
Opaganib Protects against Radiation Toxicity: Implications for Homeland Security and Antitumor Radiotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
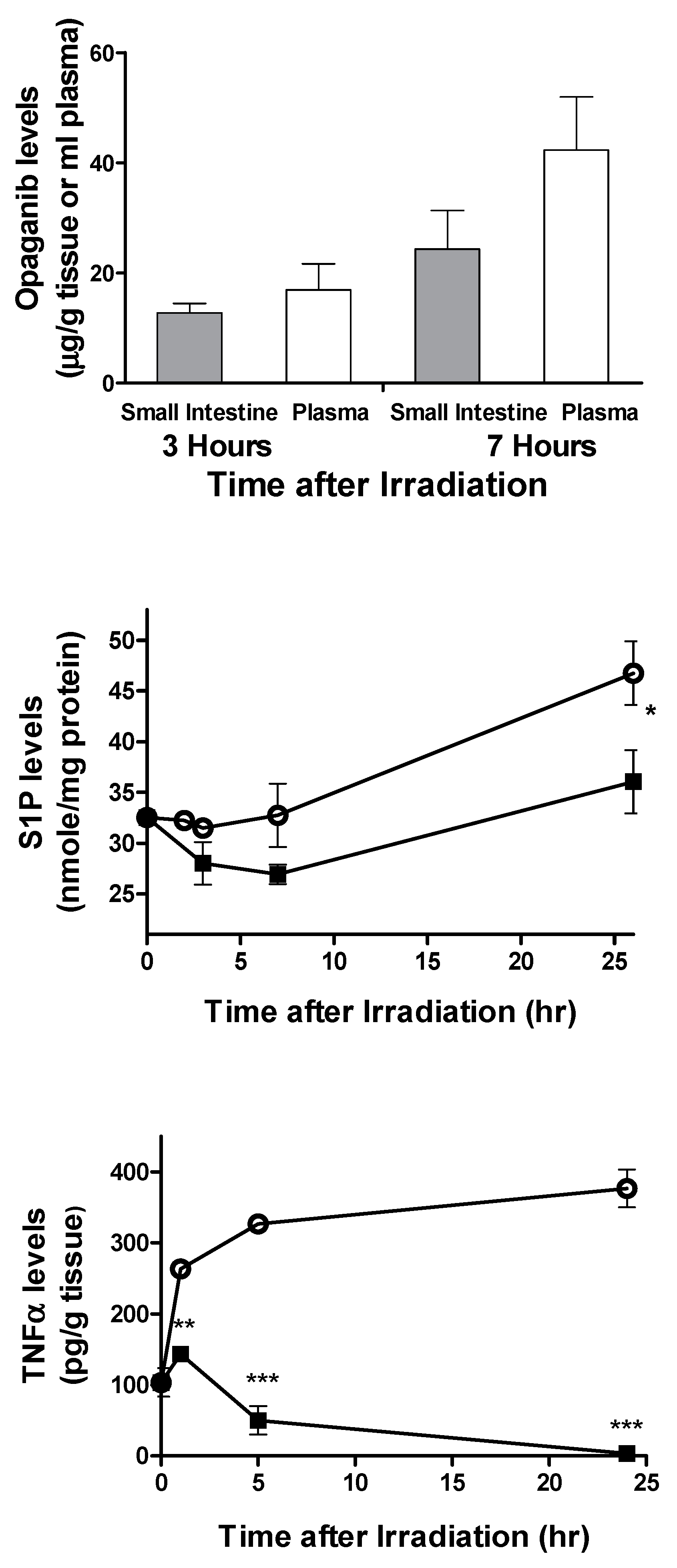
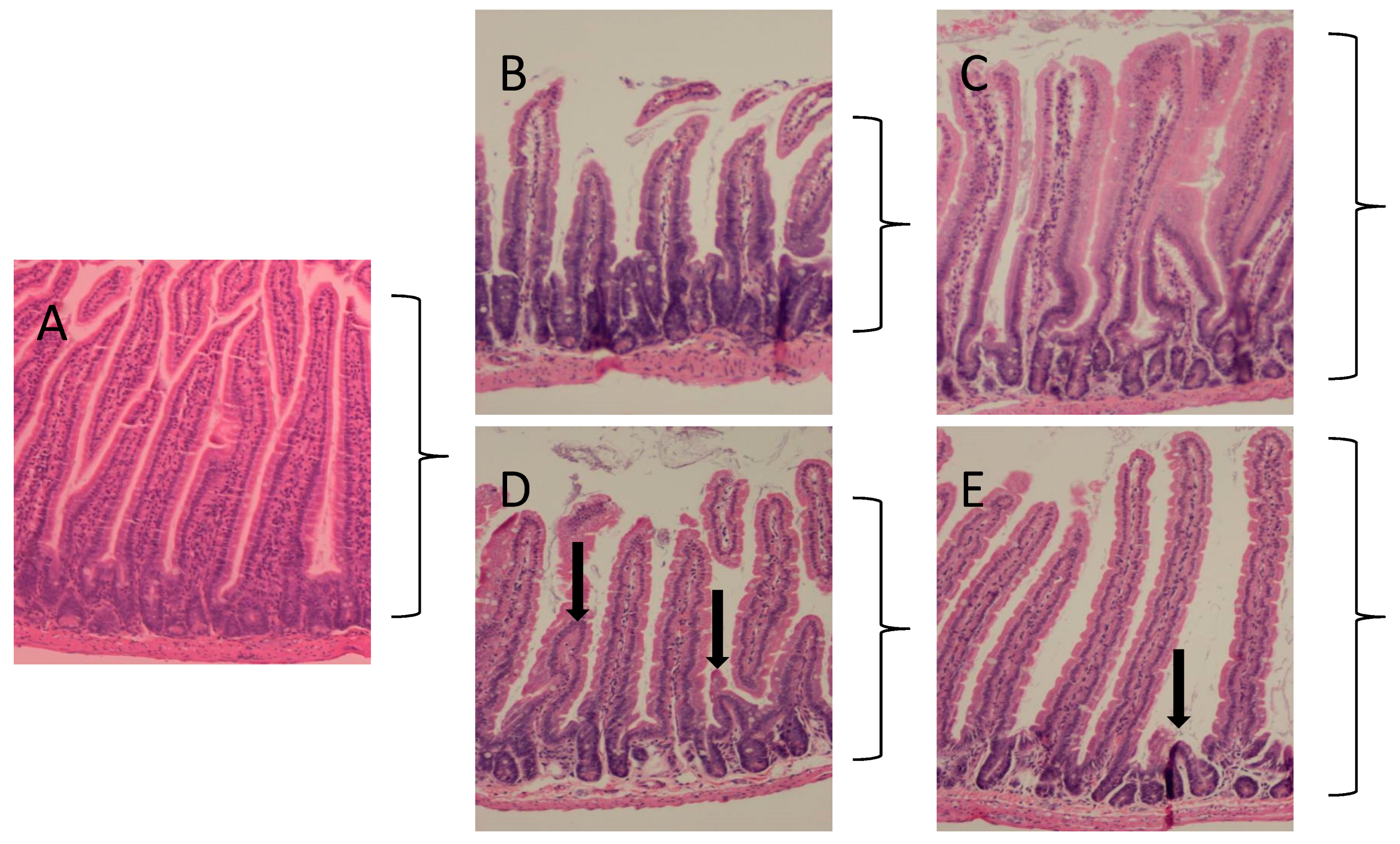
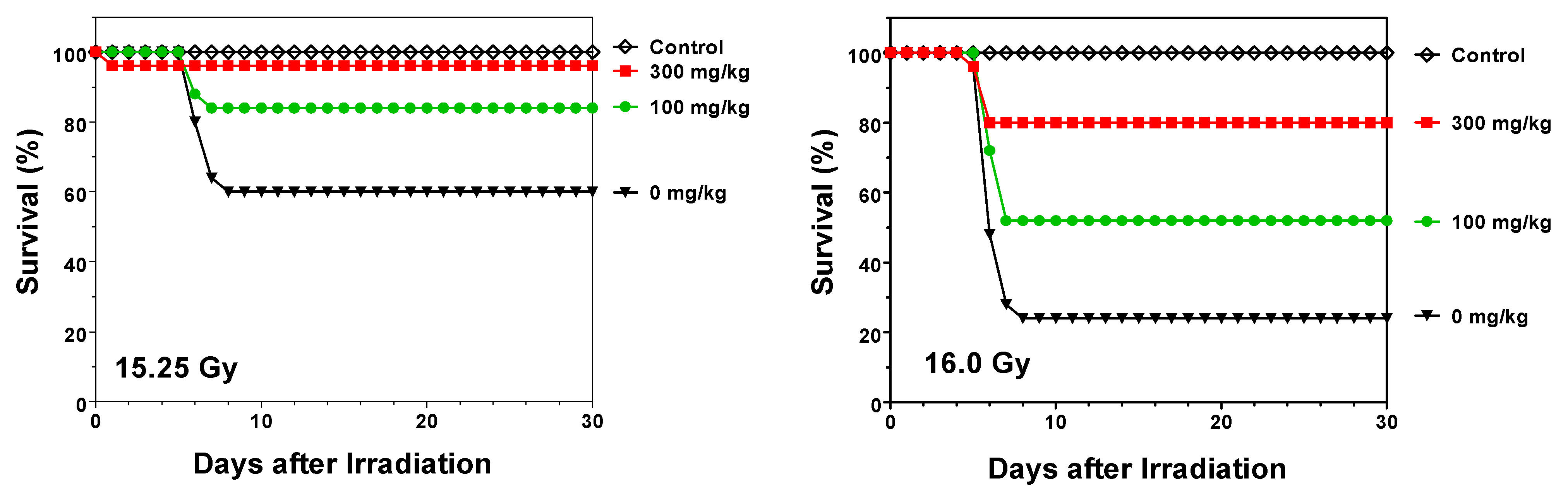
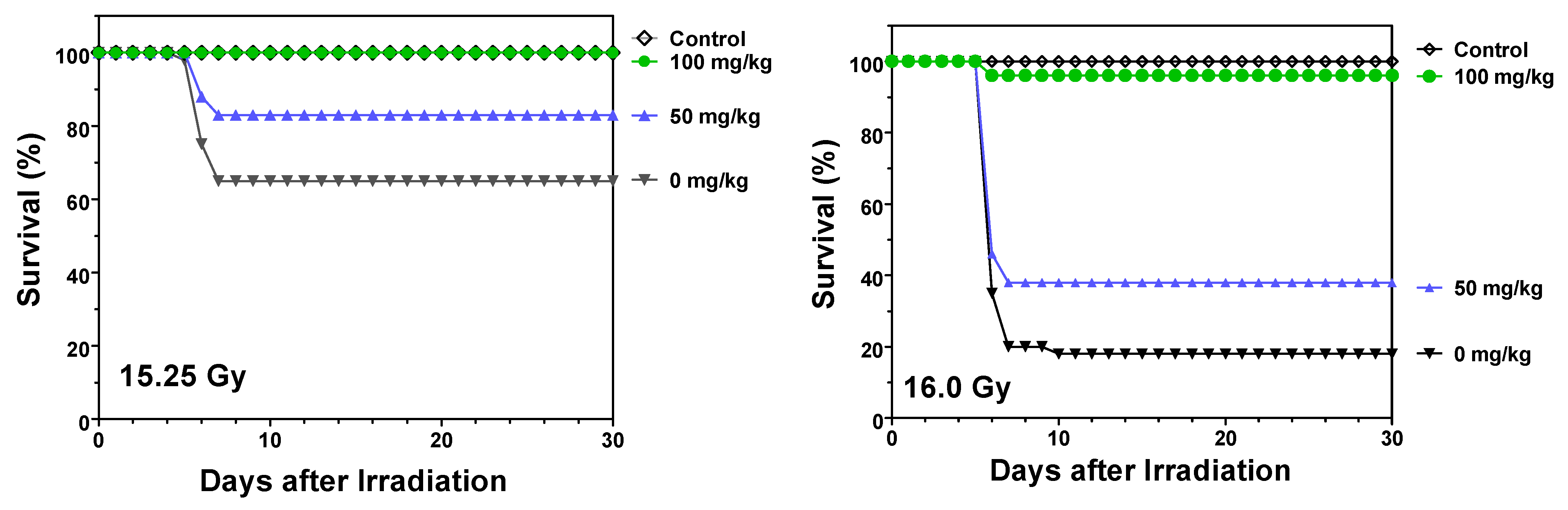
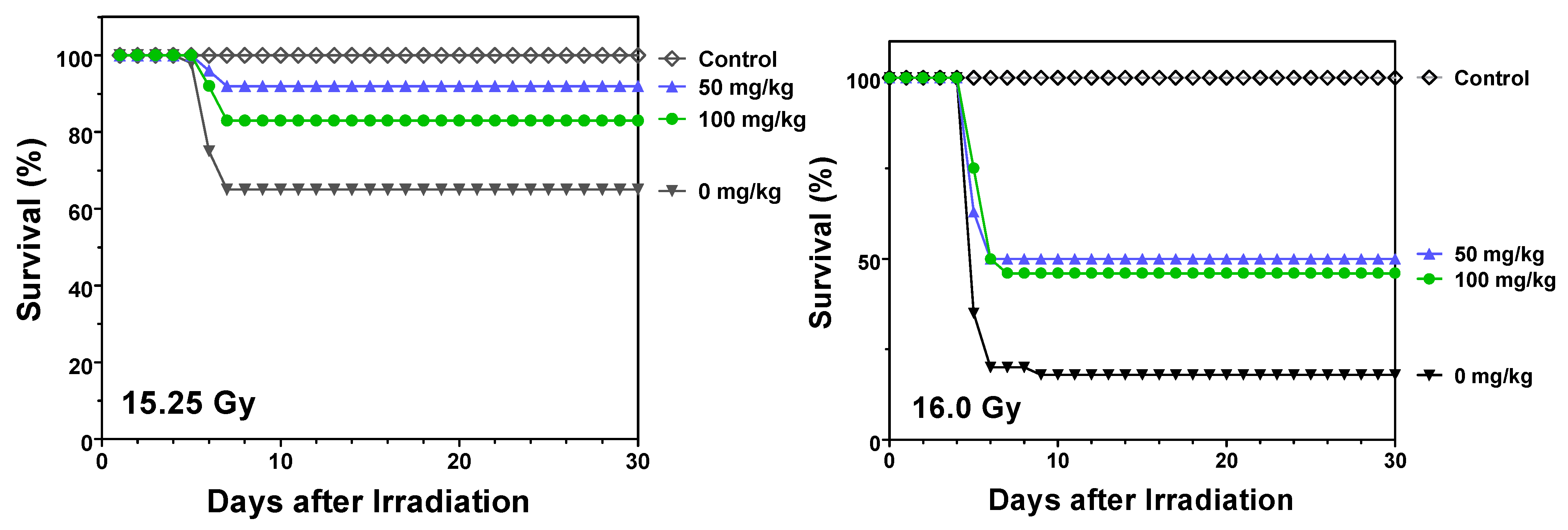
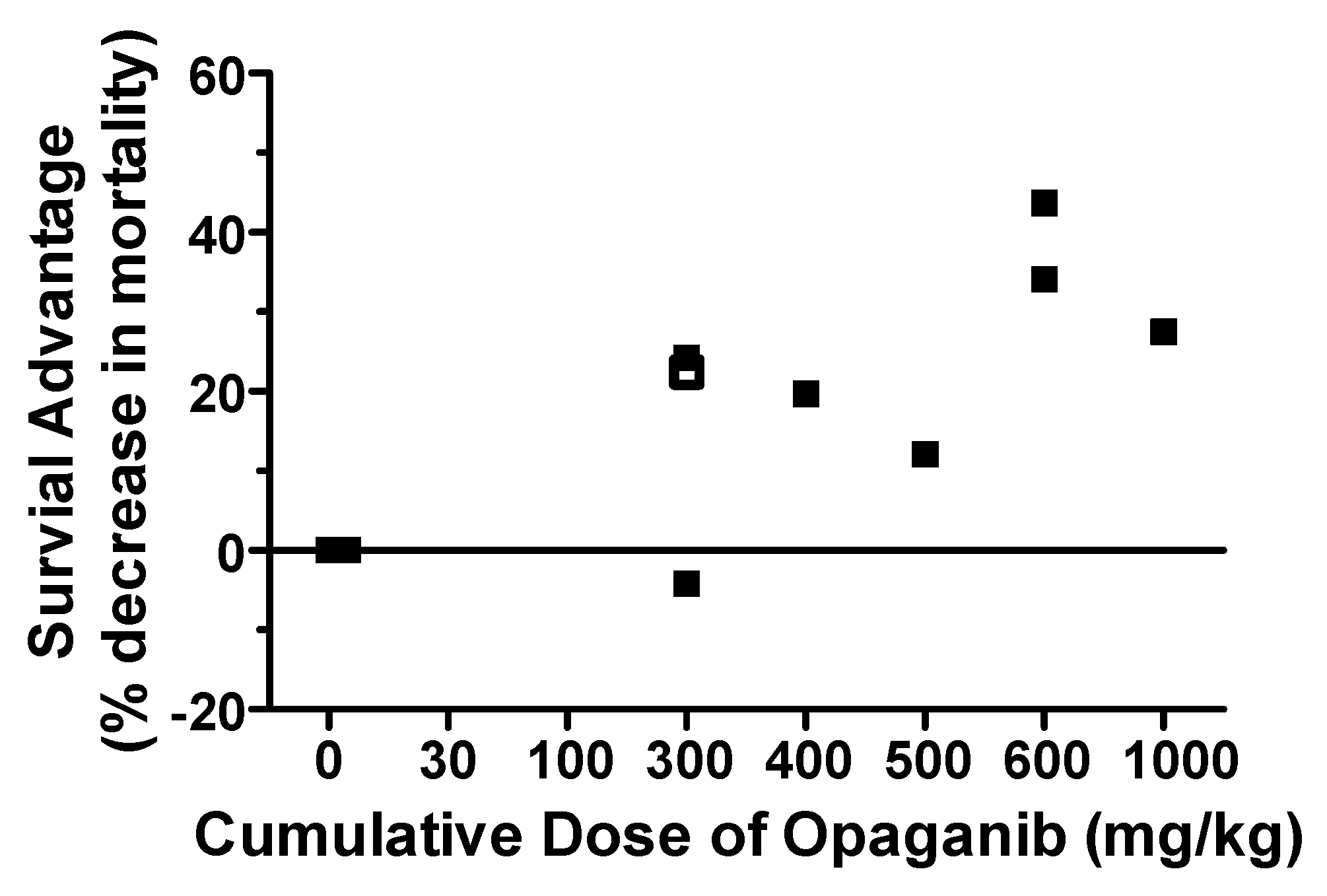
2.1. Protection against Radiation Toxicity Studies
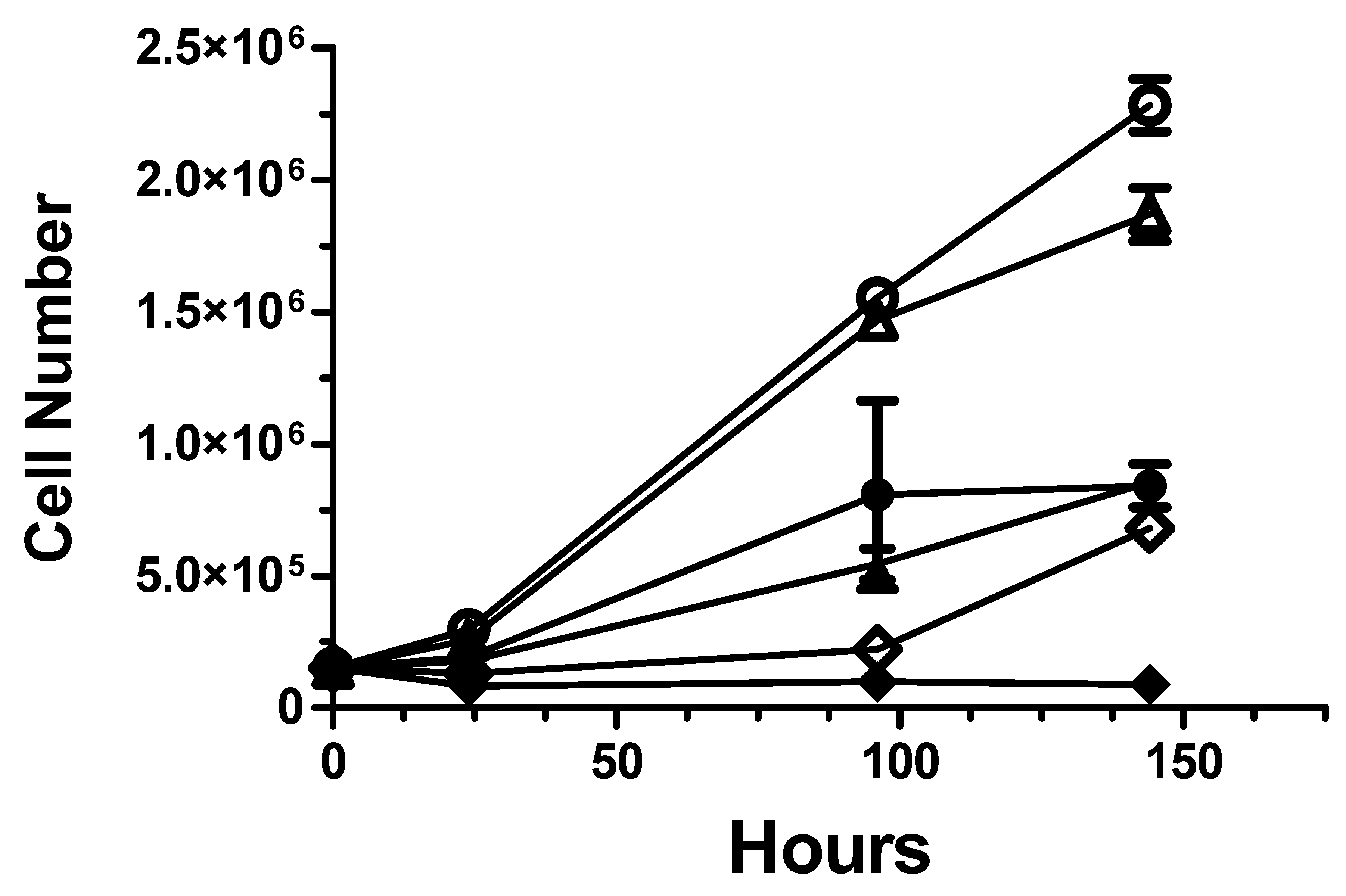
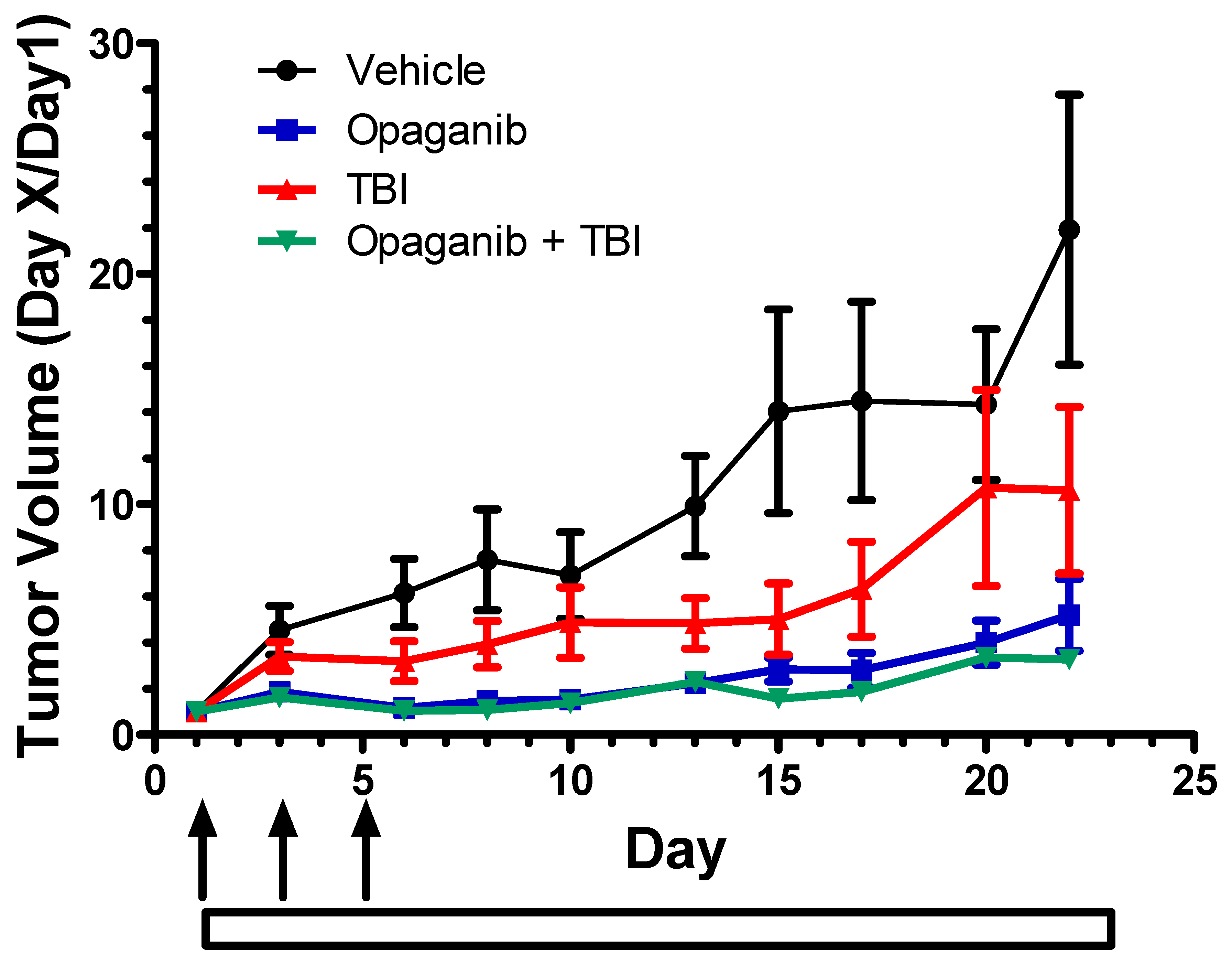
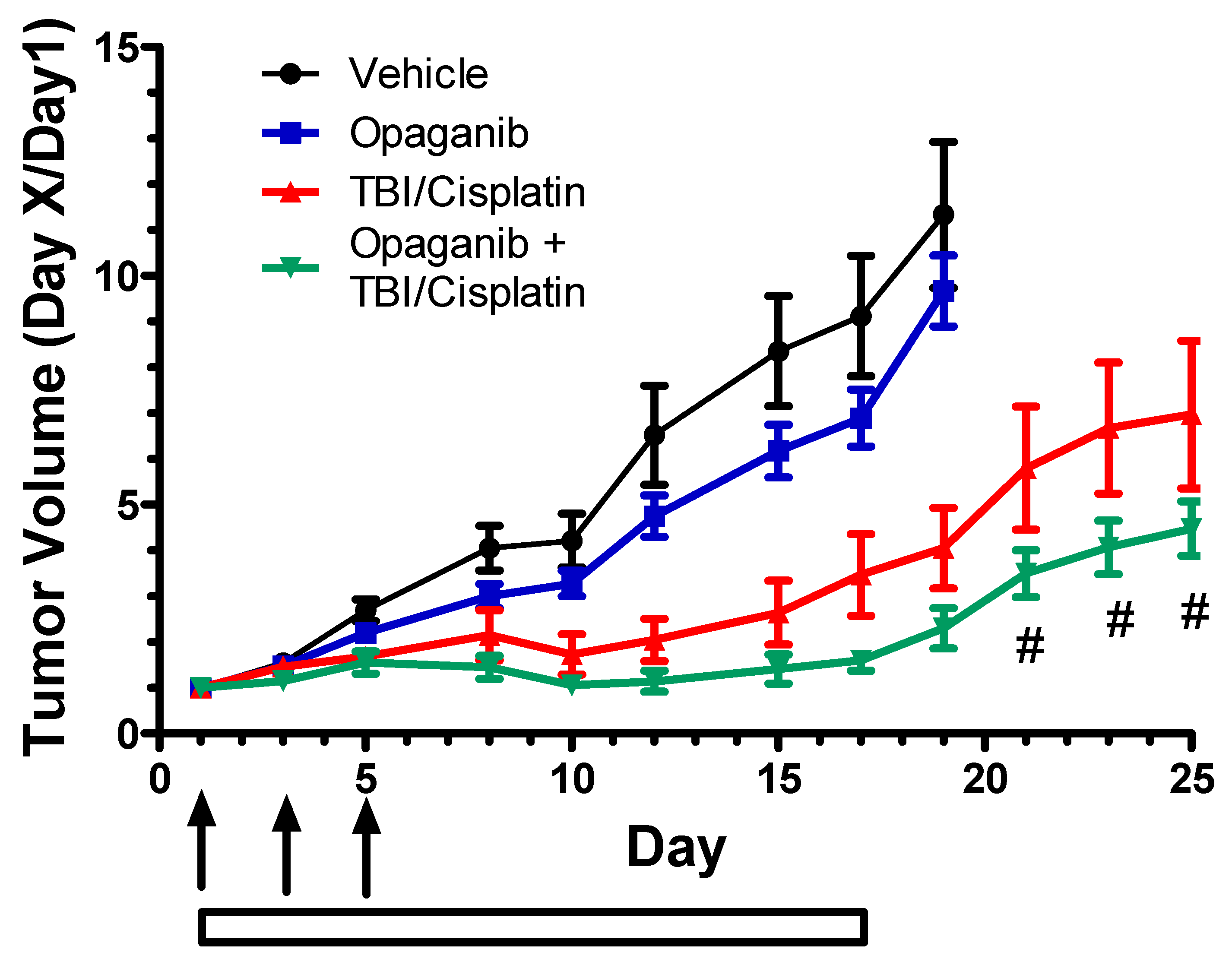
2.2. Cancer Radiotherapy Studies
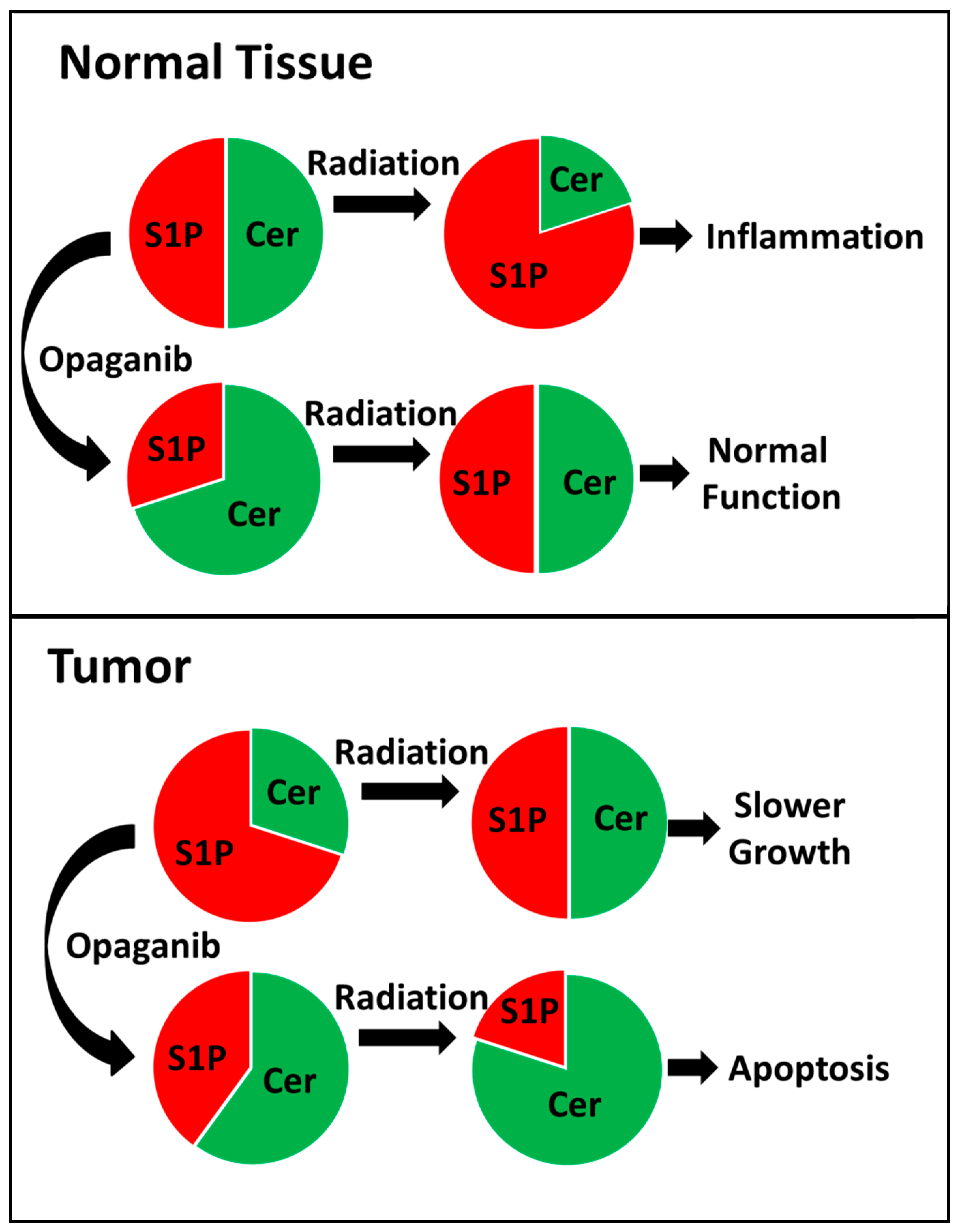
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
IACUC Statement
Acknowledgments
Conflicts of Interest
References
- Hekim, N.; Cetin, Z.; Nikitaki, Z.; Cort, A.; Saygili, E.I. Radiation triggering immune response and inflammation. Cancer Lett. 2015, 368, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Coates, P.J.; Lorimore, S.A.; Wright, E.G. Responses to ionizing radiation mediated by inflammatory mechanisms. J. Pathol. 2014, 232, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Motevaseli, E.; Shirazi, A.; Geraily, G.; Rezaeyan, A.; Norouzi, F.; Rezapoor, S.; Abdollahi, H. Mechanisms of inflammatory responses to radiation and normal tissues toxicity: Clinical implications. Int. J. Radiat. Biol. 2018, 94, 335–356. [Google Scholar] [CrossRef]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravatà, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamm. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Gupta, D.; Arora, R. NF-kB as a key player in regulation of cellular radiation responses and identification of radiation countermeasures. Discoveries 2015, 3, e35. [Google Scholar] [CrossRef]
- Yahyapour, R.; Amini, P.; Rezapoor, S.; Rezaeyan, A.; Farhood, B.; Cheki, M.; Fallah, H.; Najafi, M. Targeting of Inflammation for Radiation Protection and Mitigation. Curr. Mol. Pharmacol. 2018, 11, 203–210. [Google Scholar] [CrossRef]
- Yahyapour, R.; Amini, P.; Rezapour, S.; Cheki, M.; Rezaeyan, A.; Farhood, B.; Shabeeb, D.; Musa, A.E.; Fallah, H.; Najafi, M. Radiation-induced inflammation and autoimmune diseases. Mil. Med. Res. 2018, 5, 9. [Google Scholar] [CrossRef]
- Chen, G.; Han, Y.; Zhang, H.; Tu, W.; Zhang, S. Radiotherapy-Induced Digestive Injury: Diagnosis, Treatment and Mechanisms. Front. Oncol. 2021, 11, 757973. [Google Scholar] [CrossRef]
- Lu, L.; Li, W.; Chen, L.; Su, Q.; Wang, Y.; Guo, Z.; Lu, Y.; Liu, B.; Qin, S. Radiation-induced intestinal damage: Latest molecular and clinical developments. Futur. Oncol. 2019, 15, 4105–4118. [Google Scholar] [CrossRef]
- McKay, M.J.; Foster, R. Pathobiology, irradiation dosimetric parameters and therapy of radiation-induced gastric damage: A narrative review. J. Gastrointest. Oncol. 2021, 12, 3115–3122. [Google Scholar] [CrossRef]
- Reda, M.; Bagley, A.F.; Zaidan, H.Y.; Yantasee, W. Augmenting the therapeutic window of radiotherapy: A perspective on molecularly targeted therapies and nanomaterials. Radiother. Oncol. J. Eur. Soc. Ther. Radiol. Oncol. 2020, 150, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.W.; Kuo, M.T. Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget 2017, 8, 62742–62758. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.K.; Poortmans, P.; Chalmers, A.J.; Faivre-Finn, C.; Hall, E.; Huddart, R.A.; Lievens, Y.; Sebag-Montefiore, D.; Coles, C.E. Practice-changing radiation therapy trials for the treatment of cancer: Where are we 150 years after the birth of Marie Curie? Br. J. Cancer 2018, 119, 389–407. [Google Scholar] [CrossRef] [PubMed]
- Bismar, M.M.; Sinicrope, F.A. Radiation enteritis. Curr. Gastroenterol. Rep. 2002, 4, 361–365. [Google Scholar] [CrossRef]
- Toomey, D.P.; Cahill, R.A.; Geraghty, J.; Thirion, P. Radiation enteropathy. Ir. Med. J. 2006, 99, 215–217. [Google Scholar] [PubMed]
- Albeituni, S.; Stiban, J. Roles of Ceramides and Other Sphingolipids in Immune Cell Function and Inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [CrossRef]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Targeting Sphingosine Kinases for the Treatment of Cancer. Adv. Cancer Res. 2018, 140, 295–325. [Google Scholar] [CrossRef]
- Quinville, B.M.; Deschenes, N.M.; Ryckman, A.E.; Walia, J.S. A Comprehensive Review: Sphingolipid Metabolism and Implications of Disruption in Sphingolipid Homeostasis. Int. J. Mol. Sci. 2021, 22, 5793. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Cancer 2017, 18, 33–50. [Google Scholar] [CrossRef]
- Bataller, M.; Sánchez-García, A.; Garcia-Mayea, Y.; Mir, C.; Rodriguez, I.; Lleonart, M.E. The Role of Sphingolipids Metabolism in Cancer Drug Resistance. Front. Oncol. 2021, 11, 807636. [Google Scholar] [CrossRef]
- Hasanifard, L.; Sheervalilou, R.; Majidinia, M.; Yousefi, B. New insights into the roles and regulation of SphK2 as a therapeutic target in cancer chemoresistance. J. Cell. Physiol. 2018, 234, 8162–8181. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. The Onus of Sphingolipid Enzymes in Cancer Drug Resistance. Adv. Cancer Res. 2018, 140, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Nava, V.E.; Cuvillier, O.; Edsall, L.C.; Kimura, K.; Milstien, S.; Gelmann, E.P.; Spiegel, S. Sphingosine enhances apoptosis of radiation-resistant prostate cancer cells. Cancer Res. 2000, 60, 4468–4474. [Google Scholar]
- Beckham, T.H.; Cheng, J.C.; Marrison, S.T.; Norris, J.S.; Liu, X. Interdiction of Sphingolipid Metabolism to Improve Standard Cancer Therapies. Adv. Cancer Res. 2013, 117, 1–36. [Google Scholar] [CrossRef]
- Hajj, C.; Haimovitz-Friedman, A. Sphingolipids’ Role in Radiotherapy for Prostate Cancer. Handb. Exp. Pharmacol. 2013, 216, 115–130. [Google Scholar] [CrossRef]
- Sharma, D.; Czarnota, G.J. Involvement of Ceramide Signalling in Radiation-Induced Tumour Vascular Effects and Vascular-Targeted Therapy. Int. J. Mol. Sci. 2022, 23, 6671. [Google Scholar] [CrossRef]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and Antitumor Activity of ABC294640, a Selective Inhibitor of Sphingosine Kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef]
- Gao, P.; Peterson, Y.K.; Smith, R.A.; Smith, C.D. Characterization of Isoenzyme-Selective Inhibitors of Human Sphingosine Kinases. PLoS ONE 2012, 7, e44543. [Google Scholar] [CrossRef]
- Beljanski, V.; Knaak, C.; Smith, C.D. A Novel Sphingosine Kinase Inhibitor Induces Autophagy in Tumor Cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting NFĸB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 2011, 11, 678–689. [Google Scholar] [CrossRef]
- Schrecengost, R.S.; Keller, S.N.; Schiewer, M.J.; Knudsen, K.E.; Smith, C.D. Downregulation of Critical Oncogenes by the Selective SK2 Inhibitor ABC294640 Hinders Prostate Cancer Progression. Mol. Cancer Res. 2015, 13, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Venant, H.; Rahmaniyan, M.; Jones, E.E.; Lu, P.; Lilly, M.B.; Garrett-Mayer, E.; Drake, R.R.; Kraveka, J.M.; Smith, C.D.; Voelkel-Johnson, C. The Sphingosine Kinase 2 Inhibitor ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of Dihydroceramides In Vitro and In Vivo. Mol. Cancer Ther. 2015, 14, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Venkata, J.K.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014, 124, 1915–1925. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget 2016, 7, 60181–60192. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Meacham, W.D.; Slaughter, E.M.; Muir, S.E.; Elliott, S.; Rhodes, L.V.; Ashe, H.B.; Wiese, T.E.; Smith, C.D.; et al. Antiestrogenic Effects of the Novel Sphingosine Kinase-2 Inhibitor ABC294640. Endocrinology 2010, 151, 5124–5135. [Google Scholar] [CrossRef]
- Beljanski, V.; Knaak, C.; Zhuang, Y.; Smith, C.D. Combined anticancer effects of sphingosine kinase inhibitors and sorafenib. Investig. New Drugs 2010, 29, 1132–1142. [Google Scholar] [CrossRef]
- Beljanski, V.; Lewis, C.; Smith, C.D. Antitumor activity of sphingosine kinase 2 inhibitor ABC294640 and sorafenib in hepatocellular carcinoma xenografts. Cancer Biol. Ther. 2011, 11, 524–534. [Google Scholar] [CrossRef]
- Qin, Z.; Dai, L.; Trillo-Tinoco, J.; Senkal, C.; Wang, W.; Reske, T.; Bonstaff, K.; Del Valle, L.; Rodriguez, P.; Flemington, E.; et al. Targeting Sphingosine Kinase Induces Apoptosis and Tumor Regression for KSHV-Associated Primary Effusion Lymphoma. Mol. Cancer Ther. 2014, 13, 154–164. [Google Scholar] [CrossRef]
- Xun, C.; Chen, M.-B.; Qi, L.; Tie-Ning, Z.; Peng, X.; Ning, L.; Zhi-Xiao, C.; Li-Wei, W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J. Exp. Clin. Cancer Res. 2015, 34, 94. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R.; Green, C.; Maines, L.W.; Smith, C.D. Experimental Osteoarthritis in Rats Is Attenuated by ABC294640, a Selective Inhibitor of Sphingosine Kinase-2. Pharmacology 2011, 87, 135–143. [Google Scholar] [CrossRef]
- Fitzpatrick, L.R.; Green, C.; Frauenhoffer, E.E.; French, K.J.; Zhuang, Y.; Maines, L.W.; Upson, J.J.; Paul, E.; Donahue, H.; Mosher, T.J.; et al. Attenuation of arthritis in rodents by a novel orally-available inhibitor of sphingosine kinase. Inflammopharmacology 2010, 19, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Maines, L.W.; Fitzpatrick, L.R.; French, K.J.; Zhuang, Y.; Xia, Z.; Keller, S.N.; Upson, J.J.; Smith, C.D. Suppression of Ulcerative Colitis in Mice by Orally Available Inhibitors of Sphingosine Kinase. Am. J. Dig. Dis. 2007, 53, 997–1012. [Google Scholar] [CrossRef] [PubMed]
- MainesLeo, L.W.; Fitzpatrick, L.R.; Green, C.L.; Zhuang, Y.; Smith, C.D. Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology 2010, 18, 73–85. [Google Scholar] [CrossRef]
- Maines, L.W.; French, K.J.; Wolpert, E.B.; Antonetti, D.; Smith, C.D. Pharmacologic Manipulation of Sphingosine Kinase in Retinal Endothelial Cells: Implications for Angiogenic Ocular Diseases. Investig. Opthalmol. Vis. Sci. 2006, 47, 5022–5031. [Google Scholar] [CrossRef]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef]
- Smith, C.D.; Maines, L.W.; Keller, S.N.; Ben-Yair, V.K.; Fathi, R.; Plasse, T.F.; Levitt, M.L. Recent Progress in the Development of Opaganib for the Treatment of COVID-19. Drug Des. Dev. Ther. 2022, 16, 2199–2211. [Google Scholar] [CrossRef]
- Winthrop, K.L.; Skolnick, A.W.; Rafiq, A.M.; Beegle, S.H.; Suszanski, J.; Koehne, G.; Barnett-Griness, O.; Bibliowicz, A.; Fathi, R.; Anderson, P.; et al. Opaganib in COVID-19 pneumonia: Results of a randomized, placebo-controlled Phase 2a trial. Open Forum. Infect. Dis. 2022, 9, ofac232. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, J.E.; Johnston, J.; Killion, J.B.; Wright, D.L. Radiation biology and protection. Radiol. Technol. 2009, 80, 421–441. [Google Scholar] [PubMed]
- Langell, J.; Jennings, R.; Clark, J.; Ward, J.B. Pharmacological Agents for the Prevention and Treatment of Toxic Radiation Exposure in Spaceflight. Aviat. Space Environ. Med. 2008, 79, 651–660. [Google Scholar] [CrossRef]
- Mell, L.K.; Movsas, B. Pharmacologic normal tissue protection in clinical radiation oncology: Focus on amifostine. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1341–1350. [Google Scholar] [CrossRef]
- Hosseinimehr, S.J. Trends in the development of radioprotective agents. Drug Discov. Today 2007, 12, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.E.; Cohen, E.P. Future Strategies for Mitigation and Treatment of Chronic Radiation-Induced Normal Tissue Injury. Semin. Radiat. Oncol. 2007, 17, 141–148. [Google Scholar] [CrossRef]
- Andreassen, C.N.; Grau, C.; Lindegaard, J.C. Chemical radioprotection: A critical review of amifostine as a cytoprotector in radiotherapy. Semin. Radiat. Oncol. 2003, 13, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Grdina, D.J.; Murley, J.S.; Kataoka, Y. Radioprotectants: Current Status and New Directions. Oncology 2002, 63, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Santini, V. Amifostine: Chemotherapeutic and radiotherapeutic protective effects. Expert Opin. Pharmacother. 2001, 2, 479–489. [Google Scholar] [CrossRef]
- Kouvaris, J.R.; Kouloulias, V.E.; Vlahos, L.J. Amifostine: The First Selective-Target and Broad-Spectrum Radioprotector. Oncologist 2007, 12, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T.; Holmes, B.C. Dosing considerations with amifostine: A review of the literature and clinical experience. Semin. Oncol. 1999, 26, 108–119. [Google Scholar]
- Xia, P.; Gamble, J.R.; Rye, K.-A.; Wang, L.; Hii, C.S.T.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor- induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, L.; Gamble, J.R.; Vadas, M.A. Activation of Sphingosine Kinase by Tumor Necrosis Factor-α Inhibits Apoptosis in Human Endothelial Cells. J. Biol. Chem. 1999, 274, 34499–34505. [Google Scholar] [CrossRef] [PubMed]
- Kamata, K.; Kamijo, S.; Nakajima, A.; Koyanagi, A.; Kurosawa, H.; Yagita, H.; Okumura, K. Involvement of TNF-Like Weak Inducer of Apoptosis in the Pathogenesis of Collagen-Induced Arthritis. J. Immunol. 2006, 177, 6433–6439. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Kozawa, O.; Matsuno, H.; Kanamori, Y.; Hara, A.; Uematsu, T. Tumor necrosis factor-alpha-mediated signal transduction in human neutrophils: Involvement of sphingomyelin metabolites in the priming effect of TNF-alpha on the fMLP-stimulated superoxide production. Life Sci. 2000, 66, 245–256. [Google Scholar] [CrossRef]
- Fueller, M.; Wang, D.A.; Tigyi, G.; Siess, W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell. Signal. 2003, 15, 367–375. [Google Scholar] [CrossRef]
- Pettus, B.J.; Bielawski, J.; Porcelli, A.M.; Reames, D.L.; Johnson, K.R.; Morrow, J.; Chalfant, C.E.; Obeid, L.M.; Hannun, Y.A. The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J. 2003, 17, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- MacKinnon, A.C.; Buckley, A.; Chilvers, E.R.; Rossi, A.G.; Haslett, C.; Sethi, T. Sphingosine Kinase: A Point of Convergence in the Action of Diverse Neutrophil Priming Agents. J. Immunol. 2002, 169, 6394–6400. [Google Scholar] [CrossRef]
- Itagaki, K.; Hauser, C.J. Sphingosine 1-Phosphate, a Diffusible Calcium Influx Factor Mediating Store-operated Calcium Entry. J. Biol. Chem. 2003, 278, 27540–27547. [Google Scholar] [CrossRef] [PubMed]
- Guss, Z.D. Head and Neck Radiation Therapy: From Consultation to Survivorship and Future Directions. Surg. Clin. N. Am. 2022, 102, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, C.M.; Migliorati, C.A.; Epstein, J.B.; Ribeiro, A.C.P.; Brandão, T.B.; Lopes, M.A.; de Goes, M.F.; Santos-Silva, A.R. Over 300 Radiation Caries Papers: Reflections From the Rearview Mirror. Front. Oral Health 2022, 3, 961594. [Google Scholar] [CrossRef] [PubMed]
- Araujo, I.K.; Muñoz-Guglielmetti, D.; Mollà, M. Radiation-induced damage in the lower gastrointestinal tract: Clinical presentation, diagnostic tests and treatment options. Best Pract. Res. Clin. Gastroenterol. 2020, 48, 101707. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Lin, B.; Fan, M.; Niu, T.; Gao, F.; Tan, B.; Du, X. Research progress on the mechanism of radiation enteritis. Front. Oncol. 2022, 12, 888962. [Google Scholar] [CrossRef] [PubMed]
- Loge, L.; Florescu, C.; Alves, A.; Menahem, B. Radiation enteritis: Diagnostic and therapeutic issues. J. Visc. Surg. 2020, 157, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Yura, Y.; Masui, A.; Hamada, M. Inhibitors of Ceramide- and Sphingosine-Metabolizing Enzymes as Sensitizers in Radiotherapy and Chemotherapy for Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2062. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maines, L.W.; Schrecengost, R.S.; Zhuang, Y.; Keller, S.N.; Smith, R.A.; Green, C.L.; Smith, C.D. Opaganib Protects against Radiation Toxicity: Implications for Homeland Security and Antitumor Radiotherapy. Int. J. Mol. Sci. 2022, 23, 13191. https://doi.org/10.3390/ijms232113191
Maines LW, Schrecengost RS, Zhuang Y, Keller SN, Smith RA, Green CL, Smith CD. Opaganib Protects against Radiation Toxicity: Implications for Homeland Security and Antitumor Radiotherapy. International Journal of Molecular Sciences. 2022; 23(21):13191. https://doi.org/10.3390/ijms232113191
Chicago/Turabian StyleMaines, Lynn W., Randy S. Schrecengost, Yan Zhuang, Staci N. Keller, Ryan A. Smith, Cecelia L. Green, and Charles D. Smith. 2022. "Opaganib Protects against Radiation Toxicity: Implications for Homeland Security and Antitumor Radiotherapy" International Journal of Molecular Sciences 23, no. 21: 13191. https://doi.org/10.3390/ijms232113191
APA StyleMaines, L. W., Schrecengost, R. S., Zhuang, Y., Keller, S. N., Smith, R. A., Green, C. L., & Smith, C. D. (2022). Opaganib Protects against Radiation Toxicity: Implications for Homeland Security and Antitumor Radiotherapy. International Journal of Molecular Sciences, 23(21), 13191. https://doi.org/10.3390/ijms232113191