019, 14, 5911–5924. [Google Scholar] [CrossRef]
- Hadad, E.; Rudnick-Glick, S.; Grinberg, I.; Kolitz-Domb, M.; Chill, J.H.; Margel, S. Synthesis and Characterization of Poly(RGD) Proteinoid Polymers and NIR Fluorescent Nanoparticles of Optimal D,L-Configuration for Drug-Delivery Applications—In Vitro Study. ACS Omega 2020, 5, 23568–23577. [Google Scholar] [CrossRef]
- Hu, H.; Zhang, Y.; Shukla, S.; Gu, Y.; Yu, X.; Steinmetz, N.F. Dysprosium-Modified Tobacco Mosaic Virus Nanoparticles for Ultra-High-Field Magnetic Resonance and Near-Infrared Fluorescence Imaging of Prostate Cancer. ACS Nano 2017, 11, 9249–9258. [Google Scholar] [CrossRef]
- Rios De La Rosa, J.M.; Spadea, A.; Donno, R.; Lallana, E.; Lu, Y.; Puri, S.; Caswell, P.; Lawrence, M.J.; Ashford, M.; Tirelli, N. Microfluidic-Assisted Preparation of RGD-Decorated Nanoparticles: Exploring Integrin-Facilitated Uptake in Cancer Cell Lines. Sci. Rep. 2020, 10, 14505. [Google Scholar] [CrossRef]
- Alday-Parejo, B.; Stupp, R.; Rüegg, C. Are Integrins Still Practicable Targets for Anti-Cancer Therapy? Cancers 2019, 11, 978. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence 1 | Binding Integrin | Cancer Type | Ref. |
|---|---|---|---|---|
| RGD, (RGD)4 | RGD, RGDRGDRGDRGDPGC | αvβ1, αvβ3, αvβ5, αvβ6, αvβ8, α5β1, α8β1, αIIbβ3 | Broad spectrum antagonist with sub-micromolar binding affinity | [21,22] |
| Cilengitide® 2 | cyclo(-RGDfV-), end-to-end cyclized | αvβ1, αvβ3, αvβ5 | Supportive therapy for breast cancer therapy, glioblastoma, pancreatic cancer | [38,39] |
| iRGD (Internalizing RGD) | cyclo(-CRGDKGPDC-)-amide | αvβ3, αvβ5 | Prostate, breast, pancreas, and colon cancers; glioma; bone metastasis | [40,41] |
| cyclo[RGDfK(C-ε-6-aminocaproic acid)] | cyclo(-RGDfK-), C-ε-6-aminocaproic acid coupled to the Lysine side chain | αvβ3, αvβ5 | Pancreatic cancer and glioblastoma | [42,43,44,45,46] |
| cyclo-(-FRGDLAFpP-) | cyclo(-FRGDLAFpP-), end-to-end cyclized | αvβ6 | Colon, lung, cervix, breast, ovaries and fallopian tube, pancreas, head and neck cancers | [47] |
| cyclo-(-GLRGDLpP-) | cyclo(-GLRGDLpP-), end-to-end cyclized | αvβ8 | Human melanoma, glioblastomas, ovarian carcinomas | [48] |
| LDV | cyclo(-GLVD-BTD), cyclo(-SlDVP-), end-to-end cyclized, cyclo(-CWLDVC) | α4β1, α4β7 | Leukemia, melanoma. Mainly used in infectious disease studies | [49,50,51,52,53] |
| ATN-161 | Ac-PHSCN-NH2 | α5β1, αvβ3 | Advanced solid tumors typical for breast cancer, prostate carcinoma, glioma; metastasis of colon cancer in liver | [54,55,56,57,58,59] |
| PhScN | Ac-PhScN-NH2 | α5β1, αvβ3 | The same as its predecessor. Demonstrates lower IC50 in breast cancer model | [57] |
| ALOS-4 | cyclo(-CSSAGSLFC-) | αvβ3 | Melanoma, glioma, prostate, and breast cancers | [23,60] |
| C16Y | DFKLFAVYIKYR | αvβ3, α5β1 | Ovarian cancer, breast cancer | [11,15] |
| YSNSG | cyclo(-YSNSG-) | αvβ3 | Solid tumors, melanoma | [61,62,63] |
| VLA4pep | YCPDC | α4β1 | Multiple myeloma disease | [64,65] |
| GFOGER | GFOGER | α2β1, α11β1, α1β1, α10β1 | Mainly used in cell adhesion studies of breast cancer cells | [18,66] |
| IKVAV | IKVAV | α3β1, α6β1 | Mainly used in cell adhesion studies of breast cancer cells | [66,67,68] |
| AG86 | LGGLPSHYRARNI | α6β4 | HPV-18 positive cervical cancer | [69] |
| PR_b | SSPHSRNSGSGSGSG-SGRGDSP | α5β1 | Prostate cancer | [19,70] |
| A20(FMDV2) | NAVPNLRGDLQVLA-QKVART | αvβ6 | Head and neck squamous cell carcinoma, pancreatic cancer | [71,72] |
| Peptide | Type of AuNPs | Observed Effects | Ref. |
|---|---|---|---|
| cycloRGD | AuNCs (1.7–2.7 nm size range) | Higher affinity towards αvβ3 was observed for targeting AuNCs. A PEG spacer between the peptide and the AuNC surface was crucial for enhanced tumor accumulation. However, no significant therapeutic effect was described, possibly due to the flawed NP design. | [125,130,159] |
| RGD | AuNPs (29, 51, and 80 nm in diameter) | The 29 nm AuNPs showed higher tumor accumulation, longer plasma half-life than larger AuNPs. | [160] |
| RGD | AuNPs (10 nm in diameter) | These smaller AuNPs showed similar tumor accumulation as the 29 nm AuNPs from [149]. | [161] |
| (RGD)4 | AuNPs (30 nm in diameter) | In in vitro PPTT cell death mechanisms depended on the laser type and dosage. Lower energy continuous wave (cw) laser treatment led to apoptosis, while higher energy cw laser treatment led to necrosis. | [162] |
| (RGD)4 | AuNRs (25 by 6 nm) | This targeted PPTT mainly induced inhibition of Rho GTPases, actin, microtubule, and kinase-related signaling pathways. | [131,163] |
| cycloRGD | AuNRs (50 by 15 nm in [164]; 55 by 15 nm in [165]) | These AuNRs were used for DOX or paclitaxel co-delivery. This strategy combined effects of chemo-phototherapy, active targeting of tumors, and multiple stimuli-responsive drug release. | [164,165] |
| cycloRGD | AuNSs | These AuNSs showed high photothermal conversion efficiency producing a 24.2 °C increase in the tumor area temperature. A single treatment was sufficient to completely inhibit the tumor progression for at least two weeks. | [166] |
| RGD | AuNSs | This treatment caused caspase-dependent apoptosis in HepG2 cells, signs of mitochondria-mediated apoptosis, loss of lysosomal membrane integrity, and enhanced autophagy. Tumor temperature increased by ~20 °C during the PPTT treatment, comparably set up as in the above-mentioned study. | [30] |
| (RGD)4 | AuNPs (20 nm in diameter) | Combined radiotherapy with these AuNPs resulted in significant downregulation of fibronectin signaling by 50%. This phenomenon was accompanied by serious suppression of invasive activity by 67% compared to radiotherapy alone. | [167] |
| isoDGR | AuNPs (25 nm in diameter) | This multicomponent system comprised a gold core, cytokines IL-12 and/or TNF, and DOX. The synergistic effect was explained by affected endothelial permeability due to the isoDRG-peptide integrin-mediated uptake, and consequential reduction in drug penetration barriers. Presence of the cytokines was to trigger a reaction from immunocompetent cells directed towards the tumor. The results suggest that the displayed TNF played a more important role in the studied tumor inhibition than the displayed IL-12. | [128,142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egorova, E.A.; Nikitin, M.P. Delivery of Theranostic Nanoparticles to Various Cancers by Means of Integrin-Binding Peptides. Int. J. Mol. Sci. 2022, 23, 13735. https://doi.org/10.3390/ijms232213735
Egorova EA, Nikitin MP. Delivery of Theranostic Nanoparticles to Various Cancers by Means of Integrin-Binding Peptides. International Journal of Molecular Sciences. 2022; 23(22):13735. https://doi.org/10.3390/ijms232213735
Chicago/Turabian StyleEgorova, Elena A., and Maxim P. Nikitin. 2022. "Delivery of Theranostic Nanoparticles to Various Cancers by Means of Integrin-Binding Peptides" International Journal of Molecular Sciences 23, no. 22: 13735. https://doi.org/10.3390/ijms232213735
APA StyleEgorova, E. A., & Nikitin, M. P. (2022). Delivery of Theranostic Nanoparticles to Various Cancers by Means of Integrin-Binding Peptides. International Journal of Molecular Sciences, 23(22), 13735. https://doi.org/10.3390/ijms232213735