
The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy


Abstract
1. Introduction
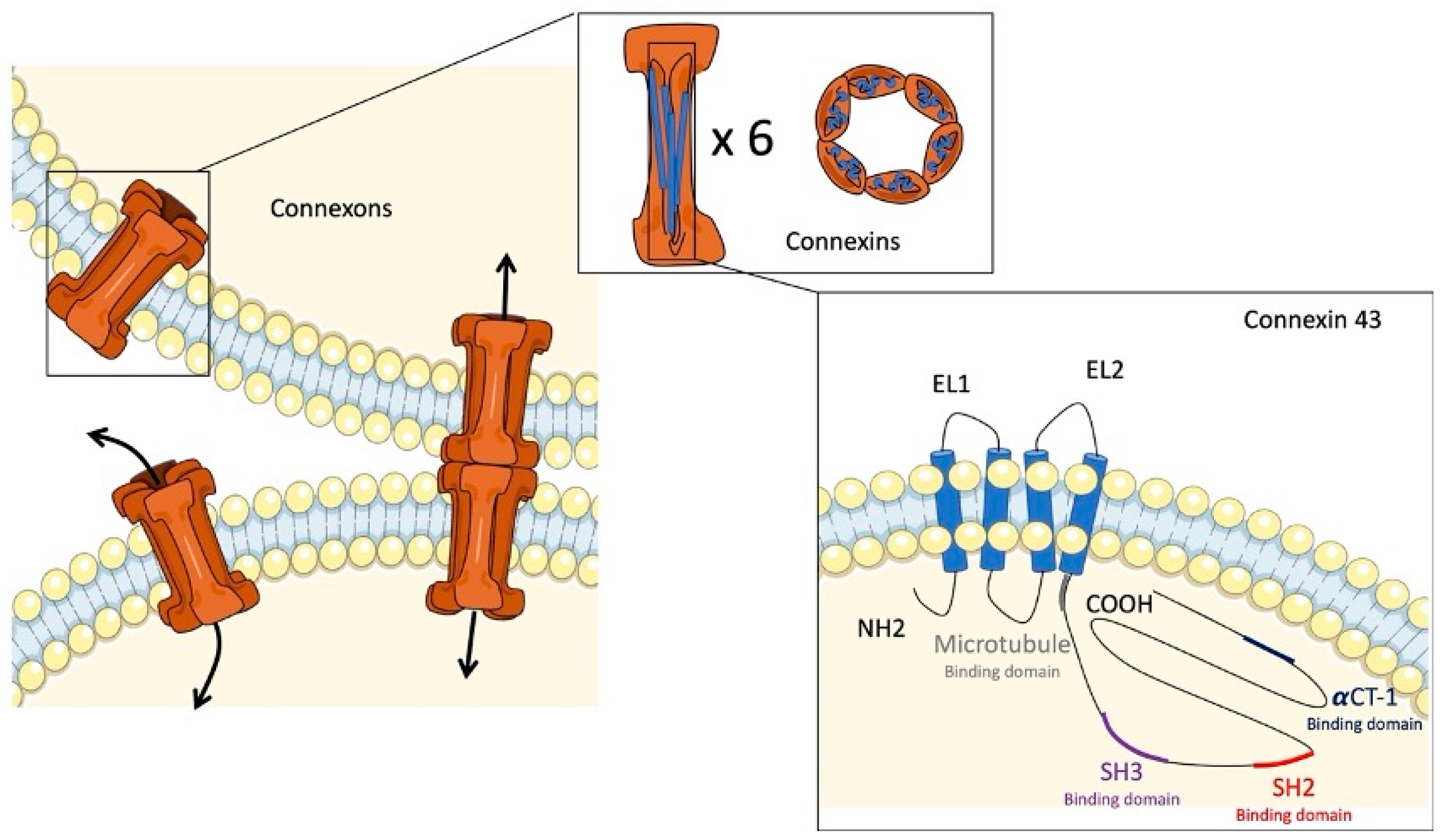
2. Gap Junctions and Connexins
3. Connexin 43
4. Connexin 43 in the Kidney
4.1. Connexin 43 in Experimental AKI
4.1.1. Uranyl Acetate
4.1.2. Lipopolysaccharide
4.1.3. Cisplatin
4.2. Connexin 43 in Experimental CKD
4.2.1. Cx43 and Glomerular Damage
Induction of Glomerulonephritis by Injection of Nephrotoxic Serum
Induction of Glomerulonephritis by Anti-Thymocyte Serum (ATS) Injection
Puromycin Aminonucleoside
Phosphate Overload-Induced Glomerulonephritis
Podocyte Injury via Aldosterone Injection
High-Fat Diet
4.2.2. Cx43 in Obstructive Nephropathy: The Unilateral Ureteral Obstruction Model
4.2.3. Cx43 and Hypertensive Nephropathy
Stimulation of Renin Secretion
Renal Injury via Angiotensin II
4.2.4. Cx43 and Diabetic Nephropathy
5. Connexin 43 and Human Nephropathy
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bao, Y.-W.; Yuan, Y.; Chen, J.-H.; Lin, W.-Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool. Res. 2018, 39, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World Incidence of AKI: A Meta-Analysis. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef]
- Harty, J. Prevention and Management of Acute Kidney Injury. Ulst. Med. J. 2014, 83, 149–157. [Google Scholar]
- Levey, A.S.; Becker, C.; Inker, L.A. Glomerular Filtration Rate and Albuminuria for Detection and Staging of Acute and Chronic Kidney Disease in Adults: A systematic review. JAMA 2015, 313, 837–846. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.-H. Gap Junctions. Compr. Physiol. 2012, 2, a002576. [Google Scholar] [CrossRef]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Jiang, J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions--an update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.; Ferdinandy, P.; Beyer, E.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Nalewajska, M.; Marchelek-Myśliwiec, M.; Opara-Bajerowicz, M.; Dziedziejko, V.; Pawlik, A. Connexins—Therapeutic Targets in Cancers. Int. J. Mol. Sci. 2020, 21, 9119. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Meda, P.; Chanson, M.; Pepper, M.; Giordano, E.; Bosco, D.; Traub, O.; Willecke, K.; El Aoumari, A.; Gros, D.; Beyer, E.C.; et al. In vivo modulation of connexin 43 gene expression and junctional coupling of pancreatic B-cells. Exp. Cell Res. 1991, 192, 469–480. [Google Scholar] [CrossRef]
- Fushiki, S.; Velazquez, J.L.P.; Zhang, L.; Bechberger, J.F.; Carlen, P.; Naus, C.C.G. Changes in Neuronal Migration in Neocortex of Connexin43 Null Mutant Mice. J. Neuropathol. Exp. Neurol. 2003, 62, 304–314. [Google Scholar] [CrossRef]
- Hanner, F.; Sorensen, C.M.; Holstein-Rathlou, N.-H.; Peti-Peterdi, J. Connexins and the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1143–R1155. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A New Therapeutic Target Against Chronic Kidney Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I. Connexins and the heart. Trends Cardiovasc. Med. 1992, 2, 50–55. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 227–246. [Google Scholar] [CrossRef]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Burger, F.; Pelli, G.; Mach, F.; Kwak, B.R. Dual Benefit of Reduced Cx43 on Atherosclerosis in LDL Receptor-Deficient Mice. Cell Commun. Adhes. 2003, 10, 395–400. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef]
- Žužul, M.; Lozić, M.; Filipović, N.; Čanović, S.; Pavičić, A.D.; Petričević, J.; Kunac, N.; Šoljić, V.; Saraga-Babić, M.; Konjevoda, S.; et al. The Expression of Connexin 37, 40, 43, 45 and Pannexin 1 in the Early Human Retina and Choroid Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 5918. [Google Scholar] [CrossRef]
- Parthasarathi, K.; Ichimura, H.; Monma, E.; Lindert, J.; Quadri, S.; Issekutz, A.; Bhattacharya, J. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J. Clin. Investig. 2006, 116, 2193–2200. [Google Scholar] [CrossRef]
- Quan, H.H.; Xu, W.X.; Qi, Y.Z.; Li, Q.R.; Zhou, H.; Huang, J. Inhibition connexin 43 by mimetic peptide Gap27 mediates protective effects on 6-hydroxydopamine induced Parkinson’s disease mouse model. Beijing Da Xue Xue Bao 2022, 54, 421–426. [Google Scholar]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.-Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting Connexin43 Expression Accelerates the Rate of Wound Repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef]
- Bobbie, M.W.; Roy, S.; Trudeau, K.; Munger, S.J.; Simon, A.M.; Roy, S. Reduced Connexin 43 Expression and Its Effect on the Development of Vascular Lesions in Retinas of Diabetic Mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Dong, L.; MacDonald, A.I.; Akbari, S.; Edward, M.; Hodgins, M.B.; Johnstone, S.R.; Graham, S.V. HPV16 E6 Controls the Gap Junction Protein Cx43 in Cervical Tumour Cells. Viruses 2015, 7, 5243–5256. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.L.; Williams, C.B.; Zambrano, J.N.; Williams, C.J.; Yeh, E.S. Connexin 43 in the development and progression of breast cancer: What’s the connection? (Review). Int. J. Oncol. 2017, 51, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Barajas, L.; Liu, L.; Tucker, M. Localization of connexin43 in rat kidney. Kidney Int. 1994, 46, 621–626. [Google Scholar] [CrossRef]
- Arensbak, B.; Mikkelsen, H.B.; Gustafsson, F.; Christensen, T.; Holstein-Rathlou, N.-H. Expression of connexin 37, 40, and 43 mRNA and protein in renal preglomerular arterioles. Histochem. Cell Biol. 2001, 115, 479–487. [Google Scholar] [CrossRef]
- Guo, R.; Liu, L.; Barajas, L. RT-PCR study of the distribution of connexin 43 mRNA in the glomerulus and renal tubular segments. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1998, 275, R439–R447. [Google Scholar] [CrossRef]
- Hillis, G.S.; Duthie, L.A.; Mlynski, R.; McKay, N.G.; Mistry, S.; MacLeod, A.M.; Simpson, J.G.; Haites, N.E. The Expression of Connexin 43 in Human Kidney and Cultured Renal Cells. Nephron 1997, 75, 458–463. [Google Scholar] [CrossRef]
- Hillis, G.S.; Duthie, L.A.; Brown, P.A.; Simpson, J.G.; MacLeod, A.M.; Haites, N.E. Upregulation and co-localization of connexin43 and cellular adhesion molecules in inflammatory renal disease. J. Pathol. 1997, 182, 373–379. [Google Scholar] [CrossRef]
- Homma-Takeda, S.; Numako, C.; Kitahara, K.; Yoshida, T.; Oikawa, M.; Terada, Y.; Kokubo, T.; Shimada, Y. Phosphorus Localization and Its Involvement in the Formation of Concentrated Uranium in the Renal Proximal Tubules of Rats Exposed to Uranyl Acetate. Int. J. Mol. Sci. 2019, 20, 4677. [Google Scholar] [CrossRef]
- Fujigaki, Y.; Muranaka, Y.; Sun, D.; Goto, T.; Zhou, H.; Sakakima, M.; Fukasawa, H.; Yonemura, K.; Yamamoto, T.; Hishida, A. Transient myofibroblast differentiation of interstitial fibroblastic cells relevant to tubular dilatation in uranyl acetate-induced acute renal failure in rats. Virchows Arch. Int. J. Pathol. 2004, 446, 164–176. [Google Scholar] [CrossRef]
- Elmas, M.; Yazar, E.; Uney, K.; Karabacak, A.E. Influence of Escherichia coli Endotoxin-Induced Endotoxaemia on the Pharmacokinetics of Enrofloxacin after Intravenous Administration in Rabbits. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2006, 53, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.; Toelboell, T.; Andersen, P.H. Dose dependency and individual variability in selected clinical, haematological and blood biochemical responses after systemic lipopolysaccharide challenge in cattle. Vet. Res. 2005, 36, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.-L.; Zhong, X.; Wu, W.-J.; Chen, J.; Ni, H.-F.; Tang, T.-T.; et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2019, 27, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cobo, M. Expression of the connexin 43 gene is increased int he kidneys and the lungs of rats injectied with bacterial lipopolysaccharide. Shock 1998, 10, 97–102. [Google Scholar] [CrossRef]
- Su, Z.; Yu, P.; Sheng, L.; Ye, J.; Qin, Z. Fangjifuling Ameliorates Lipopolysaccharide-Induced Renal Injury via Inhibition of Inflammatory and Apoptotic Response in Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 2124–2137. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, Z.; Zhang, Z.; Obata, F.; Yang, X.; Zhang, X.; Huang, Y.; Mitsui, T.; Fan, J.; Takeda, M.; et al. Connexin43 Contributes to Inflammasome Activation and Lipopolysaccharide-Initiated Acute Renal Injury via Modulation of Intracellular Oxidative Status. Antioxid. Redox Signal. 2019, 31, 1194–1212. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Hyland, C.; Falk, M.M.; Iovine, M.K. Connexin 43 gap junctional intercellular communication inhibits evx1 expression and joint formation in regenerating fins. Development 2020, 147, dev190512. [Google Scholar] [CrossRef]
- Yu, M.; Lin, Z.; Tian, X.; Chen, S.; Liang, X.; Qin, M.; Zhu, Q.; Wu, Y.; Zhong, S. Downregulation of Cx43 reduces cisplatin-induced acute renal injury by inhibiting ferroptosis. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 158, 112672. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chadban, S.; Atkins, R. Glomerulonephritis. Lancet 2005, 365, 1797–1806. [Google Scholar] [CrossRef]
- Thorner, P.S.; Ho, M.; Eremina, V.; Sado, Y.; Quaggin, S. Podocytes Contribute to the Formation of Glomerular Crescents. J. Am. Soc. Nephrol. JASN 2008, 19, 495–502. [Google Scholar] [CrossRef]
- Salant, D.J.; Cybulsky, A.V. Experimental glomerulonephritis. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1988; Volume 162, pp. 421–461. [Google Scholar] [CrossRef]
- Toubas, J.; Beck, S.; Pageaud, A.-L.; Huby, A.-C.; Mael-Ainin, M.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Alteration of connexin expression is an early signal for chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F24–F32. [Google Scholar] [CrossRef] [PubMed]
- Kavvadas, P.; Abed, A.; Poulain, C.; Authier, F.; Labéjof, L.-P.; Calmont, A.; Afieri, C.; Prakoura, N.; Dussaule, J.-C.; Chatziantoniou, C.; et al. Decreased Expression of Connexin 43 Blunts the Progression of Experimental GN. J. Am. Soc. Nephrol. JASN 2017, 28, 2915–2930. [Google Scholar] [CrossRef]
- Morioka, T.; Okada, S.; Nameta, M.; Kamal, F.; Yanakieva-Georgieva, N.T.; Yao, J.; Sato, A.; Piao, H.; Oite, T. Glomerular expression of connexin 40 and connexin 43 in rat experimental glomerulonephritis. Clin. Exp. Nephrol. 2012, 17, 191–204. [Google Scholar] [CrossRef]
- Diamond, J.R.; Karnovsky, M.J. Focal and segmental glomerulosclerosis following a single intravenous dose of puromycin aminonucleoside. Am. J. Pathol. 1986, 122, 481–487. [Google Scholar] [PubMed]
- Zhou, Y.; Kim, C.; Pablo, J.L.B.; Zhang, F.; Jung, J.Y.; Xiao, L.; Bazua-Valenti, S.; Emani, M.; Hopkins, C.R.; Weins, A.; et al. TRPC5 Channel Inhibition Protects Podocytes in Puromycin-Aminonucleoside Induced Nephrosis Models. Front. Med. 2021, 8, 721865. [Google Scholar] [CrossRef]
- Yaoita, E.; Yao, J.; Yoshida, Y.; Morioka, T.; Nameta, M.; Takata, T.; Kamiie, J.-I.; Fujinaka, H.; Oite, T.; Yamamoto, T. Up-Regulation of Connexin43 in Glomerular Podocytes in Response to Injury. Am. J. Pathol. 2002, 161, 1597–1606. [Google Scholar] [CrossRef]
- Sekiguchi, S.; Suzuki, A.; Asano, S.; Nishiwaki-Yasuda, K.; Shibata, M.; Nagao, S.; Yamamoto, N.; Matsuyama, M.; Sato, Y.; Yan, K.; et al. Phosphate overload induces podocyte injury via type III Na-dependent phosphate transporter. Am. J. Physiol. Ren. Physiol. 2011, 300, F848–F856. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, B.; Li, M.; Jiang, B. Connexin 43 is Involved in Aldosterone-Induced Podocyte Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 34, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, G.; Wang, Y.; Liu, Z. Alteration of Connexin43 expression in a rat model of obesity-related glomerulopathy. Exp. Mol. Pathol. 2018, 104, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.; Eismann, U.; Deuther-Conrad, W.; Wendt, T.; Mohorn, T.; Fünfstück, R.; Stein, G. Time Course of Cytokine mRNA Expression in Kidneys of Rats with Unilateral Ureteral Obstruction. Nephron 2000, 84, 49–57. [Google Scholar] [CrossRef]
- Abed, A.; Toubas, J.; Kavvadas, P.; Authier, F.; Cathelin, D.; Alfieri, C.; Boffa, J.-J.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Targeting connexin 43 protects against the progression of experimental chronic kidney disease in mice. Kidney Int. 2014, 86, 768–779. [Google Scholar] [CrossRef]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2017, 10, a029348. [Google Scholar] [CrossRef]
- Xu, H.; Wang, M.; Li, Y.; Shi, M.; Wang, Z.; Cao, C.; Hong, Y.; Bin Hu, B.; Zhu, H.; Zhao, Z.; et al. Blocking connexin 43 and its promotion of ATP release from renal tubular epithelial cells ameliorates renal fibrosis. Cell Death Dis. 2022, 13, 511. [Google Scholar] [CrossRef]
- Haefliger, J.-A.; Demotz, S.; Braissant, O.; Suter, E.; Waeber, B.; Nicod, P.; Meda, P. Connexins 40 and 43 are differentially regulated within the kidneys of rats with renovascular hypertension. Kidney Int. 2001, 60, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pinto, A.B.; de Matos, V.S.; Rocha, V.; Moraes-Teixeira, J.; Carvalho, J.J. Low-Intensity physical activity beneficially alters the ultrastructural renal morphology of spontaneously hypertensive rats. Clinics 2011, 66, 855–863. [Google Scholar] [CrossRef]
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. Int. J. Mol. Sci. 2019, 20, 4408. [Google Scholar] [CrossRef]
- Satriano, J.; Mansoury, H.; Deng, A.; Sharma, K.; Vallon, V.; Blantz, R.C.; Thomson, S.C. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am. J. Physiol. Cell Physiol. 2010, 299, C374–C380. [Google Scholar] [CrossRef]
- Hu, C.; Cong, X.D.; Dai, D.-Z.; Zhang, Y.; Zhang, G.L.; Dai, Y. Argirein alleviates diabetic nephropathy through attenuating NADPH oxidase, Cx43, and PERK in renal tissue. Naunyn-Schmiedebergs Arch. Pharmakol. 2011, 383, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, Y.; Ma, Z.; Li, Y.; Guo, J.; Meng, Q.; Bian, H. A novel formula from mulberry leaf ameliorates diabetic nephropathy in rats via inhibiting the TGF-β1 pathway. Food Funct. 2015, 6, 3307–3315. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-β1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2016, 102, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Huang, K.; Haiming, X.; Lin, Z.; Yang, Y.; Zhang, M.; Liu, P.; Huang, H. Connexin 43 prevents the progression of diabetic renal tubulointerstitial fibrosis by regulating the SIRT1-HIF-1α signaling pathway. Clin. Sci. 2020, 134, 1573–1592. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine Prevents Renal Alterations in Diabetic Rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef]
- Sawai, K.; Mukoyama, M.; Mori, K.; Yokoi, H.; Koshikawa, M.; Yoshioka, T.; Takeda, R.; Sugawara, A.; Kuwahara, T.; Saleem, M.A.; et al. Redistribution of connexin43 expression in glomerular podocytes predicts poor renal prognosis in patients with type 2 diabetes and overt nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.–Eur. Ren. Assoc. 2006, 21, 2472–2477. [Google Scholar] [CrossRef]
- Guo, Y.-N.; Wang, J.-C.; Cai, G.-Y.; Hu, X.; Cui, S.-Y.; Lv, Y.; Yin, Z.; Fu, B.; Hong, Q.; Chen, X.-M. AMPK-mediated downregulation of connexin43 and premature senescence of mesangial cells under high-glucose conditions. Exp. Gerontol. 2014, 51, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, L.; Madsen, K.; Kurt, B.; Jensen, B.L.; Walter, S.; Banas, B.; Wagner, C.; Kurtz, A. High-Level Connexin Expression in the Human Juxtaglomerular Apparatus. Nephron Physiol. 2010, 116, p1–p8. [Google Scholar] [CrossRef]
- Potter, J.; Price, G.; Cliff, C.; Green, C.; Squires, P.; Hills, C. Collagen I Modifies Connexin-43 Hemichannel Activity via Integrin α2β1 Binding in TGFβ1-Evoked Renal Tubular Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 3644. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Renal Compartment | Models | Effects | |
|---|---|---|---|
| AKI | Proximal Tubules | Uranyl Acetate |  Cx43 expression Cx43 expression |
| Tubules | Lipopolysaccharide |  Cx43 expression Cx43 expression  Tubular damage, inflammation, BUN and proteinuria Tubular damage, inflammation, BUN and proteinuria  NLRP3 NLRP3  ROS ROS | |
| Cisplatin |  Cx43 expression Cx43 expression  Tubular injury Tubular injury | ||
| CKD | Glomerular Compartment | Nephrotoxic Serum (NTS) |  Cx43 expression Cx43 expression  Proteinuria and glomerular damage (crescents) Proteinuria and glomerular damage (crescents) Renal fibrosis Renal fibrosis  Proinflammatory and/or profibrotic cytokines Proinflammatory and/or profibrotic cytokines |
| Anti-thymocyte Serum (ATS) | No change in Cx 43 expression | ||
| Puromycin aminonucleoside |  Cx43 expression Cx43 expression Podocytes injury Podocytes injury | ||
| Phosphate Overload |  Severe proteinuria Severe proteinuria  Cx43 expression in podocytes Cx43 expression in podocytes | ||
| Aldosterone injection |  Cx43 expression in podocytes Cx43 expression in podocytes ROS production and Bax/Bcl-2 ratio ROS production and Bax/Bcl-2 ratio | ||
| High Fat Diet |  Cx43 expression Cx43 expression  Inflammation Inflammation | ||
| Tubulo-interstial Compartment | Unilateral Ureteral Obstruction (UUO) |  Cx43 expression Cx43 expression Renal fibrosis and inflammation (NLRP3) Renal fibrosis and inflammation (NLRP3) ATP release ATP release N-cadherin and β-catenin N-cadherin and β-catenin E-cadherin and ZO-1 E-cadherin and ZO-1 | |
| Vascular Compartment | Stimulation of renin secretion |  Cx43 expression Cx43 expression Structural damage and chronic inflammation Structural damage and chronic inflammation | |
| Stimulation of Angiotensin II |  Cx43 expression Cx43 expression Proinflammatory cytokines Proinflammatory cytokines RhoA/ROCK pathway RhoA/ROCK pathway | ||
| Diabetic Mice |  Glucose levels Glucose levels Cx43 expression Cx43 expression |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roger, E.; Boutin, L.; Chadjichristos, C.E. The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy. Int. J. Mol. Sci. 2022, 23, 13090. https://doi.org/10.3390/ijms232113090
Roger E, Boutin L, Chadjichristos CE. The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy. International Journal of Molecular Sciences. 2022; 23(21):13090. https://doi.org/10.3390/ijms232113090
Chicago/Turabian StyleRoger, Elena, Louis Boutin, and Christos E. Chadjichristos. 2022. "The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy" International Journal of Molecular Sciences 23, no. 21: 13090. https://doi.org/10.3390/ijms232113090
APA StyleRoger, E., Boutin, L., & Chadjichristos, C. E. (2022). The Role of Connexin 43 in Renal Disease: Insights from In Vivo Models of Experimental Nephropathy. International Journal of Molecular Sciences, 23(21), 13090. https://doi.org/10.3390/ijms232113090