4.1. Chemistry
All commercial reagents and solvents were used without further purification. Organic layers obtained after the extraction of aqueous solutions were dried over MgSO
4 and filtered before evaporation. Reaction yields were not optimized. Column chromatography was performed using Macherey-Nagel silica gel (230–400 mesh).
1H and
13C NMR spectra were obtained using a Bruker DRX 300 spectrometer (Division BioSpin, Wissembourg, France) operating at 300.13 MHz for proton, operating at 300 MHz for
1H and 75 MHz for
13C, equipped with a BBFO 5 mm probe and a sample XpressLite. The data were processed using software TOPSPIN 4. Chemical shifts (δ) were expressed in ppm relative to either TMS or the residual proton signal in deuterated solvents. Mass spectra were recorded with an LC-MS (Waters Alliance Micromass ZQ 2000, Waters Corporation, Milford, MA, USA) using electrospray ionization. The purity of the final compounds was verified by two types of high-pressure liquid chromatography (HPLC) columns: C18 Interchrom UPTISPHERE and C4 Interchrom UPTISPHERE. Analytical HPLC was performed on a Shimadzu LC-2010AHT system equipped with a UV detector set at 254 nm and 215 nm. The following eluent systems were used: buffer A (H
2O/TFA, 100:0.1) and buffer B (CH
3CN/H
2O/TFA, 80:20:0.1). Compounds were dissolved in 50 μL of buffer B and 950 μL of buffer A and injected into the system. HPLC retention times (HPLC
tR) were obtained at a flow rate of 0.2 mL/min using a gradient run from 100% of buffer A to 100% of buffer B over 30 min. The spectra and chromatograms of final compounds can be found in
Supplementary Materials.
4.1.1. General Procedure A
Dimethylformamide (81.5 mmol) was cooled to 0 °C with a salt/ice bath. Phosphorus oxychloride (22.5 mmol) was added dropwise with the temperature maintained below 0 °C. The mixture was then stirred for 40 min at 0 °C. Hydrazone (5.39 mmol) was added and the reaction mixture was allowed to warm to room temperature. After 2 h, the temperature was increased to 50 °C. The reaction was stirred at this temperature for 4 h. The mixture was then added to crushed ice and stirred for 1 h. Potassium carbonate was added until pH = 8 and the mixture was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1).
4.1.2. General Procedure B
To a solution of alkylamine (3.73 mmol), 37% formaldehyde in water (22.4 mmol) and acetic acid (22.4 mmol) in methanol (20 mL) was slowly added sodium triacetoxyborohydride (18.7 mmol) over 30 min. The mixture was stirred until the completion of the reaction. The solution was an aqueous carbonate potassium (10%) and ethyl acetate was added to the residue and the mixture stirred for 10 min. The layers were separated, and the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 95:5).
4.1.3. General Procedure C
LiAlH4 (1 M in THF, 2.82 mmol) was added to 20 mL of anhydrous THF under nitrogen. The solution was cooled to 0 °C with an ice bath and a solution of ester (1.88 mmol) in 20 mL of anhydrous THF was added dropwise. The reaction was stirred at 0 °C for 20 min and rt for 1 h. The mixture was then cooled with an ice bath and 0.11 mL of H2O was added, followed by 0.11 mL of 15% NaOH and 0.33 mL of H2O. The solid was isolated by filtration and washed with THF. The filtrate was evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1).
4.1.4. General Procedure D
To a solution of the desired compound (1.42 mmol) in chloroform (25 mL) was added manganese(IV) oxide (14.2 mmol). This was stirred at rt for 24 h. More manganese(IV) oxide (14.2 mmol) was added, and the mixture was stirred for 24 h. The solid was filtered off and washed with methylene chloride. The filtrate was washed with 10% K2CO3 and with brine. The organic layer was dried and evaporated to give the aldehyde that was used in the next step without further purification.
4.1.5. General Procedure E
To a solution of benzaldehyde (0.265 mmol), amine (0.45 mmol), and acetic acid (0.53 mmol) in DCE (4 mL) was added sodium triacetoxyborohydride (0.53 mmol). The reaction mixture was stirred under nitrogen for 24 h. A total of 10% K2CO3 was added, and the layers were separated. The aqueous layer was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 9:1).
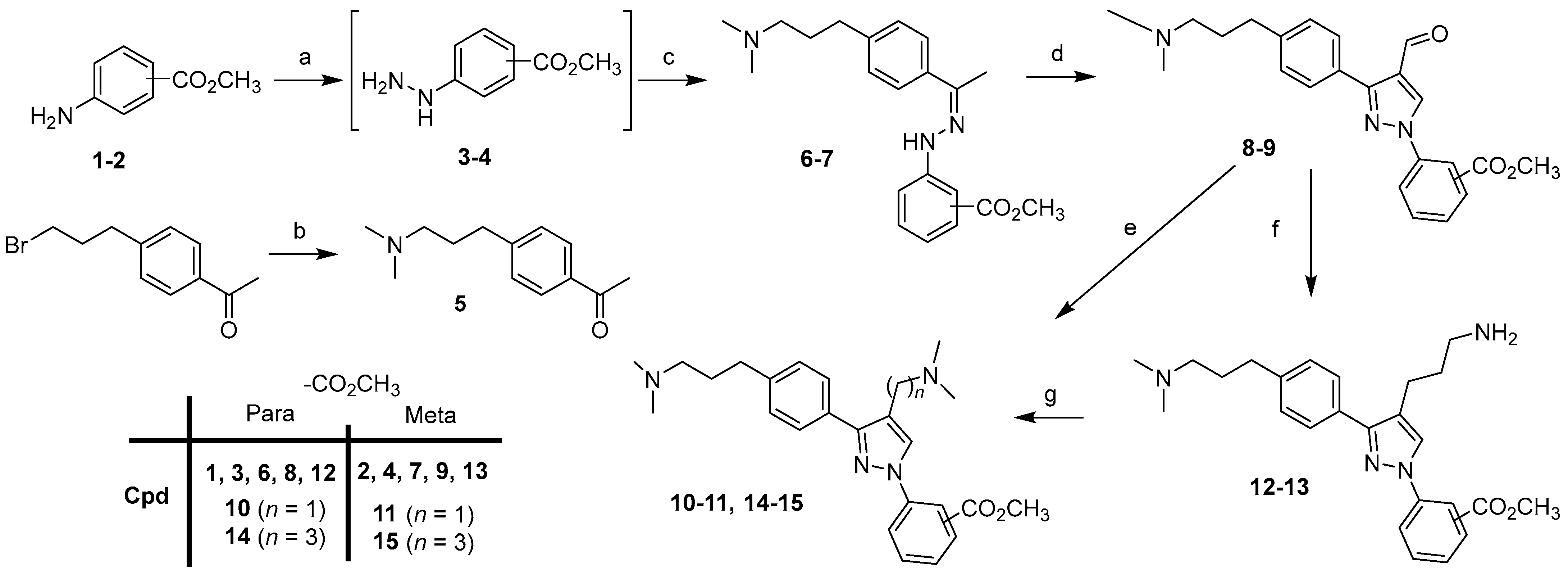
4.1.6. Methyl 4-Hydrazinobenzoate Hydrochloride (3)
A solution of 1 (6 g, 39.7 mmol) in HCl 37% (40 mL) was brought to −5 °C with a salt/ice bath. A solution of sodium nitrite (3 g, 43.5 mmol) in water (22 mL) was slowly added over 1 h while maintaining the temperature below 0 °C. The solution was then stirred at 0 °C for 40 min and a solution of tin(II) chloride (13.86 g, 48.7 mmol) in 37% HCl (20 mL) was added dropwise while maintained at 0 °C. The mixture was stirred for another 20 min at 0 °C and 2 h 30 min at room temperature. The precipitate was collected by filtration, washed with 40 mL of ice-cold water, and dried to give 8.6 g of a white solid, which was used for the next step without further purification.
4.1.7. Methyl 3-Hydrazinobenzoate Hydrochloride (4)
A solution of 2 (4 g, 26.5 mmol) in 37% HCl (40 mL) was brought to −5 °C with a salt/ice bath. A solution of sodium nitrite (2 g, 29 mmol) in water (15 mL) was slowly added over 1 h while maintaining the temperature below 3 °C. The solution was then stirred at 0 °C for 30 min and a solution of tin(II) chloride (9.24 g, 48.7 mmol) in 37% HCl (20 mL) was added dropwise while maintaining the temperature at 0 °C. The mixture was stirred for another 30 min at 0 °C and 2 h at room temperature. The precipitate was collected by filtration, washed subsequently with 15 mL of ice-cold water and with ether, and dried to give 6.55 g of a white solid, which was used for the next step without further purification.
4.1.8. 1-[4-[3-(Dimethylamino)propyl]phenyl]ethanone (5)
In a sealed tube, a mixture of 1-[4-(3-bromopropyl)phenyl]ethanone (5.6 g, 23.2 mmol) and dimethylamine (2 M in methanol, 34.8 mL, 69.6 mmol) was heated at 65 °C for 15 h. The solvent was evaporated and 10% K2CO3 (100 mL) was added. The mixture was extracted twice with ethyl acetate and the combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH = 9:1) to give 3.66 g (59%) of the product as a yellow oil. 1H NMR (CDCl3, 300 Mz): δ 7.89 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.58 (s, 3H), 2.29 (t, J = 7.1 Hz, 2H), 2.23 (s, 6H), 1.81 (quint, J = 7.5 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 197.9, 148.2, 135.1, 128.7, 128.6, 59.0, 45.5, 33.7, 29.1, 26.6. MS (ESI) m/z 206 [M + H]+.
4.1.9. Methyl 4-[2-[1-[4-[3-(Dimethylamino)ropyl]phenyl]ethylidene]hydrazino] Benzoate (6)
A solution of 3 (8.5 g) in methanol (220 mL) was added to 5 (3.45 g, 16.81 mmol). The mixture was stirred at rt for 48 h. The precipitate was collected by filtration and washed with methanol to give 4.08 g (62%) of the product as a yellowish solid. 1H NMR (CD3SOCD3, 300 Mz): δ 10.88 (br, 1H), 9.89 (s, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.75 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 8.8 Hz, 1H), 7.27 (d, J = 8.3 Hz, 1H), 3.78 (s, 3H), 3.02–2.99 (m, 2H), 2.71–2.63 (m, 8H), 2.30 (s, 3H), 2.05–1.95 (m, 2H). 13C NMR (CD3SOCD3, 75 MHz): δ 166.2, 150.0, 143.5, 140.5, 136.8, 130.8, 128.3, 125.6, 119.1, 112.0, 56.0, 51.4, 41.9, 31.6, 25.1, 13.3. MS (ESI) m/z 354 [M + H]+.
4.1.10. Methyl 3-[2-[1-[4-[3-(Dimethylamino)propyl]phenyl]ethylidene]hydrazino] Benzoate Hydrochloride (7)
A solution of 4 (10.9 g) in methanol (105 mL) was added to 5 (3.45 g, 16.81 mmol). The mixture was stirred at rt for 48 h. The precipitate was collected by filtration and washed with methanol to give 6.19 g (94%) of the product as a light brown solid. 1H NMR (CD3SOCD3, 300 Mz): δ 10.30 (br, 1H), 9.52 (s, 1H), 7.84–7.83 (m, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.53–7.49 (m, 1H), 7.36–7.34 (m, 2H), 7.26 (d, J = 8.3 Hz, 2H), 3.85 (s, 3H), 3.05–3.00 (m, 2H), 2.73 (s, 6H), 2.65 (t, J = 7.7 Hz, 2H), 2.26 (s, 3H), 2.03–1.93 (m, 2H). 13C NMR (CD3SOCD3, 75 MHz): δ 166.6, 146.4, 141.9, 140.1, 137.1, 130.3, 129.3, 128.3, 125.4, 119.3, 117.1, 113.3, 56.1, 52.0, 42.0, 31.6, 25.2, 13.1. MS (ESI) m/z 354 [M + H]+.
4.1.11. Methyl 4-[3-[4-[3-(Dimethylamino)propyl]phenyl]-4-formyl-pyrazol-1-yl] Benzoate (8)
General procedure A: 87% yield (white solid). 1H NMR (CDCl3, 300 Mz): δ 10.03 (s, 1H), 8.59 (s, 1H), 8.14 (d, J = 8.8 Hz, 2H), 7.86 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.31 (d, J = 8.1 Hz, 2H), 3.92 (s, 3H), 2.70 (t, J = 7.5 Hz, 2H), 2.32 (t, J = 7.1 Hz, 2H), 2.23 (s, 6H), 1.82 (quint, J = 7.4 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 185.1, 166.0, 155.2, 144.0, 142.1, 131.3, 131.2, 129.3, 128.9, 128.6, 123.0, 119.0, 59.1, 52.4, 45.5, 33.5, 29.3. MS (ESI) m/z 392 [M + H]+.
4.1.12. Methyl 3-[3-[4-[3-(Dimethylamino)propyl]phenyl]-4-formyl-pyrazol-1-yl] Benzoate (9)
General procedure A: 67% yield (white solid). 1H NMR (CDCl3, 300 Mz): δ 10.06 (s, 1H), 8.61 (s, 1H), 8.44–8.42 (m, 1H), 8.05–8.02 (m, 1H), 7.76 (d, J = 8.1 Hz, 2H), 7.58 (t, J = 7.9 Hz, 1H), 7.33 (d, J = 8.1 Hz, 2H), 3.96 (s, 3H), 2.72 (t, J = 7.5 Hz, 2H), 2.33 (t, J = 7.1 Hz, 2H), 2.23 (s, 6H), 1.85 (quint, J = 7.3 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 185.1, 165.9, 154.9, 143.9, 139.2, 131.8, 131.2, 129.9, 128.9, 128.9, 128.7, 128.7, 123.8, 122.8, 120.3, 59.2, 52.5, 45.5, 33.5, 29.3. MS (ESI) m/z 392 [M + H]+.
4.1.13. Methyl 4-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzoate (10)
A mixture of 8 (0.9 g, 2.3 mmol), dimethylamine (2 M in THF, 2.3 mL, 4.6 mmol), sodium triacetoxyborohydride (0.88 g, 4.14 mmol), and acetic acid (0.24 mL, 4.14 mmol) in DCE (10 mL) was stirred at rt under nitrogen for 4 h. A total of 10% K2CO3 and methylene chloride were added. The layers were separated, and the aqueous layer was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.05) to give 0.82 g (84%) of the product as a white solid. 1H NMR (CD3OD, 300 Mz): δ 8.22 (s, 1H), 8.03 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.72 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 8.1 Hz, 2H), 3.86 (s, 3H), 3.43 (s, 2H), 2.64 (t, J = 7.6 Hz, 2H), 2.37–2.32 (m, 2H), 2.22 (s, 6H), 2.20 (s, 6H), 1.86–1.79 (m, 2H). 13C NMR (CD3OD, 75 MHz): δ 167.6, 154.5, 144.4, 143.3, 132.0, 131.9, 130.3, 129.5 (2C), 128.5, 119.8, 118.9, 60.1, 54.1, 52.6, 45.4, 45.2, 34.3, 29.9. MS (ESI) m/z 421 [M + H]+.
4.1.14. Methyl 3-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzoate (11)
To a solution of 9 (0.7 g, 1.79 mmol), dimethylamine (2 M in THF, 1.79 mL, 3.58 mmol) and acetic acid (0.18 mL, 3.22 mmol) in DCE (8 mL) was added sodium triacetoxyborohydride (0.68 g, 3.22 mmol). The reaction mixture was stirred at rt for 4 h. A total of 10% K2CO3 was added, and the layers were separated. The organic layer was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH = 95:5 to 8:2) to give 0.64 g (85%) of the product as a colorless oil. 1H NMR (CD3OD, 300 Mz): δ 8.37 (s, 1H), 8.19 (s, 1H), 7.97–7.94 (m, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.49 (t, J = 7.9 Hz, 1H), 7.25 (d, J = 8.1 Hz, 2H), 3.88 (s, 3H), 3.44 (s, 2H), 2.62 (t, J = 7.6 Hz, 2H), 2.36 (t, J = 7.4 Hz, 2H), 2.24 (s, 6H), 2.20 (s, 6H), 1.81 (quint, J = 7.8 Hz, 2H). 13C NMR (CD3OD, 75 MHz): δ 167.4, 154.0, 143.0, 141.3, 132.6, 132.0, 130.8, 130.1, 129.5 (2C), 127.9, 123.7, 120.2, 119.2, 59.9, 54.0, 52.8, 45.3, 45.2, 34.2, 29.7. MS (ESI) m/z 421 [M + H]+.
4.1.15. Methyl 4-[4-(3-Aminopropyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzoate (12)
To a suspension of NaH (60% dispersion in mineral oil, 0.17 g, 4.32 mmol) in anhydrous THF (15 mL) at 0 °C under nitrogen was added dropwise diethyl cyanomethylphosphonate (0.65 mL, 3.99 mmol). The mixture was stirred at 0 °C for 30 min and a solution of 8 (1.3 g, 3.32 mmol) in anhydrous THF (20 mL) was added dropwise. The reaction mixture was stirred at 0 °C for 10 min and then allowed to warm to rt. After 2 h, the solvent was evaporated, and the residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.02) to give 1.175 g (85%) of a white solid. MS (ESI) m/z 415 [M + H]+.
A mixture of the intermediate (1.16 g, 2.80 mmol), Raney Nickel (0.12 g), and 10% Pd/C (0.12 g) in methanol saturated with ammonia (110 mL) and THF (10 mL) was stirred under a hydrogen atmosphere for 30 h. The catalyst was filtered off and the filtrate was evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1) to give 0.795 g (68%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 8.12 (d, J = 8.9 Hz, 2H), 7.88 (s, 1H), 7.82 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.28 (d, J = 8.1 Hz, 2H), 3.93 (s, 3H), 2.81–2.67 (m, 6H), 2.37–2.32 (m, 2H), 2.26 (s, 6H), 2.00 (br, 2H), 1.90–1.76 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 166.6, 152.6, 143.4, 142.3, 131.2, 131.0, 128.8, 128.0, 127.3, 126.2, 122.2, 117.7, 59.3, 52.3, 45.5, 41.8, 33.8, 33.5, 29.3, 22.1. MS (ESI) m/z 421 [M + H]+.
4.1.16. Methyl 3-[4-(3-Aminopropyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzoate (13)
To a suspension of NaH (60% dispersion in mineral oil, 0.1 g, 2.5 mmol) in 12 mL of anhydrous THF at 0 °C under nitrogen was added dropwise a solution of diethyl cyanomethylphosphonate (0.38 mL, 2.32 mmol) in anhydrous THF (3 mL). The mixture was stirred at 0 °C for 30 min and 9 (0.7 g, 1.79 mmol) was slowly added. The reaction mixture was allowed to warm to room temperature. After 2 h, water and ethyl acetate were added, and the layers were separated. The aqueous layer was extracted with ethyl acetate. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 9:1) to give 527 mg (71%) of a colorless oil. MS (ESI) m/z 415 [M + H]+.
A mixture of the intermediate (0.5 g, 1.21 mmol), Raney Nickel (50 mg), and 10% Pd/C (50 mg) in methanol saturated with ammonia (60 mL) was stirred under a hydrogen atmosphere for 30 h. The catalyst was filtered off and the filtrate was evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1) to give 317 mg (62%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 8.28–8.27 (m, 1H), 7.96–7.92 (m, 1H), 7.86–7.82 (m, 1H), 7.81 (s, 1H), 7.60 (d, J = 8.1 Hz, 2H), 7.43 (t, J = 7.9 Hz, 1H), 7.22 (d, J = 8.1 Hz, 2H), 3.88 (s, 3H), 2.73–2.60 (m, 6H), 2.26 (t, J = 7.1 Hz, 2H), 2.18 (s, 6H), 1.90 (br, 2H), 1.82–1.68 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 166.4, 151.8, 142.0, 140.1, 131.3, 131.0, 129.5, 128.5, 127.7, 126.6, 125.9, 122.7, 121.5, 118.9, 59.1, 52.2, 45.4, 41.6, 33.9, 33.3, 29.2, 22.0. MS (ESI) m/z 421 [M + H]+.
4.1.17. Methyl 4-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino) Propyl]phenyl]pyrazol-1-yl]benzoate (14)
General procedure B: 90% yield (white solid). 1H NMR (CDCl3, 300 Mz): δ 8.11 (d, J = 8.8 Hz, 2H), 7.88 (s, 1H), 7.82 (d, J = 8.8 Hz, 2H), 7.65 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 8.1 Hz, 2H), 3.92 (s, 3H), 2.74–2.66 (m, 4H), 2.37–2.32 (m, 4H), 2.26 (s, 6H), 2.23 (s, 6H), 1.89–1.77 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 166.6, 152.6, 143.4, 142.2, 131.2, 131.0, 128.7, 128.0, 127.2, 126.2, 122.3, 117.7, 59.3, 59.2, 52.3, 45.5, 45.5, 33.5, 29.3, 28.2, 22.6. MS (ESI) m/z 449 [M + H]+.
4.1.18. Methyl 3-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl] Phenyl]pyrazol-1-yl]benzoate (15)
General procedure B: 82% yield (colorless oil). 1H NMR (CD3OD, 300 Mz): δ 8.37–8.36 (m, 1H), 8.10 (s, 1H), 7.98–7.94 (m, 1H), 7.85–7.83 (m, 1H), 7.61 (d, J = 8.1 Hz, 2H), 7.50 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 8.1 Hz, 2H), 3.89 (s, 3H), 2.66–2.61 (m, 4H), 2.39–2.31 (m, 4H), 2.24 (s, 6H), 2.20 (s, 6H), 1.87–1.74 (m, 4H). 13C NMR (CD3OD, 75 MHz): δ 167.6, 153.0, 143.0, 141.5, 132.6, 132.5, 130.8, 129.6, 129.0, 128.2, 127.7, 123.6, 122.7, 120.1, 60.1, 60.0, 52.9, 45.3 (2C), 34.3, 29.9, 28.6, 23.5. MS (ESI) m/z 449 [M + H]+.
4.1.19. [4-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methanol (16)
General procedure C: 88% yield (colorless oil). 1H NMR (CD3OD, 300 Mz): δ 8.18 (s, 1H), 7.76 (d, J = 8.6 Hz, 2H), 7.68 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.6 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 4.63 (s, 2H), 3.50 (s, 2H), 2.66 (t, J = 7.6 Hz, 2H), 2.39–2.34 (m, 2H), 2.24 (s, 6H), 2.22 (s, 6H), 1.89–1.78 (m, 2H). 13C NMR (CD3OD, 75 MHz): δ 153.9, 143.2, 141.3, 140.3, 132.1, 130.4, 129.6, 129.6, 129.1, 119.9, 118.6, 64.5, 60.1, 53.9, 45.4, 45.1, 34.3, 30.0. MS (ESI) m/z 393 [M + H]+.
4.1.20. [4-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methanol (17)
General procedure C: 89% yield (white solid). 1H NMR (CD3OD, 300 Mz): δ 8.08 (s, 1H), 7.74 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.7 Hz, 2H), 7.29 (d, J = 8.2 Hz, 2H), 4.62 (s, 2H), 2.69–2.64 (m, 4H), 2.38–2.30 (m, 4H), 2.23 (s, 6H), 2.19 (s, 6H), 1.88–1.72 (m, 4H). 13C NMR (CD3OD, 75 MHz): δ 152.7, 143.0, 141.0, 140.4, 132.6, 129.6, 129.1, 129.1, 128.4, 122.2, 119.7, 64.6, 60.2, 60.1, 45.4 (2C), 34.3, 30.0, 28.8, 23.4. MS (ESI) m/z 421 [M + H]+.
4.1.21. [3-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl]pyrazol-1-yl]phenyl]methanol (18)
General procedure C: 92% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 7.94 (s, 1H), 7.76–7.73 (m, 3H), 7.59 (d, J = 8.1 Hz, 1H), 7.31 (t, J = 7.7 Hz, 1H), 7.23–7.16 (m, 3H), 5.89 (s, 1H), 4.63 (s, 2H), 3.42 (s, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.28 (t, J = 7.0 Hz, 2H), 2.22 (s, 6H), 2.18 (s, 6H), 1.79 (quint, J = 7.3 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 151.8, 143.5, 141.3, 139.7, 130.7, 129.0, 128.2, 128.0, 127.8, 123.9, 117.8, 116.9, 116.4, 63.6, 58.7, 53.4, 44.9, 44.8, 33.1, 28.6. MS (ESI) m/z 393 [M + H]+.
4.1.22. [3-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl]Pyrazol-1-yl]phenyl]methanol (19)
General procedure C: 88% yield (colorless oil). 1H NMR (CD3OD, 300 Mz): δ 8.08 (s, 1H), 7.79 (s, 1H), 7.67–7.60 (m, 3H), 7.42 (t, J = 7.7 Hz, 1H), 7.29–7.27 (m, 3H), 4.67 (s, 2H), 2.69–2.63 (m, 4H), 2.38–2.30 (m, 4H), 2.23 (s, 6H), 2.19 (s, 6H), 1.88–1.72 (m, 4H). 13C NMR (CD3OD, 75 MHz): δ 152.8, 144.7, 143.1, 141.4, 132.6, 130.5, 129.6, 129.1, 128.5, 125.6, 122.3, 118.6, 118.1, 64.8, 60.2, 60.1, 45.4 (2C), 34.3, 30.0, 28.8, 23.4. MS (ESI) m/z 421 [M + H]+.
4.1.23. 4-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl] benzaldehyde (20)
General procedure D: 72% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 9.96 (s, 1H), 8.01 (s, 1H), 7.95–7.89 (m, 4H), 7.78 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 3.44 (s, 2H), 2.67 (t, J = 7.6 Hz, 2H), 2.32–2.27 (m, 8H), 2.21 (s, 6H), 1.86–1.76 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 191.0, 153.5, 144.2, 142.5, 133.7, 131.3, 130.4, 128.6, 128.3, 128.0, 120.3, 118.2, 59.2, 53.9, 45.5, 45.3, 33.5, 29.3. MS (ESI) m/z 391 [M + H]+.
4.1.24. 4-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzaldehyde (21)
General procedure D: 76% yield (yellow oil). 1H NMR (50 °C, CD3OD, 300 Mz): δ 9.92 (s, 1H), 8.16 (s, 1H), 7.95 (s, 4H), 7.64–7.61 (m, 2H), 7.29–7.27 (m, 2H), 2.70–2.64 (m, 4H), 2.39–2.31 (m, 4H), 2.24 (s, 6H), 2.20 (s, 6H), 1.86–1.75 (m, 4H). 13C NMR (50°C, CD3OD, 75 MHz): δ 192.7, 154.2, 145.6, 143.4, 135.3, 132.3, 132.2, 129.6, 129.1, 128.5, 123.7, 119.3, 60.2, 60.2, 45.4 (2C), 34.4, 29.8, 28.7, 23.5. MS (ESI) m/z 419 [M + H]+.
4.1.25. 3-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl] Benzaldehyde (22)
General procedure D: 60% yield (yellow oil). 1H NMR (CDCl3, 300 Mz): δ 10.04 (s, 1H), 8.24–8.23 (m, 1H), 8.08–8.04 (m, 1H), 8.01 (s, 1H), 7.82 (d, J = 8.1 Hz, 2H), 7.72 (d, J = 7.6 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 8.2 Hz, 2H), 3.45 (s, 2H), 2.69 (t, J = 7.6 Hz, 2H), 2.35–2.29 (m, 8H), 2.24 (s, 6H), 1.84 (quint, J = 7.4 Hz, 2H). 13C NMR (CD3OD, 75 MHz): δ 193.2, 154.3, 143.3, 141.9, 139.1, 132.0, 130.3, 129.6, 129.5, 128.2, 125.0, 119.9, 119.4, 118.7, 60.1, 54.0, 45.4, 45.2, 34.3, 29.9. MS (ESI) m/z 391 [M + H]+.
4.1.26. 3-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]benzaldehyde (23)
General procedure D: 66% yield (colorless oil). 1H NMR (CD3OD, 50 °C, 300 Mz): δ 10.03 (s, 1H), 8.28–8.27 (m, 1H), 8.16 (s, 1H), 8.09–8.04 (m, 1H), 7.79–7.76 (m, 1H), 7.66–7.59 (m, 3H), 7.31–7.27 (m, 2H), 2.73–2.66 (m, 4H), 2.49–2.40 (m, 4H), 2.31 (s, 6H), 2.25 (s, 6H), 1.89–1.78 (m, 4H). 13C NMR (CD3OD, 50 °C, 75 MHz): δ 193.3, 153.5, 143.1, 142.1, 139.3, 132.5, 131.4, 129.6, 129.2, 128.4, 128.0, 125.1, 122.9, 119.8, 60.0, 60.0, 45.2, 45.2, 34.2, 29.5, 28.5, 23.3. MS (ESI) m/z 419 [M + H]+.
4.1.27. 3-[4-[4-(Dimethylaminomethyl)-1-[4-(dimethylaminomethyl)phenyl] Pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (24)
General procedure E: 80% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.21 (s, 1H), 7.77 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 7.30 (t, J = 8.2 Hz, 2H), 3.52 (s, 2H), 3.49 (s, 2H), 2.68 (t, J = 7.6 Hz, 2H), 2.40–2.35 (m, 2H), 2.25 (s, 12H), 2.23 (s, 6H), 1.87–1.82 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 154.0, 143.2, 140.5, 137.2, 132.2, 131.8, 130.4, 129.6, 129.6, 119.8, 118.8, 64.2, 60.1, 54.0, 45.4, 45.2, 45.1, 34.3, 30.0. MS (ESI) m/z 420 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 8.4 min, PHPLC 99%; HPLC (C18, 35 min): tR 10.8 min, PHPLC 97%.
4.1.28. 3-[4-[1-[4-(Dimethylaminomethyl)phenyl]-4-[3-(dimethylamino)propyl] Pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (25)
General procedure E: 89% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.13 (s, 1H), 7.76 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 3.50 (s, 2H), 2.72–2.65 (m, 4H), 2.48–2.40 (m, 4H), 2.31 (s, 6H), 2.26 (s, 6H), 2.25 (s, 6H), 1.92–1.76 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.7, 142.8, 140.7, 136.7, 132.6, 131.8, 129.6, 129.1, 128.4, 122.1, 119.6, 64.1, 60.0, 59.9, 45.2 (3C), 34.2, 29.7, 28.5, 23.3. MS (ESI) m/z 448 [M + H]+. PHPLC > 96%. HPLC (C4, 35 min): tR 8.7 min, PHPLC 99%; HPLC (C18, 35 min): tR 11.8 min, PHPLC 96%.
4.1.29. 3-[4-[4-(Dimethylaminomethyl)-1-[3-(dimethylaminomethyl)phenyl] Pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (26)
General procedure E: 72% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CDCl3, 300 Mz): δ 7.97 (s, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.71 (s, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.25 (d, J = 8.3 Hz, 2H), 7.20 (d, J = 7.6 Hz, 1H), 3.46 (s, 2H), 3.46 (s, 2H), 2.68 (t, J = 7.6 Hz, 2H), 2.37 (t, J = 7.2 Hz, 2H), 2.28 (s, 6H), 2.27 (s, 6H), 2.25 (s, 6H), 1.86 (quint, J = 7.4 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 152.2, 141.6, 140.6, 140.1, 131.1, 129.3, 128.5, 128.3, 128.1, 126.7, 119.1, 118.4, 117.6, 64.2, 59.1, 53.9, 45.5, 45.2, 45.2, 33.4, 29.0. MS (ESI) m/z 420 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 7.6 min, PHPLC 98%; HPLC (C18, 35 min): tR 11.1 min, PHPLC 97%.
4.1.30. 3-[4-[1-[3-(Dimethylaminomethyl)phenyl]-4-[3-(dimethylamino)propyl] Pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (27)
General procedure E: 68% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.12 (s, 1H), 7.77 (s, 1H), 7.73–7.69 (m, 1H), 7.63 (d, J = 8.2 Hz, 2H), 7.43 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 8.1 Hz, 2H), 7.23 (d, J = 7.6 Hz, 1H), 3.52 (s, 2H), 2.72–2.65 (m, 4H), 2.42–2.34 (m, 4H), 2.26 (s, 12H), 2.22 (s, 6H), 1.90–1.77 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 143.0, 141.4, 140.7, 132.6, 130.5, 129.6, 129.1, 128.4, 128.3, 122.3, 120.7, 118.8, 64.7, 60.2, 60.1, 45.3 (3C), 34.3, 29.9, 28.8, 23.4. MS (ESI) m/z 448 [M + H]+. PHPLC > 98%. HPLC (C4, 35 min): tR 8.9 min, PHPLC 98%; HPLC (C18, 35 min): tR 12.1 min, PHPLC 98%.
4.1.31. 3-[4-[4-(Dimethylaminomethyl)-1-[4-[(4-methylpiperazin-1-yl)methyl] Phenyl]pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (28)
General procedure E: 75% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.21 (s, 1H), 7.76 (d, J = 8.6 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.6 Hz, 2H), 7.30 (t, J = 8.2 Hz, 2H), 3.55 (s, 2H), 3.52 (s, 2H), 2.68 (t, J = 7.6 Hz, 2H), 2.49 (br, 8H), 2.42–2.37 (m, 2H), 2.26 (s, 9H), 2.24 (s, 6H), 1.90–1.80 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 153.9, 143.2, 140.4, 137.1, 132.2, 131.7, 130.4, 129.6, 129.6, 119.8, 118.7, 63.1, 60.1, 55.7, 54.0, 53.5, 46.0, 45.3, 45.1, 34.3, 29.9. MS (ESI) m/z 475 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 7.6 min, PHPLC 98%; HPLC (C18, 35 min): tR 10.5 min, PHPLC 97%.
4.1.32. 3-[4-[4-[3-(Dimethylamino)propyl]-1-[4-[(4-methylpiperazin-1-yl) Methyl]phenyl] pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (29)
General procedure E: 77% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.10 (s, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.63 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8.1 Hz, 2H), 3.53 (s, 2H), 2.71–2.65 (m, 4H), 2.49 (br, 8H), 2.42–2.34 (m, 4H), 2.27 (s, 6H), 2.26 (s, 3H), 2.22 (s, 6H), 1.90–1.74 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.7, 143.0, 140.6, 136.7, 132.6, 131.6, 129.6, 129.1, 128.4, 122.2, 119.6, 63.1, 60.1, 60.0, 55.7, 53.5, 46.0, 45.3 (2C), 34.3, 29.9, 28.7, 23.4. MS (ESI) m/z 503 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 6.6 min, PHPLC 98%; HPLC (C18, 35 min): tR 11.3 min, PHPLC 97%.
4.1.33. 3-[4-[4-(Dimethylaminomethyl)-1-[3-[(4-methylpiperazin-1-yl)methyl] Phenyl]pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (30)
General procedure E: 74% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.23 (s, 1H), 7.81 (s, 1H), 7.73–7.69 (m, 3H), 7.43 (t, J = 7.8 Hz, 1H), 7.31 (d, J = 8.1 Hz, 2H), 7.27 (d, J = 8.0 Hz, 1H), 3.58 (s, 2H), 3.52 (s, 2H), 2.68 (t, J = 7.6 Hz, 2H), 2.49–2.43 (m, 10H), 2.32 (s, 6H), 2.26 (s, 3H), 2.24 (s, 6H), 1.92–1.82 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 153.9, 143.0, 141.3, 140.6, 132.2, 130.5, 130.4, 129.6, 129.6, 128.5, 120.8, 118.9, 118.7, 63.4, 59.9, 55.7, 54.0, 53.5, 45.9, 45.2, 45.1, 34.2, 29.7. MS (ESI) m/z 475 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 5.1 min, PHPLC 97%; HPLC (C18, 35 min): tR 10.9 min, PHPLC 98%.
4.1.34. 3-[4-[4-[3-(Dimethylamino)propyl]-1-[3-[(4-methylpiperazin-1-yl)methyl] Phenyl] pyrazol-3-yl]phenyl]-N,N-dimethyl-propan-1-amine (31)
General procedure E: 82% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.12 (s, 1H), 7.78 (s, 1H), 7.70–7.67 (m, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.41 (t, J = 7.8 Hz, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 7.6 Hz, 1H), 3.56 (s, 2H), 2.72–2.65 (m, 4H), 2.50 (br, 8H), 2.43–2.35 (m, 4H), 2.27 (s, 6H), 2.25 (s, 3H), 2.23 (s, 6H), 1.90–1.77 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 142.9, 141.4, 140.5, 132.6, 130.5, 129.6, 129.1, 128.4, 128.2, 122.2, 120.6, 118.7, 63.5, 60.1, 60.0, 55.7, 53.6, 46.0, 45.3 (2C), 34.3, 29.9, 28.7, 23.4. MS (ESI) m/z 503 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 7.9 min, PHPLC 97%; HPLC (C18, 35 min): tR 11.6 min, PHPLC 97%.
4.1.35. N′-[[4-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methyl]-N,N,N′-trimethyl-propane-1,3-diamine (32)
General procedure E: 62% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.23 (s, 1H), 7.78 (d, J = 8.5 Hz, 2H), 7.71 (d, J = 8.2 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.32 (t, J = 8.1 Hz, 2H), 3.57 (s, 2H), 3.55 (s, 2H), 2.70 (t, J = 7.6 Hz, 2H), 2.52–2.44 (m, 6H), 2.36 (s, 6H), 2.34 (s, 6H), 2.25 (s, 9H), 1.94–1.83 (m, 2H), 1.82–1.72 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 154.0, 143.0, 140.4, 137.8, 132.2, 131.6, 130.5, 129.7, 129.6, 119.9, 118.6, 62.4, 59.9, 58.6, 56.0, 53.9, 45.1, 45.1, 45.1, 42.3, 34.2, 29.6, 25.2. MS (ESI) m/z 491 [M + H]+. PHPLC > 95%. HPLC (C4, 35 min): tR 8.1 min, PHPLC 99%; HPLC (C18, 35 min): tR 10.3 min, PHPLC 95%.
4.1.36. N′-[[4-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methyl]-N,N,N′-trimethyl-propane-1,3-diamine (33)
General procedure E: 41% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.11 (s, 1H), 7.74 (d, J = 8.6 Hz, 2H), 7.63 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 8.2 Hz, 2H), 3.54 (s, 2H), 2.72–2.65 (m, 4H), 2.44–2.33 (m, 8H), 2.25 (s, 12H), 2.22 (s, 3H), 2.20 (s, 6H), 1.90–1.67 (m, 6H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 143.0, 140.5, 137.5, 132.6, 131.6, 129.6, 129.1, 128.4, 122.3, 119.6, 62.4, 60.2, 60.1, 58.6, 56.2, 45.4 (3C), 42.3, 34.3, 30.0, 28.8, 25.7, 23.4. MS (ESI) m/z 519 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 8.3 min, PHPLC 97%; HPLC (C18, 35 min): tR 11.3 min, PHPLC 97%.
4.1.37. N′-[[3-[4-(Dimethylaminomethyl)-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methyl]-N,N,N′-trimethyl-propane-1,3-diamine (34)
General procedure E: 60% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.22 (s, 1H), 7.81 (s, 1H), 7.73–7.68 (m, 3H), 7.43 (t, J = 7.8 Hz, 1H), 7.29 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 7.8 Hz, 1H), 3.58 (s, 2H), 3.52 (s, 2H), 2.67 (t, J = 7.6 Hz, 2H), 2.45–2.32 (m, 6H), 2.24–2.22 (m, 21H), 1.89–1.69 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 153.9, 143.2, 141.6, 141.3, 132.2, 130.5, 130.3, 129.6, 129.5, 128.4, 120.7, 118.8, 118.7, 62.9, 60.1, 58.6, 56.3, 54.0, 45.4 (2C), 45.2, 42.5, 34.4, 30.0, 25.8. MS (ESI) m/z 491 [M + H]+. PHPLC > 96%. HPLC (C4, 35 min): tR 8.0 min, PHPLC 96%; HPLC (C18, 35 min): tR 10.6 min, PHPLC 96%.
4.1.38. N′-[[3-[4-[3-(Dimethylamino)propyl]-3-[4-[3-(dimethylamino)propyl]phenyl] Pyrazol-1-yl]phenyl]methyl]-N,N,N′-trimethyl-propane-1,3-diamine (35)
General procedure E: 62% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.12 (s, 1H), 7.79 (s, 1H), 7.70–7.67 (m, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.42 (t, J = 7.8 Hz, 1H), 7.30 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 7.6 Hz, 1H), 3.57 (s, 2H), 2.72–2.65 (m, 4H), 2.45–2.31 (m, 8H), 2.23 (s, 9H), 2.21 (s, 6H), 2.19 (s, 6H), 1.89–1.67 (m, 6H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 143.1, 141.5, 141.4, 132.7, 130.4, 129.6, 129.1, 128.4, 128.1, 122.3, 120.6, 118.5, 62.9, 60.2, 60.1, 58.6, 56.3, 45.4 (3C), 42.5, 34.4, 30.1, 28.9, 25.8, 23.5. MS (ESI) m/z 519 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 7.5 min, PHPLC 98%; HPLC (C18, 35 min): tR 11.4 min, PHPLC 97%.
4.1.39. Methyl 3-[2-[1-(4-Cyanophenyl)ethylidene]hydrazino]benzoate (38)
A mixture of 4′-cyanoacetophenone 36 (5 g, 34.4 mmol) and 4 (10 g, 49.3 mmol) in methanol (90 mL) was refluxed for 6 h. The reaction mixture was then stirred at rt for 18 h. The solid was collected by filtration, washed with methanol, and dried to give 7.34 g (73%) of the product as a yellow solid. 1H NMR (CD3SOCD3, 300 Mz): δ 9.80 (s, 1H), 7.94 (d, J = 8.6 Hz, 2H), 7.87–7.86 (m, 1H), 7.82 (d, J = 8.6 Hz, 2H), 7.58–7.54 (m, 1H), 7.42–7.35 (m, 2H), 3.85 (s, 3H), 2.27 (s, 3H). 13C NMR (CD3SOCD3, 75 MHz): δ 166.5, 145.8, 143.3, 139.7, 132.2, 130.4, 129.4, 125.8, 120.1, 119.1, 117.4, 113.8, 109.5, 52.1, 12.6. MS (ESI) m/z 294 [M + H]+.
4.1.40. Methyl 3-[2-[1-(3-Cyanophenyl)ethylidene]hydrazino]benzoate (39)
A solution of 3’-cyanoacetophenone 37 (3.5 g, 24.1 mmol) and 4 (5.8 g, 28.6 mmol) in methanol (30 mL) was stirred at rt for 1 h, heated at reflux for 6 h and then stirred at rt for 18 h. The solid was collected by filtration, washed with methanol, and dried to give 5.29 g (75%) of the product as a white solid. 1H NMR (CD3SOCD3, 300 Mz): δ 9.71 (s, 1H), 8.16–8.10 (m, 2H), 7.84 (dd, J = 1.1, 1.1 Hz, 1H), 7.75 (ddd, J = 7.7, 1.2, 1.2 Hz, 1H), 7.62–7.56 (m, 2H), 7.39–7.38 (m, 2H), 3.85 (s, 3H), 2.29 (s, 3H). 13C NMR (CD3SOCD3, 75 MHz): δ 166.5, 145.9, 140.2, 139.7, 131.0, 130.3, 129.7, 129.6, 129.5, 128.7, 119.9, 118.9, 117.3, 113.7, 111.6, 52.1, 12.8. MS (ESI) m/z 292 [M − H]+.
4.1.41. Methyl 3-[3-(4-Cyanophenyl)-4-formyl-pyrazol-1-yl]benzoate (40)
Dimethylformamide (26.9 mL, 348 mmol) was cooled to −5 °C with a salt/ice bath. Phosphorus oxychloride (15.3 g, 9.28 mL, 99.6 mmol) was added dropwise while maintaining the temperature below 0 °C. The mixture was stirred at −5 °C for 40 min and 38 (7.3 g, 24.9 mmol) was slowly added. The reaction mixture was allowed to warm to rt. After 1 h, the mixture was heated at 50 °C for 4 h. The mixture was then poured on water and stirred for 2 h. The solid was collected by filtration, washed with a mixture of methanol and ether (1/3), and dried to give 7.84 g (95%) of the product as a white solid. 1H NMR (CF3COOD, 300 Mz): δ 9.90 (s, 1H), 9.81 (s, 1H), 8.44 (s, 1H), 8.20 (d, J = 7.7 Hz, 1H), 7.97–7.94 (m, 3H), 7.86 (d, J = 7.9 Hz, 2H), 7.70–7.65 (m, 1H), 4.04 (s, 3H). 13C NMR (CF3COOD, 75 MHz): δ 190.7, 171.2, 156.9, 140.4, 138.7, 136.6, 135.1, 133.5, 133.2, 132.7, 132.2, 128.6, 124.9, 123.8, 55.3. MS (ESI) m/z 332 [M + H]+.
4.1.42. Methyl 3-[3-(3-Cyanophenyl)-4-formyl-pyrazol-1-yl]benzoate (41)
General procedure A: 95% yield (white solid). 1H NMR (CDCl3, 300 Mz): δ 10.07 (s, 1H), 8.64 (s, 1H), 8.43 (dd, J = 1.8, 1.8 Hz, 1H), 8.29 (dd, J = 1.4, 1.4 Hz, 1H), 8.22 (ddd, J = 7.9, 1.3, 1.3 Hz, 1H), 8.09 (ddd, J = 7.9, 1.3, 1.3 Hz, 1H), 8.07–8.03 (m, 1H), 7.75 (ddd, J = 7.8, 1.6, 1.6 Hz, 1H), 7.66–7.59 (m, 2H), 3.99 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 183.6, 165.9, 151.8, 139.0, 133.6, 133.3, 132.8, 132.7, 132.5, 132.1, 130.2, 129.6, 129.3, 124.0, 123.1, 120.4, 118.6, 113.1, 52.8. MS (ESI) m/z 332 [M + H]+.
4.1.43. Methyl 3-[3-(4-Cyanophenyl)-4-methyl-pyrazol-1-yl]benzoate (42)
A mixture of 40 (7.76 g, 23.4 mmol), triethylsilane (9.45 mL, 58.5 mmol), and trifluoroacetic acid (26.1 mL, 351 mmol) was stirred vigorously at rt for 24 h. The reaction mixture was then evaporated to dryness. The residue was purified by column chromatography (DCM/MeOH = 95:5) to give 6.44 g (87%) of the product as a white solid. 1H NMR (CDCl3, 300 Mz): δ 8.34–8.33 (m, 1H), 8.01–7.93 (m, 4H), 7.89–7.88 (m, 1H), 7.75–7.72 (m, 2H), 7.54 (dd, J = 8.0, 8.0 Hz, 1H), 3.96 (s, 3H), 2.35 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 166.4, 149.9, 140.1, 138.3, 132.5, 131.7, 129.8, 127.9, 127.8, 127.5, 123.1, 119.4, 119.1, 117.4, 111.2, 52.6, 10.6. MS (ESI) m/z 318 [M + H]+.
4.1.44. Methyl 3-[3-(3-Cyanophenyl)-4-methyl-pyrazol-1-yl]benzoate (43)
A mixture of 41 (2.5 g, 7.55 mmol), triethylsilane (2.1 g, 2.92 mL, 18.1 mmol), and trifluoroacetic acid (12.8 g, 8.33 mL, 112 mmol) was vigorously stirred at rt for 24 h. It was then evaporated to dryness. The residue was purified by column chromatography (DCM) to give 2.07 g (86%) of the product as a white solid. 1H NMR (CDCl3, 300 Mz): δ 8.33 (s, 1H), 8.11 (s, 1H), 8.05 (d, J = 7.8 Hz, 1H), 8.00–7.94 (m, 2H), 7.88 (s, 1H), 7.64 (d, J = 7.7 Hz, 1H), 7.58–7.52 (m, 2H), 3.97 (s, 3H), 2.34 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 166.3, 149.5, 139.9, 134.9, 131.5, 131.5, 131.0, 130.8, 129.7, 129.4, 127.6, 127.3, 122.9, 119.2, 118.9, 116.9, 112.7, 52.4, 10.4. MS (ESI) m/z 318 [M + H]+.
4.1.45. Methyl 3-[3-[4-(Aminomethyl)phenyl]-4-methyl-pyrazol-1-yl]benzoate (44)
To a solution of 42 (6.37 g, 20.1 mmol) in anhydrous THF (90 mL) under nitrogen was added BH3-THF (1 M in THF, 30.1 mL, 30.1 mmol). The reaction mixture was refluxed for 3 h. Methanol (25 mL) was then slowly added. HCl (4 M in dioxane, 27.6 mL, 110 mmol) was then added and the mixture was refluxed for 90 min. The solvent was evaporated. Ethyl acetate and water were added to the residue and the pH of the mixture was brought to 10 by the addition of potassium carbonate. The layers were separated. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 95:5) to give 3.3 g (51%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 8.26–8.25 (m, 1H), 7.89–7.85 (m, 1H), 7.82–7.79 (m, 1H), 7.71–7.68 (m, 3H), 7.38 (dd, J = 7.9, 7.9 Hz, 1H), 7.31 (d, J = 8.2 Hz, 2H), 3.85 (s, 3H), 3.82 (s, 2H), 2.21 (s, 3H), 1.53 (s, 2H). 13C NMR (CDCl3, 75 MHz): δ 166.1, 151.5, 142.7, 139.9, 131.8, 131.1, 129.2, 127.4, 126.9, 126.8, 126.4, 122.4, 118.6, 116.4, 52.1, 46.0, 10.1. MS (ESI) m/z 322 [M + H]+.
4.1.46. Methyl 3-[3-[3-(Aminomethyl)phenyl]-4-methyl-pyrazol-1-yl]benzoate (45)
To a solution of 43 (3.03 g, 9.53 mmol) in anhydrous THF (60 mL) under nitrogen was added BH3-THF (1 M in THF, 13.3 mL, 13.3 mmol). The reaction mixture was refluxed for 2 h 30. Methanol (25 mL) was then slowly added, followed by HCl (4 M in dioxane, 13.1 mL, 52.4 mmol), and the mixture was refluxed for 90 min. The solvent was evaporated. Ethyl acetate and water were added to the residue and the pH of the mixture was brought to 10 by the addition of potassium carbonate. The layers were separated. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 95:5) to give 2.31 g (75%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 8.32 (dd, J = 1.8, 1.8 Hz, 1H), 7.98–7.95 (m, 1H), 7.89 (ddd, J = 7.8, 1.3, 1.3 Hz, 1H), 7.81 (s, 1H), 7.75 (s, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.48 (dd, J = 7.9, 7.9 Hz, 1H), 7.40 (dd, J = 7.6, 7.6 Hz, 1H), 7.30 (d, J = 7.6 Hz, 1H), 3.92 (s, 5H), 2.29 (s, 3H), 1.69 (s, 2H). 13C NMR (CDCl3, 75 MHz): δ 166.4, 152.0, 143.6, 140.2, 133.8, 131.4, 129.5, 128.7, 127.1, 126.8, 126.6, 126.3, 126.1, 122.9, 119.1, 116.8, 52.3, 46.6, 10.3. MS (ESI) m/z 322 [M + H]+.
4.1.47. Methyl 3-[3-[4-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl]benzoate (46)
General procedure B: 46% yield (colorless oil). 1H NMR (CD3OD, 300 Mz): δ 8.24–8.22 (m, 1H), 7.83–7.83 (m, 1H), 7.82–7.78 (m, 1H), 7.74–7.71 (m, 1H), 7.64 (d, J = 8.2 Hz, 2H), 7.37 (dd, J = 7.9, 7.9 Hz, 1H), 7.28 (d, J = 8.2 Hz, 2H), 3.82 (s, 3H), 3.38 (s, 2H), 2.27 (s, 6H), 2.14 (s, 3H). 13C NMR (CD3OD, 75 MHz): δ 167.4, 152.5, 141.2, 138.2, 133.9, 132.4, 130.6, 128.7, 128.4, 127.5, 123.3, 119.8, 117.8, 65.5, 52.8, 45.2, 10.5. MS (ESI) m/z 350 [M + H]+.
4.1.48. Methyl 3-[3-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl] Benzoate (47)
General procedure B: 93% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 8.31 (dd, J = 1.8, 1.8 Hz, 1H), 7.96–7.92 (m, 1H), 7.88–7.85 (m, 1H), 7.78–7.77 (m, 1H), 7.73–7.72 (m, 1H), 7.66–7.64 (m, 1H), 7.44 (dd, J = 7.9, 7.9 Hz, 1H), 7.38 (dd, J = 7.6, 7.6 Hz, 1H), 7.33–7.30 (m, 1H), 3.90 (s, 3H), 3.48 (s, 2H), 2.27–2.25 (m, 9H). 13C NMR (CDCl3, 75 MHz): δ 166.3, 151.9, 140.1, 139.1, 133.4, 131.3, 129.4, 128.5, 128.4, 128.3, 126.9, 126.6, 126.3, 122.7, 118.9, 116.7, 64.3, 52.2, 45.3, 10.3. MS (ESI) m/z 350 [M + H]+.
4.1.49. [3-[3-[4-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl]phenyl] Methanol (48)
General procedure C: 85% yield (colorless oil). 1H NMR (CD3OD, 300 Mz): δ 7.98–7.98 (m, 1H), 7.76–7.75 (m, 1H), 7.72–7.69 (m, 2H), 7.63–7.60 (m, 1H), 7.42–7.35 (m, 3H), 7.26–7.24 (m, 1H), 4.66 (s, 2H), 3.49 (s, 2H), 2.24 (s, 9H). 13C NMR (CD3OD, 75 MHz): δ 152.5, 144.6, 141.3, 138.0, 134.2, 130.8, 130.5, 129.2, 128.6, 125.5, 118.4, 118.0, 117.5, 64.7, 64.5, 45.1, 10.3. MS (ESI) m/z 322 [M + H]+.
4.1.50. [3-[3-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl]phenyl] Methanol (49)
General procedure C: 88% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 7.72 (s, 1H), 7.67–7.64 (m, 3H), 7.58–7.55 (m, 1H), 7.39 (dd, J = 7.6, 7.6 Hz, 1H), 7.33–7.28 (m, 2H), 7.13 (d, J = 7.6 Hz, 1H), 4.62 (s, 2H), 4.48 (s, 1H), 3.48 (s, 2H), 2.26 (s, 3H), 2.23 (s, 6H). 13C NMR (CDCl3, 75 MHz): δ 151.4, 143.2, 140.1, 138.4, 133.7, 129.3, 128.6, 128.5, 128.5, 127.2, 126.5, 124.1, 117.3, 116.7, 116.2, 64.2, 45.2 (2C), 10.3. MS (ESI) m/z 322 ([M + H]+.
4.1.51. 3-[3-[4-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl] Benzaldehyde (50)
General procedure D: 56% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 9.99 (s, 1H), 8.15–8.14 (m, 1H), 7.99–7.95 (m, 1H), 7.78–7.77 (m, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.68–7.65 (m, 1H), 7.52 (dd, J = 7.8, 7.8 Hz, 1H), 7.36 (d, J = 8.2 Hz, 2H), 3.44 (s, 2H), 2.26 (s, 3H), 2.23 (s, 6H). 13C NMR (CDCl3, 75 MHz): δ 191.5, 152.0, 140.6, 138.4, 137.3, 132.2, 130.1, 129.3, 127.4, 126.9 (2C), 123.8, 118.3, 117.0, 64.0, 45.3, 10.3. MS (ESI) m/z 320 [M + H]+.
4.1.52. 3-[3-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl]benzaldehyde (51)
General procedure D: 61% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 10.03 (s, 1H), 8.18 (dd, J = 1.8, 1.8 Hz, 1H), 8.03–7.99 (m, 1H), 7.82–7.81 (m, 1H), 7.73 (dd, J = 1.5, 1.5 Hz, 1H), 7.70 (ddd, J = 7.6, 1.2, 1.2 Hz, 1H), 7.65 (ddd, J = 7.5, 1.5, 1.5 Hz, 1H), 7.55 (dd, J = 7.8, 7.8 Hz, 1H), 7.40 (dd, J = 7.6, 7.6 Hz, 1H), 7.33 (ddd, J = 7.7, 1.5, 1.5 Hz, 1H), 3.49 (s, 2H), 2.29–2.27 (m, 9H). 13C NMR (CDCl3, 75 MHz): δ 191.6, 152.3, 140.8, 139.2, 137.5, 133.3, 130.1, 128.7, 128.5, 128.3, 127.0, 126.9, 126.4, 123.9, 118.5, 117.1, 64.4, 45.4, 10.3. MS (ESI) m/z 320 [M + H]+.
4.1.53. 3-(Dimethylamino)propanoic Acid Hydrochloride (52)
A mixture of beta-alanine (5.4 g, 60.6 mmol), formic acid (40 mL, 1060 mmol), and 37% formaldehyde in water (13 mL, 173 mmol) was refluxed for 15 h. A total of 37% HCl (12 mL) was added, and the reaction mixture was evaporated. The residue was washed with a mixture of ethyl acetate and methanol (4:1), collected by filtration, and dried to give 8.07 g (87%) of the product as a white solid. 1H NMR (CD3OD, 300 Mz): δ 3.44 (t, J = 6.9 Hz, 2H), 2.93 (s, 6H), 2.88 (t, J = 6.9 Hz, 2H). 13C NMR (CD3OD, 75 MHz): δ 173.3, 54.6, 43.7, 29.8. MS (ESI) m/z 117 [M]+.
4.1.54. Methyl 3-[3-(4-{[3-(Dimethylamino)propanamido]methyl}phenyl)-4-methyl- 1H-pyrazol-1-yl] Benzoate (53)
To a mixture of 44 (1.87 g, 5.83 mmol), 52 (0.98 g, 6.41 mmol), hydroxybenzotriazole hydrate (1.02 g, 7.58 mmol) and triethylamine (3.25 mL, 23.3 mmol) in methylene chloride (60 mL) under nitrogen at rt was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (1.34 g, 7 mmol). The mixture was stirred for 20 h. A total of 10% K2CO3 was added, and the layers were separated. The aqueous layer was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 475:25:1) to give 1.77 g (72%) of the product as a white solid. 1H NMR (CDCl3, 300 Mz): δ 8.75 (br, 1H), 8.33–8.32 (m, 1H), 8.01–7.97 (m, 1H), 7.93–7.89 (m, 1H), 7.85–7.84 (m, 1H), 7.75 (d, J = 8.2 Hz, 2H), 7.51 (dd, J = 7.9, 7.9 Hz, 1H), 7.34 (d, J = 8.2 Hz, 2H), 4.49 (d, J = 5.7 Hz, 2H), 3.95 (s, 3H), 2.59 (t, J = 5.9 Hz, 2H), 2.44 (t, J = 6.2 Hz, 2H), 2.31 (s, 3H), 2.25 (s, 6H). 13C NMR (CDCl3, 75 MHz): δ 172.6, 166.6, 151.8, 140.3, 138.5, 132.5, 131.5, 129.7, 127.9, 127.6, 127.2, 126.9, 123.0, 119.2, 116.9, 55.4, 52.5, 44.7, 42.9, 33.0, 10.4. MS (ESI) m/z 421 [M + H]+.
4.1.55. Methyl 3-[3-[3-[[3-(Dimethylamino)propanoylamino]methyl]phenyl]-4-methyl-pyrazol-1-yl]benzoate (54)
To a mixture of 45 (0.5 g, 1.56 mmol), 52 (0.26 g, 1.71 mmol), hydroxybenzotriazole hydrate (0.27 g, 2.02 mmol) and triethylamine (0.86 mL, 6.22 mmol) in methylene chloride (15 mL) under nitrogen at rt was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.36 g, 1.87 mmol). The mixture was stirred for 39 h. A total of 10% K2CO3 was added, and the layers were separated. The aqueous layer was extracted with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 95:5:0.3) to give 535 mg (82%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 8.69–8.68 (m, 1H), 8.27 (dd, J = 1.9, 1.9 Hz, 1H), 7.91–7.87 (m, 1H), 7.84 (ddd, J = 7.8, 1.3, 1.3 Hz, 1H), 7.78–7.77 (m, 1H), 7.67 (s, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.43 (dd, J = 7.9, 7.9 Hz, 1H), 7.34 (dd, J = 7.6, 7.6 Hz, 1H), 7.22 (d, J = 7.7 Hz, 1H), 4.47 (d, J = 5.7 Hz, 2H), 3.88 (s, 3H), 2.56 (t, J = 6.0 Hz, 2H), 2.39 (t, J = 6.3 Hz, 2H), 2.24 (s, 3H), 2.19 (s, 6H). 13C NMR (CDCl3, 75 MHz): δ 172.3, 166.3, 151.7, 140.1, 139.1, 133.7, 131.3, 129.4, 128.6, 127.0, 126.7 (2C), 126.4, 126.2, 122.6, 118.9, 116.7, 55.1, 52.2, 44.5, 42.9, 32.9, 10.2. MS (ESI) m/z 421 [M + H]+.
4.1.56. (3-{3-[4-({[3-(Dimethylamino)propyl]amino}methyl)phenyl]-4-methy-1H-pyrazol-1-yl}phenyl)methanol (55)
To a suspension of aluminum chloride (2.39 g, 18 mmol) in anhydrous THF (50 mL) at 0 °C under nitrogen was added dropwise lithium aluminum hydride (1 M in THF, 18 mL, 18 mmol). The mixture was stirred for 20 min and a solution of 53 (1.68 g, 3.99 mmol) in anhydrous THF (50 mL) was added dropwise. The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to rt. After 20 h, the solution was poured on ice. Ethyl acetate and K2CO3 were added, and the mixture was stirred for 15 min. The solid was filtered off. The layers were separated, and the organic layer was washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1) to give 740 mg (49%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 7.75–7.70 (m, 4H), 7.61–7.58 (m, 1H), 7.40–7.34 (m, 3H), 7.22–7.19 (m, 1H), 4.69 (s, 2H), 3.79 (s, 2H), 3.00 (br, 2H), 2.66 (t, J = 7.0 Hz, 2H), 2.30 (t, J = 7.0 Hz, 2H), 2.27 (s, 3H), 2.20 (s, 6H), 1.68 (quint, J = 7.2 Hz, 2H). 13C NMR (CDCl3, 75 MHz): δ 151.5, 143.2, 140.3, 139.3, 132.6, 129.5, 128.4, 127.7, 127.2, 124.2, 117.5, 117.0, 116.3, 64.6, 58.2, 53.7, 47.9, 45.5, 27.6, 10.4. MS (ESI) m/z 379 [M + H]+.
4.1.57. [3-[3-[3-[[3-(Dimethylamino)propylamino]methyl]phenyl]-4-methyl-pyrazol-1-yl]phenyl]methanol (56)
To a suspension of aluminum chloride (1.53 g, 11.5 mmol) in anhydrous THF (40 mL) at 0 °C under nitrogen was added dropwise LAH (1 M in THF, 11.5 mL, 11.5 mmol). The mixture was stirred for 20 min and a solution of 54 (1.07 g, 2.54 mmol) in anhydrous THF (40 mL) was added dropwise. The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to rt. After 20 h, the solution was slowly poured on ice. Ethyl acetate and K2CO3 were added, and the mixture was stirred for 15 min. The solid was filtered off. The layers were separated, and the organic layer was washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 9:1:0.1) to give 737 mg (77%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 7.71–7.70 (m, 3H), 7.63–7.60 (m, 1H), 7.57–7.54 (m, 1H), 7.39–7.26 (m, 3H), 7.17 (d, J = 7.7 Hz, 1H), 4.64 (s, 2H), 3.80 (s, 2H), 3.41 (br, 2H), 2.65 (t, J = 7.0 Hz, 2H), 2.29 (t, J = 7.1 Hz, 2H), 2.25 (s, 3H), 2.16 (s, 6H), 1.71–1.61 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 151.4, 143.5, 140.2, 133.9, 129.3, 128.6, 127.5, 127.4, 127.2, 126.3, 124.1, 117.3, 116.8, 116.2, 64.2, 58.2, 53.9, 47.8, 45.4, 27.6, 10.3. MS (ESI) m/z 379 [M + H]+.
4.1.58. (3-{3-[4-({[3-(Dimethylamino)propyl](methyl)amino}methyl)phenyl]-4-methyl-1H-pyrazol-1-yl}phenyl)methanol (57)
To a mixture of 55 (0.72 g, 1.9 mmol), 37% formaldehyde in water (0.85 mL, 11.4 mmol) and acetic acid (0.65 mL, 11.4 mmol) in methanol (15 mL) were slowly added over 40 min STAB (2.42 g, 11.4 mmol). The mixture was stirred for 1 h and the solvent was evaporated. Ethyl acetate and 10% K2CO3 were added. After 10 min of stirring, the layers were separated. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH-NH3 sat = 95:5 to 92:8) to give 0.55 g (74%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 7.77–7.70 (m, 4H), 7.63–7.60 (m, 1H), 7.42–7.35 (m, 3H), 7.23–7.21 (m, 1H), 4.72 (s, 2H), 3.51 (s, 2H), 2.92 (br, 1H), 2.40 (t, J = 7.3 Hz, 2H), 2.33–2.28 (m, 5H), 2.22 (s, 6H), 2.20 (s, 3H), 1.76–1.66 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 151.7, 143.0, 140.4, 138.6, 132.5, 129.6, 129.3, 127.5, 127.2, 124.2, 117.6, 117.0, 116.3, 64.8, 62.2, 57.9, 55.6, 45.5, 42.3, 25.6, 10.4. MS (ESI) m/z 393 [M + H]+.
4.1.59. [3-[3-[3-[[3-(Dimethylamino)propyl-methyl-amino]methyl]phenyl]-4-methyl-pyrazol-1-yl]phenyl]methanol (58)
To a solution of 56 (0.79 g, 2.08 mmol), 37% formaldehyde in water (0.47 mL, 6.23 mmol) and acetic acid (0.36 mL, 6.23 mmol) in methanol (15 mL) were slowly added over 15 min STAB (1.1 g, 5.19 mmol). The mixture was stirred at rt for 15 h and the solvent was evaporated. Methylene chloride and water were added to the residue. The mixture was brought to pH = 10 with ammonium hydroxide and the layers were separated. The aqueous layer was extracted twice with methylene chloride. The combined organic layers were washed with brine, dried, and evaporated. The residue was purified by column chromatography (DCM/MeOH/NH4OH = 95:5:0.5) to give 591 mg (73%) of the product as a colorless oil. 1H NMR (CDCl3, 300 Mz): δ 7.73–7.70 (m, 3H), 7.65–7.62 (m, 1H), 7.57–7.54 (m, 1H), 7.39–7.27 (m, 3H), 7.16 (d, J = 7.6 Hz, 1H), 4.95 (br, 1H), 4.64 (s, 2H), 3.52 (s, 2H), 2.39 (t, J = 7.2 Hz, 2H), 2.33–2.28 (m, 2H), 2.26 (s, 3H), 2.19 (s, 3H), 2.18 (s, 6H), 1.74–1.64 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 151.5, 143.5, 140.1, 139.1, 133.6, 129.2, 128.3, 128.2, 127.1, 126.2, 124.0, 117.1, 116.7, 116.1, 64.1, 62.3, 57.7, 55.4, 45.2, 42.2, 25.2, 10.3. MS (ESI) m/z 393 [M + H]+
4.1.60. 3-{3-[4-({[3-(Dimethylamino)propyl](methyl)amino}methyl)phenyl]-4-methyl-1H-pyrazol-1-yl}benzaldehyde (59)
General procedure D: 66% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 10.03 (s, 1H), 8.18–8.17 (m, 1H), 8.03–7.99 (m, 1H), 7.82–7.82 (m, 1H), 7.73–7.68 (m, 3H), 7.56 (dd, J = 7.8, 7.8 Hz, 1H), 7.38 (d, J = 8.2 Hz, 2H), 3.51 (s, 2H), 2.41 (t, J = 7.3 Hz, 2H), 2.34–2.29 (m, 5H), 2.22 (s, 6H), 2.20 (s, 3H), 1.75–1.65 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 191.6, 152.2, 140.8, 139.0, 137.5, 132.0, 130.2, 129.2, 127.4, 127.0, 126.9, 123.9, 118.4, 117.1, 62.2, 57.9, 55.5, 45.5, 42.3, 25.7, 10.4. MS (ESI) m/z 391 [M + H]+.
4.1.61. 3-[3-[3-[[3-(Dimethylamino)propyl-methyl-amino]methyl]phenyl]-4-methyl-pyrazol-1-yl]benzaldehyde (60)
General procedure D: 55% yield (colorless oil). 1H NMR (CDCl3, 300 Mz): δ 10.00 (s, 1H), 8.15 (dd, J = 1.7, 1.7 Hz, 1H), 8.00–7.96 (m, 1H), 7.79–7.78 (m, 1H), 7.71–7.70 (m, 1H), 7.67 (ddd, J = 7.6, 1.2, 1.2 Hz, 1H), 7.62 (ddd, J = 7.5, 1.5, 1.5 Hz, 1H), 7.52 (dd, J = 7.9, 7.9 Hz, 1H), 7.36 (dd, J = 7.6, 7.6 Hz, 1H), 7.31–7.29 (m, 1H), 3.52 (s, 2H), 2.41 (t, J = 7.2 Hz, 2H), 2.32–2.27 (m, 2H), 2.27 (s, 3H), 2.19 (s, 9H), 1.73–1.63 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 191.5, 152.3, 140.7, 139.6, 137.4, 133.2, 130.1, 128.5, 128.4, 128.1, 126.9, 126.2, 123.8, 118.3, 117.0, 62.3, 57.8, 55.6, 45.4, 42.2, 25.6, 10.3. MS (ESI) m/z 391 [M + H]+.
4.1.62. {[4-(1-{3-[(Dimethylamino)methyl]phenyl}-4-methyl-1H-pyrazol-3-yl)phenyl]methyl}dimethylamine (61)
General procedure E: 79% yield. The compound was converted to its 2HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.04–8.03 (m, 1H), 7.74–7.72 (m, 3H), 7.69–7.65 (m, 1H), 7.43–7.37 (m, 3H), 7.22–7.20 (m, 1H), 3.49 (s, 2H), 3.48 (s, 2H), 2.28–2.24 (m, 15H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.6, 141.4, 140.7, 138.3, 134.2, 130.8, 130.5, 129.2, 128.6, 128.2, 120.6, 118.7, 117.6, 64.7, 64.6, 45.3, 45.2, 10.3. MS (ESI) m/z 349 [M + H]+. PHPLC > 96%. HPLC (C4, 35 min): tR 13.4 min, PHPLC 99%; HPLC (C18, 35 min): tR 17.6 min, PHPLC 96%.
4.1.63. Dimethyl({[4-(4-methyl-1-{3-[(4-methylpiperazin-1-yl)methyl]phenyl}-1H-pyrazol-3-yl)phenyl]methyl})amine (62)
General procedure E: 61% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.04–8.04 (m, 1H), 7.76–7.71 (m, 3H), 7.67–7.64 (m, 1H), 7.42–7.38 (m, 3H), 7.24–7.22 (m, 1H), 3.56 (s, 2H), 3.50 (s, 2H), 2.49 (m, 8H), 2.28–2.25 (m, 12H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.6, 141.4, 140.5, 138.3, 134.2, 130.8, 130.4, 129.2, 128.7, 128.2, 120.6, 118.7, 117.6, 64.6, 63.5, 55.7, 53.6, 46.0, 45.2, 10.3. MS (ESI) m/z 404 [M + H]+. PHPLC > 96%. HPLC (C4, 30 min): tR 12.1 min, PHPLC 96%; HPLC (C18, 30 min): tR 17.3 min, PHPLC 97%.
4.1.64. [(4-{1-[3-({[3-(Dimethylamino)propyl](methyl)amino}methyl)phenyl]-4-methyl-1H-pyrazol-3-yl}phenyl)methyl]dimethylamine (63)
General procedure E: 73% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.05–8.05 (m, 1H), 7.77–7.72 (m, 3H), 7.67–7.64 (m, 1H), 7.43–7.38 (m, 3H), 7.24–7.21 (m, 1H), 3.56 (s, 2H), 3.50 (s, 2H), 2.44–2.39 (m, 2H), 2.36–2.31 (m, 2H), 2.29 (s, 3H), 2.25 (s, 6H), 2.23 (s, 3H), 2.22 (s, 6H), 1.76–1.66 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.6, 141.4, 141.4, 138.3, 134.2, 130.8, 130.4, 129.2, 128.7, 128.1, 120.5, 118.5, 117.5, 64.7, 62.9, 58.6, 56.2, 45.4, 45.2, 42.5, 25.7, 10.4. MS (ESI) m/z 420 [M + H]+. PHPLC > 95%. HPLC (C4, 35 min): tR 12.7 min, PHPLC 98%; HPLC (C18, 35 min): tR 16.6 min, PHPLC 95%.
4.1.65. 1-[3-[1-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-3-yl]phenyl]-N,N-dimethylmethanamine (64)
General procedure E: 80% yield. The compound was converted to its 2HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.05 (s, 1H), 7.76–7.72 (m, 2H), 7.70–7.66 (m, 2H), 7.45–7.39 (m, 2H), 7.33–7.30 (m, 1H), 7.23–7.21 (m, 1H), 3.53 (s, 2H), 3.51 (s, 2H), 2.29–2.25 (m, 15H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 141.4, 140.7, 139.0, 135.0, 130.5, 130.1, 129.9, 129.6, 129.2, 128.2, 127.9, 120.7, 118.8, 117.6, 64.9, 64.7, 45.3, 45.2, 10.3. MS (ESI) m/z 349 [M + H]+. PHPLC > 97%. HPLC (C4, 35 min): tR 9.1 min, PHPLC 98%; HPLC (C18, 35 min): tR 13.7 min, PHPLC 97%.
4.1.66. N,N-dimethyl-1-[3-[4-methyl-1-[3-[(4-methylpiperazin-1-yl)methyl]phenyl]pyrazol-3-yl]phenyl]methanamine (65)
General procedure E: 27% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.08–8.07 (m, 1H), 7.79–7.73 (m, 2H), 7.70–7.66 (m, 2H), 7.47–7.40 (m, 2H), 7.36–7.32 (m, 1H), 7.27–7.25 (m, 1H), 3.60 (s, 2H), 3.58 (s, 2H), 2.52 (br, 8H), 2.31–2.28 (m, 12H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 141.5, 140.5, 138.8, 135.1, 130.5, 130.1, 130.0, 129.6, 129.3, 128.3, 128.1, 120.7, 118.7, 117.6, 64.8, 63.5, 55.7, 53.5, 45.9, 45.2, 10.2. MS (ESI) m/z 404 [M + H]+. PHPLC > 98%. HPLC (C4, 35 min): tR 12.1 min, PHPLC 98%; HPLC (C18, 35 min): tR 18.3 min, PHPLC 98%.
4.1.67. N′-[[3-[3-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-1-yl]phenyl] methyl]-N,N,N’-trimethyl-propane-1,3-diamine (66)
General procedure E: 78% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (salt, CD3OD, 300 Mz): δ 8.28–8.26 (m, 2H), 8.09 (s, 1H), 7.97–7.91 (m, 2H), 7.65–7.51 (m, 4H), 4.61–4.45 (m, 4H), 3.40–3.26 (m, 4H), 2.94 (s, 6H), 2.92 (s, 6H), 2.91 (s, 3H), 2.37 (br, 5H). 13C NMR (salt, CD3OD, 75 MHz): δ 152.1, 141.9, 136.1, 132.3, 131.7, 131.6, 131.3, 131.1, 130.6, 130.2, 129.8, 129.6, 122.2, 120.9, 118.3, 62.1, 60.9, 55.6, 53.9, 43.6, 43.1, 40.2, 21.1, 10.4. MS (ESI) m/z 420 [M + H]+. PHPLC > 99%. HPLC (C4, 35 min): tR 12.8 min, PHPLC 100%; HPLC (C18, 35 min): tR 17.9 min, PHPLC 99%.
4.1.68. {[4-(1-{3-[(Dimethylamino)methyl]phenyl}-4-methyl-1H-pyrazol-3-yl)phenyl] methyl} [3-(Dimethylamino)propyl]methylamine (67)
General procedure E: 63% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.05–8.05 (m, 1H), 7.75–7.67 (m, 4H), 7.44–7.39 (m, 3H), 7.23–7.21 (m, 1H), 3.54 (s, 2H), 3.51 (s, 2H), 2.41 (t, J = 7.4 Hz, 2H), 2.36–2.31 (m, 2H), 2.29–2.22 (m, 18H), 1.77–1.67 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.7, 141.4, 140.7, 138.9, 133.9, 130.6, 130.5, 129.2, 128.6, 128.2, 120.6, 118.7, 117.5, 64.7, 62.9, 58.6, 56.2, 45.4, 45.3, 42.4, 25.7, 10.4. MS (ESI) m/z 420 [M + H]+. PHPLC > 98%. HPLC (C4, 30 min): tR 12.6 min, PHPLC 98%; HPLC (C18, 30 min): tR 16.6 min, PHPLC 100%.
4.1.69. [3-(Dimethylamino)propyl](methyl){[4-(4-methyl-1-{3-[(4-methylpiperazin-1-yl) methyl]phenyl}-1H-pyrazol-3-yl)phenyl]methyl}amine (68)
General procedure E: 49% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.06–8.06 (m, 1H), 7.78–7.77 (m, 1H), 7.74–7.71 (m, 2H), 7.69–7.65 (m, 1H), 7.45–7.39 (m, 3H), 7.26–7.24 (m, 1H), 3.59 (s, 2H), 3.57 (s, 2H), 2.51 (br, 8H), 2.46–2.41 (m, 2H), 2.40–2.34 (m, 2H), 2.30–2.24 (m, 15H), 1.79–1.69 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 141.4, 140.4, 138.9, 134.0, 130.7, 130.5, 129.3, 128.7, 128.3, 120.7, 118.7, 117.6, 63.5, 62.9, 58.6, 56.2, 55.7, 53.6, 46.0, 45.3, 42.4, 25.6, 10.3. MS (ESI) m/z 475 [M + H]+. PHPLC > 99%. HPLC (C4, 30 min): tR 11.7 min, PHPLC 100%; HPLC (C18, 30 min): tR 16.2 min, PHPLC 99%.
4.1.70. [3-(Dimethylamino)propyl][(4-{1-[3-({[3-(dimethylamino)propyl](methyl) amino}methyl)phenyl]-4-methyl-1H-pyrazol-3-yl}phenyl)methyl]methylamine (69)
General procedure E: 59% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.06–8.06 (m, 1H), 7.77–7.76 (m, 1H), 7.73 (d, J = 8.2 Hz, 2H), 7.68–7.64 (m, 1H), 7.44–7.38 (m, 3H), 7.25–7.22 (m, 1H), 3.57 (s, 2H), 3.55 (s, 2H), 2.42 (t, J = 7.4 Hz, 4H), 2.37–2.29 (m, 7H), 2.24–2.22 (m, 18H), 1.78–1.67 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.7, 141.4, 141.4, 139.0, 134.0, 130.6, 130.4, 129.2, 128.6, 128.1, 120.5, 118.5, 117.5, 62.9, 62.9, 58.6 (2C), 56.2 (2C), 45.4 (2C), 42.5, 42.4, 25.7, 25.7, 10.4. MS (ESI) m/z 491 [M + H]+. PHPLC > 98%. HPLC (C4, 30 min): tR 11.9min, PHPLC 98%; HPLC (C18, 30 min): tR 15.9 min, PHPLC 98%.
4.1.71. N′-[[3-[1-[3-(Dimethylaminomethyl)phenyl]-4-methyl-pyrazol-3-yl]phenyl] Methyl]-N,N,N′-trimethyl-propane-1,3-diamine (70)
General procedure E: 84% yield. The compound was converted to its 3HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.05 (s, 1H), 7.76–7.73 (m, 2H), 7.71–7.64 (m, 2H), 7.44–7.39 (m, 2H), 7.40 (ddd, J = 7.7, 1.3, 1.3 Hz, 1H), 7.23–7.21 (m, 1H), 3.57 (s, 2H), 3.51 (s, 2H), 2.46–2.35 (m, 4H), 2.29–2.24 (m, 18H), 1.78–1.68 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 141.4, 140.7, 139.7, 135.0, 130.5, 129.9, 129.7, 129.5, 129.2, 128.2, 127.7, 120.6, 118.7, 117.6, 64.7, 63.1, 58.6, 56.2, 45.3 (2C), 42.4, 25.5, 10.4. MS (ESI) m/z 420 [M + H]+. PHPLC > 96%. HPLC (C4, 35 min): tR 7.9 min, PHPLC 99%; HPLC (C18, 35 min): tR 12.7 min, PHPLC 96%.
4.1.72. N,N,N′-Trimethyl-N′-[[3-[4-methyl-1-[3-[(4-methylpiperazin-1-yl)methyl] phenyl]pyrazol-3-yl]phenyl]methyl]propane-1,3-diamine (71)
General procedure E: 89% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.05 (s, 1H), 7.78–7.73 (m, 2H), 7.69–7.64 (m, 2H), 7.44–7.38 (m, 2H), 7.33–7.31 (m, 1H), 7.25–7.22 (m, 1H), 3.57 (s, 2H), 3.56 (s, 2H), 2.49 (br, 8H), 2.45–2.33 (m, 4H), 2.29–2.23 (m, 15H), 1.77–1.67 (m, 2H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.8, 141.4, 140.5, 139.7, 135.0, 130.4, 129.9, 129.7, 129.5, 129.2, 128.2, 127.7, 120.6, 118.7, 117.6, 63.5, 63.1, 58.6, 56.2, 55.7, 53.6, 46.0, 45.4, 42.5, 25.6, 10.4. MS (ESI) m/z 475 [M + H]+. PHPLC > 99%. HPLC (C4, 35 min): tR 4.5 min, PHPLC 100%; HPLC (C18, 35 min): tR 12.5 min, PHPLC 99%.
4.1.73. N′-[[3-[1-[3-[[3-(Dimethylamino)propyl-methyl-amino]methyl]phenyl]-4-methyl-pyrazol-3-yl]phenyl]methyl]-N,N,N′-trimethyl-propane-1,3-diamine (72)
General procedure E: 66% yield. The compound was converted to its 4HCl salt (white solid). 1H NMR (free amine, CD3OD, 300 Mz): δ 8.07 (s, 1H), 7.78–7.73 (m, 2H), 7.69–7.65 (m, 2H), 7.42 (dd, J = 7.7, 7.7 Hz, 2H), 7.35–7.32 (m, 1H), 7.26–7.24 (m, 1H), 3.59 (s, 2H), 3.58 (s, 2H), 2.47–2.41 (m, 4H), 2.39–2.34 (m, 4H), 2.3–2.24 (m, 21H), 1.79–1.69 (m, 4H). 13C NMR (free amine, CD3OD, 75 MHz): δ 152.9, 141.4, 139.7, 135.0, 130.4, 129.9, 129.8, 129.5, 129.2, 128.1, 127.7, 120.6, 118.6, 117.6, 63.1, 62.9, 58.6 (2C), 56.3, 56.2, 45.4 (2C), 42.5 (2C), 25.7 (2C), 10.4. MS (ESI) m/z 491 [M + H]+. PHPLC > 98%. HPLC (C4, 35 min): tR 7.7 min, PHPLC 99%; HPLC (C18, 35 min): tR 12.1 min, PHPLC 98%.



 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




