

Phosphonate Inhibitors of Pyruvate Dehydrogenase Perturb Homeostasis of Amino Acids and Protein Succinylation in the Brain
 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
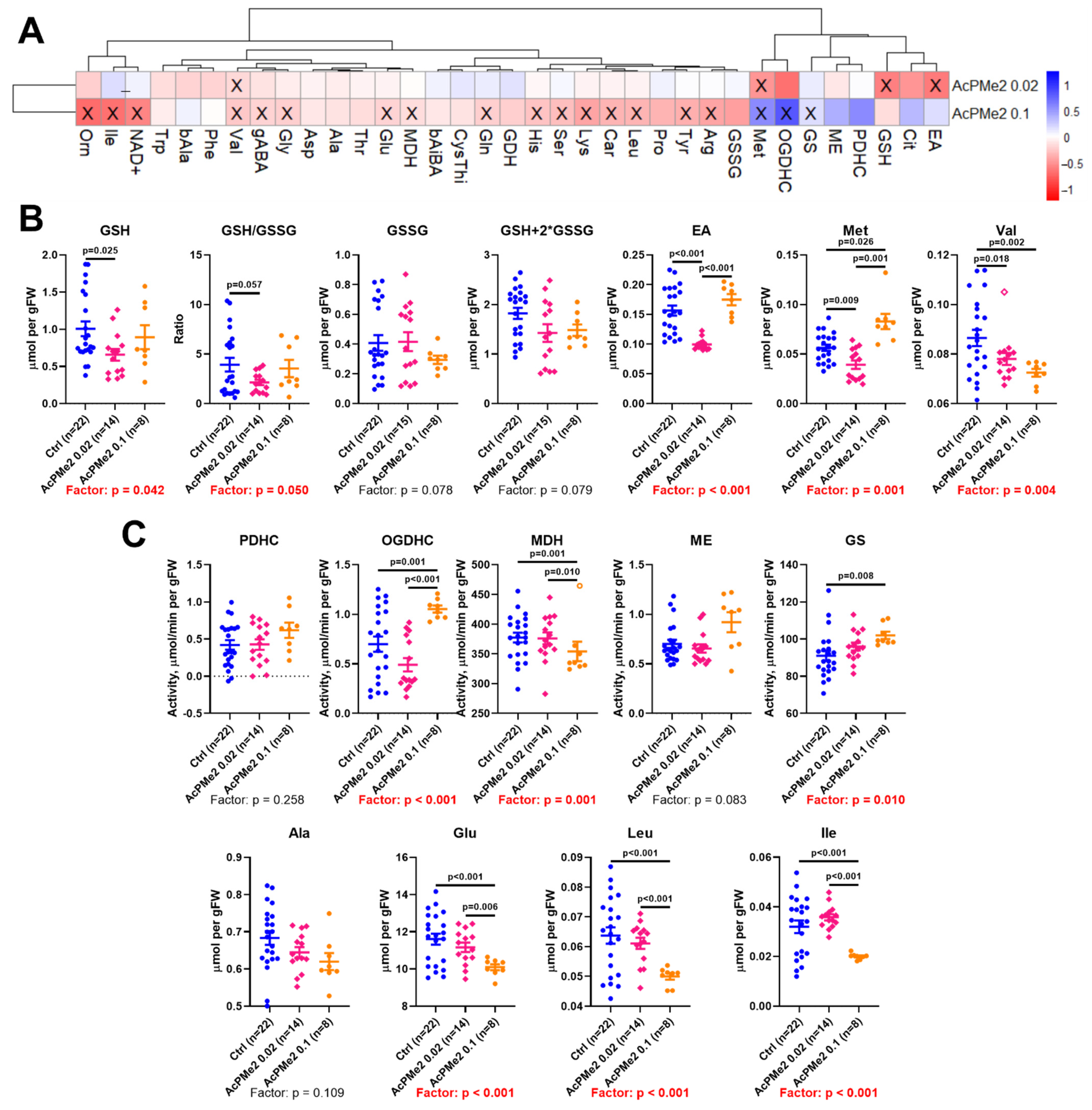
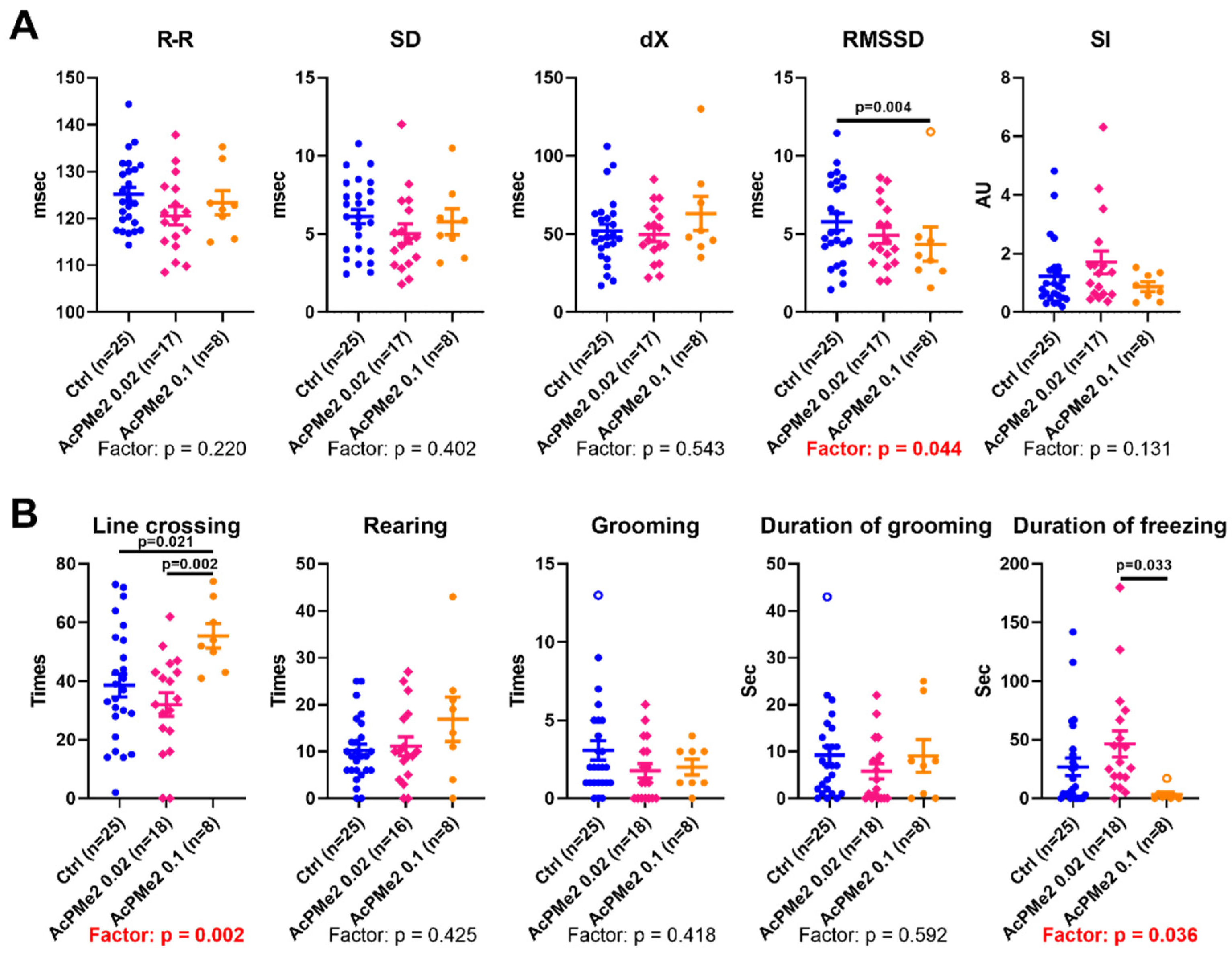
2.1. Dose-Dependent Effects of Dimethyl Ester of Acetyl Phosphonate on Metabolism and Physiology
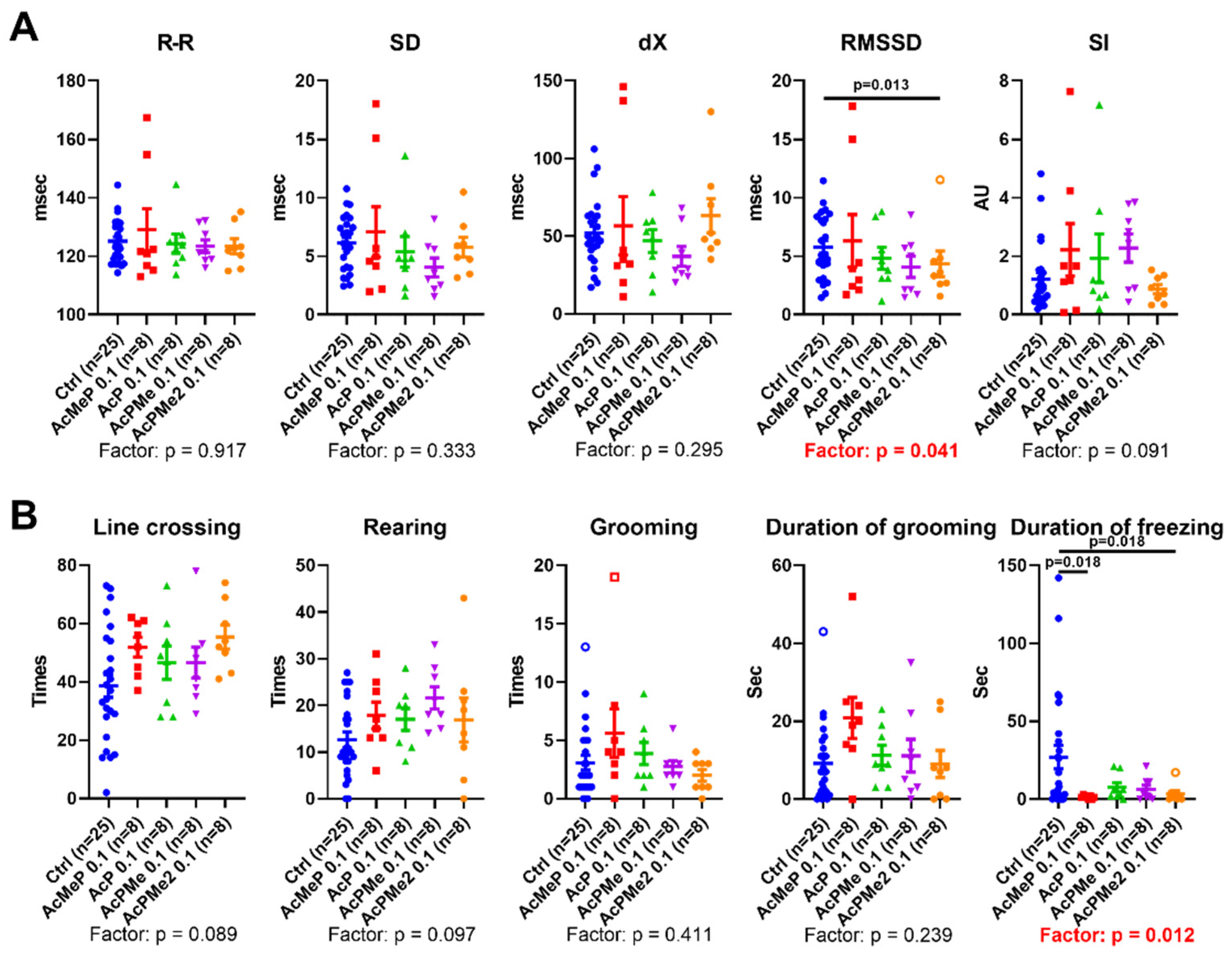
2.2. Comparison of the Biochemical and Physiological Effects of the Phosphinate and Phosphonate Inhibitors of PDHC
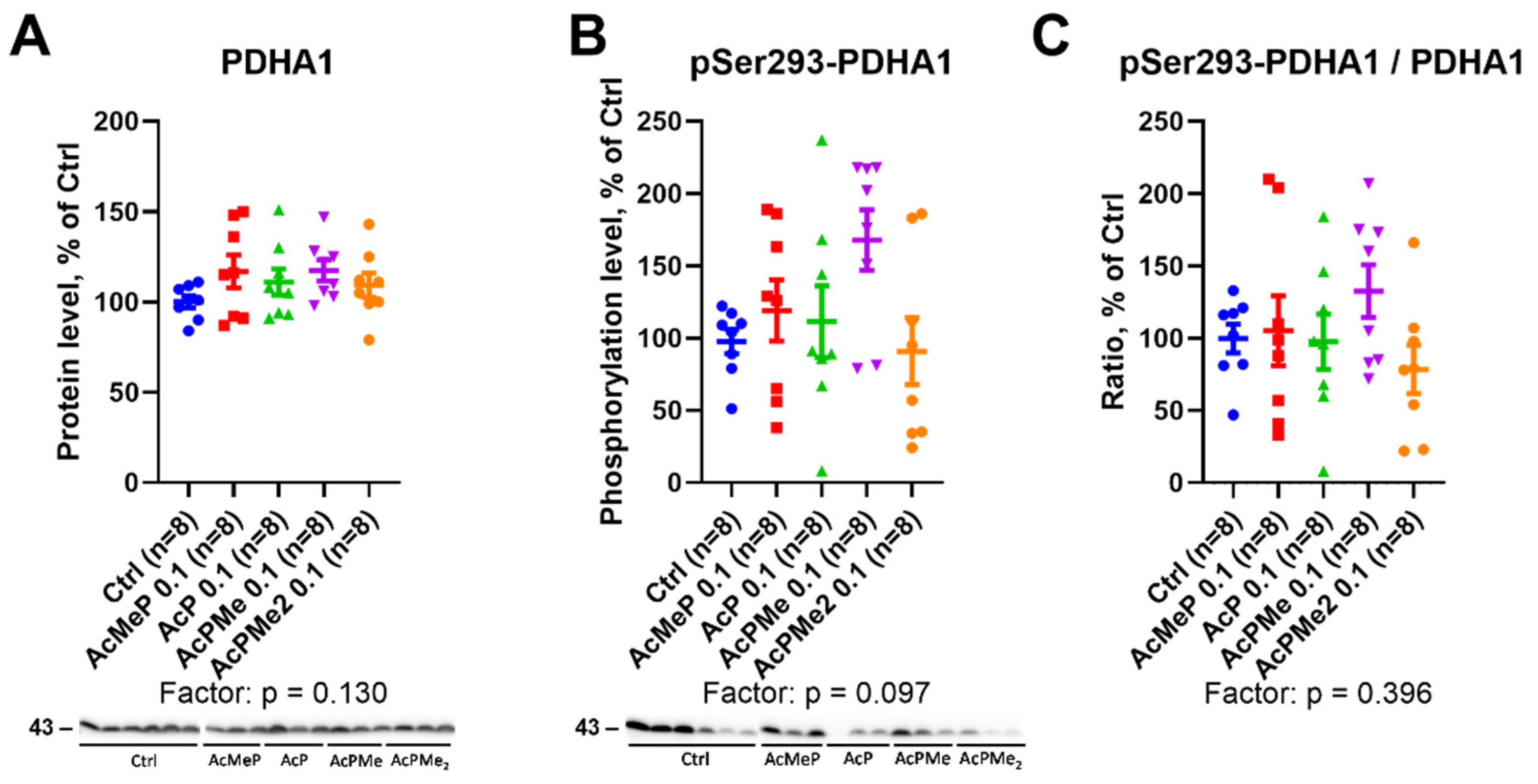
2.3. Action of the PDHC Inhibitors on the PDHA Expression and Its Active Site Phosphorylation at Ser293
2.4. Changes in Total Levels of the Brain Protein Acylations Induced by the PDHC Inhibitors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animal Experiments
4.3. Physiological Tests
4.4. Metabolite and Enzyme Activity Assays
4.5. Western Blotting
4.6. Statistics and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bunik, V. Vitamin-Dependent Complexes of 2-oxo Acid Dehydrogenases: Structure, Function, Regulation and Medical Implications; Nova Science Publishers: New York, NY, USA, 2017; p. 203. [Google Scholar]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.J.; Islam, R.; Kim, J.G.; Dogsom, O.; Cap, K.C.; Park, J.B. Pyruvate Dehydrogenase A1 Phosphorylated by Insulin Associates with Pyruvate Kinase M2 and Induces LINC00273 through Histone Acetylation. Biomedicines 2022, 10, 1256. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Jiang, H.; Sun, X.; Xu, H.; Wei, X.; Wei, Y.; Xiao, G.; Song, Z.; Zhou, F. Mitochondrial dysfunction induces radioresistance in colorectal cancer by activating [Ca(2+)]m-PDP1-PDH-histone acetylation retrograde signaling. Cell Death Dis. 2021, 12, 837. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Bunik, V.I. Mechanisms of Non-coenzyme Action of Thiamine: Protein Targets and Medical Significance. Biochem. Biokhimiia 2019, 84, 829–850. [Google Scholar] [CrossRef]
- Bunik, V.I.; Aleshin, V.A. Analysis of the protein binding sites for thiamin and its derivatives to elucidate molecular mechanisms of the non-coenzyme action of thiamin (vitamin B1). Stud. Nat. Prod. Chem. 2017, 53, 375–429. [Google Scholar]
- Bunik, V.I.; Tylicki, A.; Lukashev, N.V. Thiamin diphosphate-dependent enzymes: From enzymology to metabolic regulation, drug design and disease models. FEBS J. 2013, 280, 6412–6442. [Google Scholar] [CrossRef]
- Bingham, P.M.; Stuart, S.D.; Zachar, Z. Lipoic acid and lipoic acid analogs in cancer metabolism and chemotherapy. Expert Rev. Clin. Pharmacol. 2014, 7, 837–846. [Google Scholar] [CrossRef]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef]
- Zachar, Z.; Marecek, J.; Maturo, C.; Gupta, S.; Stuart, S.D.; Howell, K.; Schauble, A.; Lem, J.; Piramzadian, A.; Karnik, S.; et al. Non-redox-active lipoate derivates disrupt cancer cell mitochondrial metabolism and are potent anticancer agents In Vivo. J. Mol. Med. 2011, 89, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Artiukhov, A.V.; Graf, A.V.; Bunik, V.I. Directed Regulation of Multienzyme Complexes of 2-Oxo Acid Dehydrogenases Using Phosphonate and Phosphinate Analogs of 2-Oxo Acids. Biochem. Biokhimiia 2016, 81, 1498–1521. [Google Scholar] [CrossRef] [PubMed]
- Bunik, V.I.; Artiukhov, A.; Kazantsev, A.; Goncalves, R.; Daloso, D.; Oppermann, H.; Kulakovskaya, E.; Lukashev, N.; Fernie, A.; Brand, M.; et al. Specific inhibition by synthetic analogs of pyruvate reveals that the pyruvate dehydrogenase reaction is essential for metabolism and viability of glioblastoma cells. Oncotarget 2015, 6, 40036–40052. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Artiukhov, A.V.; Oppermann, H.; Kazantsev, A.V.; Lukashev, N.V.; Bunik, V.I. Mitochondrial Impairment May Increase Cellular NAD(P)H: Resazurin Oxidoreductase Activity, Perturbing the NAD(P)H-Based Viability Assays. Cells 2015, 4, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Tsepkova, P.M.; Artiukhov, A.V.; Boyko, A.I.; Aleshin, V.A.; Mkrtchyan, G.V.; Zvyagintseva, M.A.; Ryabov, S.I.; Ksenofontov, A.L.; Baratova, L.A.; Graf, A.V.; et al. Thiamine Induces Long-Term Changes in Amino Acid Profiles and Activities of 2-Oxoglutarate and 2-Oxoadipate Dehydrogenases in Rat Brain. Biochem. Biokhimiia 2017, 82, 723–736. [Google Scholar] [CrossRef]
- Graf, A.; Ksenofontov, A.; Bunik, V. Inhibition of 2-oxoglutarate dehydrogenase as a chemical model of acute hypobaric hypoxia. Front. Med. 2021, 8, 751639. [Google Scholar] [CrossRef]
- Nemeria, N.S.; Korotchkina, L.G.; Chakraborty, S.; Patel, M.S.; Jordan, F. Acetylphosphinate is the most potent mechanism-based substrate-like inhibitor of both the human and Escherichia coli pyruvate dehydrogenase components of the pyruvate dehydrogenase complex. Bioorg. Chem. 2006, 34, 362–379. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Graf, A.V.; Kazantsev, A.V.; Boyko, A.I.; Aleshin, V.A.; Ksenofontov, A.L.; Bunik, V.I. Increasing Inhibition of the Rat Brain 2-Oxoglutarate Dehydrogenase Decreases Glutathione Redox State, Elevating Anxiety and Perturbing Stress Adaptation. Pharmaceuticals 2022, 15, 182. [Google Scholar] [CrossRef]
- Bakovic, M.; Fullerton, M.D.; Michel, V. Metabolic and molecular aspects of ethanolamine phospholipid biosynthesis: The role of CTP:phosphoethanolamine cytidylyltransferase (Pcyt2). Biochem. Cell Biol. 2007, 85, 283–300. [Google Scholar] [CrossRef]
- Patel, D.; Witt, S.N. Ethanolamine and Phosphatidylethanolamine: Partners in Health and Disease. Oxid. Med. Cell. Longev. 2017, 2017, 4829180. [Google Scholar] [CrossRef]
- Jang, S.; Chapa-Dubocq, X.R.; Tyurina, Y.Y.; St Croix, C.M.; Kapralov, A.A.; Tyurin, V.A.; Bayir, H.; Kagan, V.E.; Javadov, S. Elucidating the contribution of mitochondrial glutathione to ferroptosis in cardiomyocytes. Redox Biol. 2021, 45, 102021. [Google Scholar] [CrossRef] [PubMed]
- Basu Ball, W.; Baker, C.D.; Neff, J.K.; Apfel, G.L.; Lagerborg, K.A.; Zun, G.; Petrovic, U.; Jain, M.; Gohil, V.M. Ethanolamine ameliorates mitochondrial dysfunction in cardiolipin-deficient yeast cells. J. Biol. Chem. 2018, 293, 10870–10883. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lou, W.; Raja, V.; Denis, S.; Yu, W.; Schmidtke, M.W.; Reynolds, C.A.; Schlame, M.; Houtkooper, R.H.; Greenberg, M.L. Cardiolipin-induced activation of pyruvate dehydrogenase links mitochondrial lipid biosynthesis to TCA cycle function. J. Biol. Chem. 2019, 294, 11568–11578. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.; Trofimova, L.; Loshinskaja, A.; Mkrtchyan, G.; Strokina, A.; Lovat, M.; Tylicky, A.; Strumilo, S.; Bettendorff, L.; Bunik, V.I. Up-regulation of 2-oxoglutarate dehydrogenase as a stress response. Int. J. Biochem. Cell Biol. 2013, 45, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Araujo, W.L.; Trofimova, L.; Mkrtchyan, G.; Steinhauser, D.; Krall, L.; Graf, A.; Fernie, A.R.; Bunik, V.I. On the role of the mitochondrial 2-oxoglutarate dehydrogenase complex in amino acid metabolism. Amino Acids 2013, 44, 683–700. [Google Scholar] [CrossRef]
- Trofimova, L.K.; Araujo, W.L.; Strokina, A.A.; Fernie, A.R.; Bettendorff, L.; Bunik, V.I. Consequences of the alpha-ketoglutarate dehydrogenase inhibition for neuronal metabolism and survival: Implications for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 5895–5906. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Graf, A.; Bettendorff, L.; Bunik, V. Cellular thiamine status is coupled to function of mitochondrial 2-oxoglutarate dehydrogenase. Neurochem. Int. 2016, 101, 66–75. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Graf, A.V.; Bunik, V.I. Daytime Dependence of the Activity of the Rat Brain Pyruvate Dehydrogenase Corresponds to the Mitochondrial Sirtuin 3 Level and Acetylation of Brain Proteins, All Regulated by Thiamine Administration Decreasing Phosphorylation of PDHA Ser293. Int. J. Mol. Sci. 2021, 22, 8006. [Google Scholar] [CrossRef]
- Graf, A.; Trofimova, L.; Ksenofontov, A.; Baratova, L.; Bunik, V. Hypoxic Adaptation of Mitochondrial Metabolism in Rat Cerebellum Decreases in Pregnancy. Cells 2020, 9, 139. [Google Scholar] [CrossRef]
- Tucek, S. Problems in the organization and control of acetylcholine synthesis in brain neurons. Prog. Biophys. Mol. Biol. 1984, 44, 1–46. [Google Scholar] [CrossRef]
- Gibson, G.E.; Jope, R.; Blass, J.P. Decreased synthesis of acetylcholine accompanying impaired oxidation of pyruvic acid in rat brain minces. Biochem. J. 1975, 148, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Ksiezak-Reding, H.; Sheu, K.F.; Mykytyn, V.; Blass, J.P. Correlation of enzymatic, metabolic, and behavioral deficits in thiamin deficiency and its reversal. Neurochem. Res. 1984, 9, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.; Barclay, L.; Blass, J. The role of the cholinergic system in thiamin deficiency. Ann. N. Y. Acad. Sci. 1982, 378, 382–403. [Google Scholar] [CrossRef]
- Bizon-Zygmanska, D.; Jankowska-Kulawy, A.; Bielarczyk, H.; Pawelczyk, T.; Ronowska, A.; Marszall, M.; Szutowicz, A. Acetyl-CoA metabolism in amprolium-evoked thiamine pyrophosphate deficits in cholinergic SN56 neuroblastoma cells. Neurochem. Int. 2011, 59, 208–216. [Google Scholar] [CrossRef]
- Jankowska-Kulawy, A.; Bielarczyk, H.; Pawelczyk, T.; Wroblewska, M.; Szutowicz, A. Acetyl-CoA and acetylcholine metabolism in nerve terminal compartment of thiamine deficient rat brain. J. Neurochem. 2010, 115, 333–342. [Google Scholar] [CrossRef]
- Ronowska, A.; Gul-Hinc, S.; Michno, A.; Bizon-Zygmanska, D.; Zysk, M.; Bielarczyk, H.; Szutowicz, A.; Gapys, B.; Jankowska-Kulawy, A. Aggravated effects of coexisting marginal thiamine deficits and zinc excess on SN56 neuronal cells. Nutr. Neurosci. 2021, 24, 432–442. [Google Scholar] [CrossRef]
- Hoshi, M.; Takashima, A.; Noguchi, K.; Murayama, M.; Sato, M.; Kondo, S.; Saitoh, Y.; Ishiguro, K.; Hoshino, T.; Imahori, K. Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc. Natl. Acad. Sci. USA 1996, 93, 2719–2723. [Google Scholar] [CrossRef]
- Ronowska, A.; Dys, A.; Jankowska-Kulawy, A.; Klimaszewska-Lata, J.; Bielarczyk, H.; Romianowski, P.; Pawelczyk, T.; Szutowicz, A. Short-term effects of zinc on acetylcholine metabolism and viability of SN56 cholinergic neuroblastoma cells. Neurochem. Int. 2010, 56, 143–151. [Google Scholar] [CrossRef]
- Baillie, A.C.; Wright, K.; Wright, B.J.; Earnshaw, C.G. Inhibitors of pyruvate dehydrogenase as herbicides. Pestic. Biochem. Physiol. 1988, 30, 103–112. [Google Scholar] [CrossRef]
- Kim, M.J.; Hennen, W.J.; Sweers, H.M.; Wong, C.H. Enzymes in carbohydrate synthesis: N-acetylneuraminic acid aldolase catalyzed reactions and preparation of N-acetyl-2-deoxy-D-neuraminic acid derivatives. J. Am. Chem. Soc. 1988, 110, 6481–6486. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Graf, A.V.; Artiukhov, A.V.; Boyko, A.I.; Ksenofontov, A.L.; Maslova, M.V.; Nogues, I.; di Salvo, M.L.; Bunik, V.I. Physiological and Biochemical Markers of the Sex-Specific Sensitivity to Epileptogenic Factors, Delayed Consequences of Seizures and Their Response to Vitamins B1 and B6 in a Rat Model. Pharmaceuticals 2021, 14, 737. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Kaehne, T.; Graf, A.V.; Maslova, M.V.; Bunik, V.I. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J. Neurochem. 2020, 153, 80–102. [Google Scholar] [CrossRef] [PubMed]
- Graf, A.V.; Maslova, M.V.; Artiukhov, A.V.; Ksenofontov, A.L.; Aleshin, V.A.; Bunik, V.I. Acute prenatal hypoxia in rats affects physiology and brain metabolism in the offspring, dependent on sex and gestational age. Int. J. Mol. Sci. 2022, 23, 2579. [Google Scholar] [CrossRef] [PubMed]
- Ksenofontov, A.L.; Boyko, A.I.; Mkrtchyan, G.V.; Tashlitsky, V.N.; Timofeeva, A.V.; Graf, A.V.; Bunik, V.I.; Baratova, L.A. Analysis of Free Amino Acids in Mammalian Brain Extracts. Biochem. Biokhimiia 2017, 82, 1183–1192. [Google Scholar] [CrossRef]
- Artiukhov, A.V.; Grabarska, A.; Gumbarewicz, E.; Aleshin, V.A.; Kahne, T.; Obata, T.; Kazantsev, A.V.; Lukashev, N.V.; Stepulak, A.; Fernie, A.R.; et al. Synthetic analogues of 2-oxo acids discriminate metabolic contribution of the 2-oxoglutarate and 2-oxoadipate dehydrogenases in mammalian cells and tissues. Sci. Rep. 2020, 10, 1886. [Google Scholar] [CrossRef]
- Ladner, C.L.; Yang, J.; Turner, R.J.; Edwards, R.A. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Artiukhov, A.V.; Aleshin, V.A.; Karlina, I.S.; Kazantsev, A.V.; Sibiryakina, D.A.; Ksenofontov, A.L.; Lukashev, N.V.; Graf, A.V.; Bunik, V.I. Phosphonate Inhibitors of Pyruvate Dehydrogenase Perturb Homeostasis of Amino Acids and Protein Succinylation in the Brain. Int. J. Mol. Sci. 2022, 23, 13186. https://doi.org/10.3390/ijms232113186
Artiukhov AV, Aleshin VA, Karlina IS, Kazantsev AV, Sibiryakina DA, Ksenofontov AL, Lukashev NV, Graf AV, Bunik VI. Phosphonate Inhibitors of Pyruvate Dehydrogenase Perturb Homeostasis of Amino Acids and Protein Succinylation in the Brain. International Journal of Molecular Sciences. 2022; 23(21):13186. https://doi.org/10.3390/ijms232113186
Chicago/Turabian StyleArtiukhov, Artem V., Vasily A. Aleshin, Irina S. Karlina, Alexey V. Kazantsev, Daria A. Sibiryakina, Alexander L. Ksenofontov, Nikolay V. Lukashev, Anastasia V. Graf, and Victoria I. Bunik. 2022. "Phosphonate Inhibitors of Pyruvate Dehydrogenase Perturb Homeostasis of Amino Acids and Protein Succinylation in the Brain" International Journal of Molecular Sciences 23, no. 21: 13186. https://doi.org/10.3390/ijms232113186
APA StyleArtiukhov, A. V., Aleshin, V. A., Karlina, I. S., Kazantsev, A. V., Sibiryakina, D. A., Ksenofontov, A. L., Lukashev, N. V., Graf, A. V., & Bunik, V. I. (2022). Phosphonate Inhibitors of Pyruvate Dehydrogenase Perturb Homeostasis of Amino Acids and Protein Succinylation in the Brain. International Journal of Molecular Sciences, 23(21), 13186. https://doi.org/10.3390/ijms232113186