
Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes


 ,
,  and
and
Abstract
:1. Introduction
2. Targeting Inflammation in Microvascular Complications of Diabetes
3. Cx43 Hemichannel Blockers and Treatment of Inflammation in Diabetes and Its Secondary Complications
4. Cx43 Hemichannels and Treatment of Inflammation in Diabetic Kidney Disease
5. The Therapeutic Potential of Blocking Cx43 in Diabetic Retinopathy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- International Diabetes Federation, Ninth Edition. 2019. Available online: https://www.diabetesatlas.org/upload/resources/material/20200302_133351_IDFATLAS9e-final-web.pdf (accessed on 23 November 2021).
- Whicher, C.A.; O’Neill, S.; Holt, R.I.G. Diabetes in the UK: 2019. Diabet. Med. 2020, 37, 242–247. [Google Scholar] [CrossRef]
- Einarson, T.R.; Acs, A.; Ludwig, C.; Panton, U.H. Prevalence of cardiovascular disease in type 2 diabetes: A systematic literature review of scientific evidence from across the world in 2007–2017. Cardiovac. Diabetol. 2018, 17, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Soyoye, D.O.; Ablodun, O.O.; Ikem, R.T.; Kolawole, B.A.; Akintomide, A.O. Diabetes and peripheral artery disease: A review. World J. Diabetes 2021, 12, 827–838. [Google Scholar] [CrossRef]
- Huang, D.; Refaat, M.; Mohammedi, K.; Jayyousi, A.; Suwaidi, J.A.; Khalil, C.A. Biomed. Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Chen, Y.; Lee, K.; Ni, Z.; He, J.C. Diabetic kidney disease: Challenges, advances, and opportunities. Kidney Dis. 2020, 6, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Keshari, K.R.; Wilson, D.M.; Sai, V.; Bok, R.; Jen, K.; Larson, P.; van Criekinge, M.; Kurhanewicz, J.; Wang, Z.J. Noninvasive in vivo imaging of diabetes-induced renal oxidative stress and response to therapy using hyperpolarized 13C dehydroascorbate magnetic resonance. Am. Diabetes Assoc. 2015, 64, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoja, C.; Xinaris, C.; Macconi, D. Diabetic nephropathy: Novel molecular mechanisms and therapeutic targets. Front. Pharmacol. 2020, 11, 2139. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A.C.Y. Diabetic retinopathy: Pathophysiology and treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef] [Green Version]
- Sabanayagam, C.; Banu, R.; Chee, M.L.; Lee, R.; Wang, Y.X.; Tan, G.T.; Jonas, J.B.; Lamourex, E.L.; Cheng, C.Y.; Klein, B.E.K.; et al. Incidence and progression of diabetic retinopathy: A systematic review. Lancet 2019, 7, 140–149. [Google Scholar] [CrossRef]
- Lan, C.C.E.; Wu, C.S.; Huang, S.M.; Wu, I.H.; Chen, G.S. High-glucose environment enhanced oxidative stress and interleukin-8 secretion from keratinocytes: New insights into impaired diabetic wound healing. Am. Diabetes Assoc. 2013, 62, 2530–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spampinato, S.F.; Caruso, G.I.; de Pasquale, R.; Sortino, M.A.; Merlo, S. The treatment of impaired wound healing in diabetes: Looking among old drugs. Pharmaceuticals 2020, 13, 60. [Google Scholar] [CrossRef] [Green Version]
- MacIsaac, R.J.; Jerums, G.; Ekinci, E.I. Effects of glycaemic management on diabetic kidney disease. World J. Diabetes 2017, 8, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Hill-Briggs, F.; Adler, N.E.; Berkowitz, S.A.; Chin, M.H.; Gary-Webb, T.L.; Navas-Acien, A.; Thornton, P.L.; Haire-Joshu, D. Social determinants of health and diabetes: A scientific review. Diabetes Care 2021, 44, 258–279. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Nam, J.H. Insight into the relationship between obesity-induced low-level chronic inflammation and COVID-19 infection. Int. J. Obes. 2020, 44, 1541–1542. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Ramirez, B.; Rotellar, F.; Pastor, C.; Silva, C.; Rodríguez, A.; Gil, M.J.; Cienfuegos, F.A.; Frühbeck, G. Proinflammatory cytokines in obesity: Impact of type 2 diabetes mellitus and gastric bypass. Obes. Surg. 2007, 17, 1464–1474. [Google Scholar] [CrossRef]
- Muniz, M.G.R.; Palfreeman, M.; Setzu, N.; Sanchez, M.A.; Portillo, P.S.; Garza, K.M.; Gosselink, K.L.; Spencer, C.T. Obesity exacerbates the cytokine storm elicited by Francisella tularensis infection of females and is associated with increased mortality. Adv. Emerg. Negl. Infect. Dis. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Edwards, M.S.; Wilson, D.B.; Craven, T.E.; Stafford, J.; Fried, L.F.; Wong, T.Y.; Klein, R.; Burke, G.L.; Hansen, K.J. Associations between retinal microvascular abnormalities and declining renal function in the elderly population: The cardiovascular health study. Am. J. Kidney Dis. 2005, 2, 214–224. [Google Scholar] [CrossRef]
- Jeng, C.J.; Hsieh, Y.T.; Yang, C.M.; Yang, C.H.; Lin, C.L.; Wang, I.J. Diabetic retinopathy in patients with diabetic nephropathy: Development and progression. PLoS ONE 2016, 11, e0161897. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.H.; Shin, J.A.; Han, J.H.; Park, Y.M.; Yim, H.W. The association between chronic kidney disease and diabetic retinopathy: The Korea national health and nutrition examination survey 2008–2010. PLoS ONE 2015, 10, e125338. [Google Scholar] [CrossRef]
- Pedro, R.A.; Ramon, S.A.; Marc, B.B.; Juan, F.B.; Isabel, M.M. Prevalence and relationship between diabetic retinopathy and nephropathy, and its risk factors in the north-east of Spain, a population-based study. Ophthalmic Epidemiol. 2010, 17, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Li, L.; Zhang, R.; Guo, R.; Li, H.; Han, Q.; Teng, G.; Liu, F. Diabetic retinopathy may predict the renal outcomes of patients with diabetic nephropathy. Ren. Fail. 2018, 40, 243–251. [Google Scholar] [CrossRef]
- Sasaki, T.; Numano, R.; Yokota-Hasimoto, H.; Matsui, S.; Kimura, N.; Takeuchi, H.; Kitamura, T. A central-acting connexin inhibitor, INI-0602, prevents high-fat diet-induced feeding pattern disturbances and obesity in mice. Mol. Brain 2018, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Codero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. Geroscience 2020, 42, 715–725. [Google Scholar] [CrossRef]
- Yi, C.; Ezan, P.; Fernández, P.; Schmitt, J.; Sáez, J.C.; Giaume, C.; Koulakoff, A. Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 2017, 65, 1607–1625. [Google Scholar] [CrossRef] [PubMed]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Carpintero-Fernández, P.; Sánchez, T.A.; Varela, E.M.; García-Yuste, A.; Yáñez, C.J.; Díez-Ulloa, A.; Caeiro, J.R.; Mayan, M.D. Connexin 43 and cellular senescence: New therapeutic strategies for treating osteoarthritis. Rev. Osteoporos. Metab. Miner. 2020, 12, 152–154. [Google Scholar] [CrossRef]
- Wagner, C. Function of connexins in the renal circulation. Kidney Int. 2008, 73, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Fiori, M.C.; Reuss, L.; Cuello, L.G.; Altenberg, G.A. Functional analysis and regulation of purified connexin hemichannels. Front. Physiol. 2014, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Beyer, E.C.; Berthoud, V.M. Gap junction structure: Unraveled, but not fully revealed. F1000Research 2017, 6, 568. [Google Scholar] [CrossRef] [Green Version]
- Evans, W.H.; Ahmad, S.; Diez, J.; George, C.H.; Kendall, J.M.; Martin, P.E. Trafficking pathways leading to the formation of gap junctions. Novartis Found. Symp. 1999, 219, 244–254. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Dong, A.; Liu, S.; Li, Y. Gap junctions in the nervous system: Probing functional connections using new imaging approaches. Front. Cell. Neurosci. 2018, 12, 320. [Google Scholar] [CrossRef]
- Retamal, M.A.; Reyes, E.P.; García, I.E.; Pinto, B.; Martínez, A.D.; González, C. Diseases associated with leaky hemichannels. Front. Cell. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girão, H. Role of connexin 43 in different forms of intercellular communication—Gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Bennett, M.V.L.; Sáez, J.C. Opening of connexin 43 hemichannels is increased by lowering intracellular redox potential. Proc. Natl. Acad. Sci. USA 2007, 104, 8322–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willebrords, J.; Yanguas, S.C.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; Green, C.R.; Cogliati, B.; Vinken, M. Connexins and their channels in inflammation. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Verselis, V.K. Connexin hemichannels and cochlear function. Neurosci. Lett. 2019, 695, 40–45. [Google Scholar] [CrossRef]
- Ma, D.; Feng, L.; Cheng, Y.; Xin, M.; You, J.; Yin, X.; Hao, Y.; Cui, L.; Feng, J. Astrocytic gap junction inhibition by carbenoxolone enhances the protective effects of ischemic preconditioning following cerebral ischemia. J. Neuroinflamm. 2018, 15, 1–12. [Google Scholar] [CrossRef]
- Yang, H.; Yan, H.; Li, X.; Liu, J.; Cao, S.; Huang, B.; Huang, D.; Wu, L. Inhibition of Connexin 43 and phosphorylated NR2B in spinal astrocytes attenuates bone cancer pain in mice. Front. Cell. Neurosci. 2018, 12, 129. [Google Scholar] [CrossRef]
- Wang, H.; Sun, X. Carbon monoxide-releasing molecule-2 inhibits Connexin 43-hemichannel activity in spinal cord astrocytes to attenuate neuropathic pain. J. Mol. Neurosci. 2017, 63, 58–69. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, S.; Sun, G.; Zhang, R.; Li, X.; Zhang, Y.; Huang, F.; Yuan, D. Hyperglycemia aggravates monocyte-endothelial adhesion in human umbilical vein endothelial cells from women with gestational diabetes mellitus by inducing Cx43 overexpression. Ann. Transl. Med. 2021, 9, 234. [Google Scholar] [CrossRef] [PubMed]
- Tien, T.; Barrette, K.F.; Chronopoulos, A.; Roy, S. Effects of high glucose-induced Cx43 downregulation on occluding and ZO-1 expression and tight junction barrier function in retinal endothelial cells. Invest. Ophthalmol. Vis. Sci. 2013, 54, 6518–6525. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lewis, C.; Sarthy, V.; Roy, S. High-glucose-induced rab20 upregulation disrupts gap junction intercellular communication and promotes apoptosis in retinal endothelial and müller cells: Implications for diabetic retinopathy. J. Clin. Med. 2020, 9, 3710. [Google Scholar] [CrossRef] [PubMed]
- Sáez, J.; Contreras-Duarte, S.; Gómez, G.; Labra, V.C.; Santibañez, C.A.; Gajardo-Gómez, R.; Avendaño, B.C.; Díaz, E.F.; Montero, T.D.; Velarde, V. Connexin 43 hemichannel activity promoted by pro-inflammatory cytokines and high glucose alters endothelial cell function. Front. Immunol. 2018, 9, 1899. [Google Scholar] [CrossRef] [Green Version]
- González-Casanova, J.; Schmachtenberg, O.; Martínez, A.D.; Sanchez, H.A.; Harcha, P.A.; Rojas-Gomez, D. An update on connexin gap junction and hemichannels in diabetic retinopathy. Int. J. Mol. Sci. 2021, 22, 3194. [Google Scholar] [CrossRef]
- Sáez, J.C.; Contreras-Duarte, S.; Labra, V.C.; Santibañez, C.A.; Mellado, L.A.; Inostroza, C.A.; Alvear, T.F.; Retamal, M.A.; Velarde, V.; Orellana, J.A. Interferon-γ and high glucose-induced opening of Cx43 hemichannels causes endothelial cell dysfunction and damage. BBA Mol. Cell Res. 2020, 1867, 118720. [Google Scholar] [CrossRef]
- Roy, S.; Jiang, J.X.; Li, A.F.; Kim, D. Connexin channel and its role in diabetic retinopathy. Prog. Retin. Eye Res. 2017, 61, 35. [Google Scholar] [CrossRef]
- Lyon, H.; Shome, A.; Rupenthal, I.; Green, C.R.; Mugisho, O.O. Tonabersat inhibits connexin43 hemichannel opening and inflammasome activation in an in vitro retinal epithelial cell model of diabetic retinopathy. Int. J. Mol. Sci. 2020, 22, 298. [Google Scholar] [CrossRef]
- Thakur, V.; Alcoreza, N.; Cazares, J.; Chattopadhyay, M. Changes in stress-mediated markers in a human cardiomyocyte cell line under hyperglycemia. Int. J. Mol. Sci. 2021, 22, 802. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S. Podocytes and microRNA-30/Cx43 axis in diabetic nephropathy. Ann. Transl. Med. 2021, 9, 828. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.; Chodock, R.; Hand, A.R.; Laird, D.W. The origin of annular junctions: A mechanism of gap junction internalization. J. Cell Sci. 2001, 114, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.; Donath, M.; LeRoith, D.; Leibowitz, G. Anti-inflammatory agents in the treatment of diabetes and its vascular complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef] [Green Version]
- Tsalamandris, S.; Antonopoulos, A.; Oikonomou, E.; Papamikroulis, G.A.; Vogiatzi, G.; Papaioannou, S.; Deftereos, S.; Tousoulis, D. The role of inflammation in diabetes: Current concepts and future perspectives. Eur. Cardiol. Rev. 2019, 14, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Teodoro, J.; Nunes, S.; Rolo, A.; Reis, F.; Palmeira, C.M. Therapeutic options targeting oxidative stress, mitochondrial dysfunction and inflammation to hinder the progression of vascular complications of diabetes. Front. Physiol. 2019, 10, 1857. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophth. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. DMSO Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.; Tchkonia, T.; Kirkland, J. Senolytics: Potential for alleviating diabetes and its complications. Endocrinology 2021, 162, bqab058. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Vandvik, P.; Lytvyn, L.; Guyatt, G.H.; Palmer, S.C.; Vermandere, M.; Rodriguez-Gutierrez, R.; Foroutan, F.; Agoritsas, T.; Siemieniuk, R.A.; et al. SGLT-2 inhibitors or GLP-1 receptor agonists for adults with type 2 diabetes: A clinical practice guideline. Br. Med. J. 2021, 8, 22. [Google Scholar] [CrossRef]
- Lee, M.; Kim, B.; Han, K.; Lee, J.H.; Kim, M.; Kim, M.K.; Baek, K.; Song, K.; Kwon, H.; Roh, Y. Sodium-glucose cotransporter 2 inhibitors and risk of retinal vein occlusion among patients with type 2 diabetes: A propensity score-matched cohort study. Diabetes Care 2021, 44, 2419–2426. [Google Scholar] [CrossRef]
- Petrie, J. SGLT2 inhibitors and renal complications in type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 803–805. [Google Scholar] [CrossRef]
- Coll, R.; O’Neill, L.; Schroder, K. Questions and controversies in innate immune research: What is the physiological role of NLRP3? Nat. Publ. Group 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice—Brief report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, V.; Kalyanasundaram, R. Therapeutic implications of inflammasome in inflammatory bowel disease. FASEB J. 2021, 35, e21439. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Brol, M.; Magdaleno, F.; Schierwagen, R.; Uschner, F.E.; Klein, S.; Ortiz, C.; Tyc, O.; Bachtler, N.; Stunden, J.; et al. The specific NLRP3 antagonist IFM-514 decreases fibrosis and inflammation in experimental murine non-alcoholic steatohepatitis. Front. Mol. Biosci. 2021, 8, 771. [Google Scholar] [CrossRef]
- Ram, C.; Jha, A.; Ghosh, A.; Gairola, S.; Syed, A.M.; Murty, U.S.; Naidu, V.G.M.; Sahu, B.D. Targeting NLRP3 inflammasome as a promising approach for treatment of diabetic nephropathy: Preclinical evidences with therapeutic approaches. Eur. J. Pharmacol. 2020, 885, 173503. [Google Scholar] [CrossRef]
- Sharma, A.; Choi, J.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E.; et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef]
- Gora, I.; Ciechanowska, A.; Ladyzynski, P. NLRP3 inflammasome at the interface of inflammation, endothelial dysfunction, and type 2 diabetes. Cells 2021, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Ge, K.; Wang, Y.; Li, P.; Li, M.; Zhang, W.; Dan, H.; Hu, X.; Zhou, J.; Yang, Q.; Wang, J.; et al. Down-expression of the NLRP3 inflammasome delays the progression of diabetic retinopathy. Microvasc. Res. 2021, 139, 104265. [Google Scholar] [CrossRef]
- Huang, W.; Jiao, J.; Liu, J.; Huang, M.; Hu, Y.; Ran, W.; Yan, L.; Li, M.; Quan, Z.; Rao, Y.; et al. MFG-E8 accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death Discov. 2020, 6, 84. [Google Scholar] [CrossRef]
- He, X.; Li, L.; Xian, W.; Li, M.Y.; Zhang, L.Y.; Xu, J.H.; Pei, Z.; Zheng, H.Q.; Hu, X.Q. Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. J. Neuroinflamm. 2021, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, J.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Surabhi, S.; Cuypers, F.; Hammerschmidt, S.; Siemens, N. The role of NLRP3 inflammasome in Pneumococcal infections. Front. Immunol. 2020, 11, 3277. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 inflammasome—A key player in antiviral responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Tan, Z.; Wang, M. Inhibition of NLRP3 inflammasome: A prospective target for the treatment of ischemic stroke. Front. Cell. Neurosci. 2020, 14, 155. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Jouadir, M. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 97. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Wohlford, G.; Tassell, B.; Billingsley, H.; Kadariya, D.; Canada, J.M.; Carbone, S.; Mihalick, V.; Bonaventurea, A.; Vecchié, A.; Chiabrando, J.G.; et al. Single-center repeat dose safety and pharmacodynamics study of the oral NLRP3 inhibitor Dapansutrile in subjects with NYHA II-III systolic heart failure. J. Cardiovasc. Pharmacol. 2019, 77, 49–60. [Google Scholar] [CrossRef]
- El-Sharkawy, L.; Brough, D.; Freeman, S. Inhibiting the NLRP3 inflammasome. Molecules 2020, 25, 5533. [Google Scholar] [CrossRef]
- Ridker, P.; MacFadyen, J.; Everett, B.; Libby, P.; Thuren, T.; Glynn, R.J. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomized controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.; Quartier, P.; Constantin, T.; Wulffraat, N.M.; Horneff, G.; Kasapcopur, O.; Schneider, R.; Anton, J.; Barash, J.; et al. Canakinumab in patients with systemic juvenile idiopathic arthritis and active systemic features: Results from the 5-year long-term extension of the phase III pivotal trials. Ann. Rheum. Dis. 2018, 77, 1710–1719. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.; Bundy, B.; Becker, D.J.; Dimeglio, L.A.; Gitelman, S.E.; Goland, R. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomized, double-blind, placebo-controlled trials. Lancet 2013, 381, 1905–1915. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.; Howard, C.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of interleukin-1ß inhibition with canakinumab on hemoglobin A1c, lipids, C-reactive protein, interleukin-6, and fibrinogen: A phase IIb randomized, placebo-controlled trial. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.; Everett, B.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Kiyoshi, M.; Tatematsu, K.; Tada, M.; Sezutsu, H.; Shibata, H.; Ishii-Watabe, A. Structural insight and stability of TNFR-Fc fusion protein (Etanercept) produced by transgenic silkworms. J. Biochem. 2021, 169, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, M.; Archer, R.; Tosh, J.; Simpson, E.; Everson-Hock, E.; Stevens, J.; Hernandez-Alava, M.; Paisley, S.; Dickinson, K.; Scott, D.; et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: Systematic review and economic evaluation. Health Technol. Assess. 2016, 35, 1–610. [Google Scholar] [CrossRef] [Green Version]
- Kawanami, D.; Matoba, K.; Takeda, Y.; Negai, Y.; Akamine, T.; Yokota, T.; Sango, K.; Utsunomiya, K. SGLT2 inhibitors as a therapeutic option for diabetic nephropathy. Int. J. Mol. Sci. 2017, 18, 1083. [Google Scholar] [CrossRef] [PubMed]
- McGuire, D.K.; Shih, W.J.; Cosentino, F.; Charbonnel, B.; Cherney, D.Z.I.; Dagogo-Jack, S.; Pratley, R.; Greenberg, M.; Wang, S.; Huyck, S.; et al. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: A meta-analysis. JAMA Cardiol. 2021, 6, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Argyropoulos, C.; Singh, N. Are the protective effects of SGLT2 inhibitors a “class-effect” or are there differences between agents? Kidney360 2021, 2, 881–885. [Google Scholar] [CrossRef]
- Liu, H.; Sridhar, V.; Boulet, J.; Dharia, A.; Khan, A.; Lawler, P.R.; Cherney, D.Z.I. Cardiorenal protection with SGLT2 inhibitors in patients with diabetes mellitus: From biomarkers to clinical outcomes in heart failure and diabetic kidney disease. Metab. Clin. Exp. 2021, 126, 154918. [Google Scholar] [CrossRef] [PubMed]
- Thirunavukarasu, S.; Jex, N.; Chowdhary, A.; Hassan, I.U.; Straw, S.; Craven, T.P.; Gorecka, M.; Broadbent, D.; Swoboda, P.; Witte, K.K.; et al. Empagliflozin treatment is associated with improvements in cardiac energetics and function and reductions in myocardial cellular volume in patients with type 2 diabetes. Diabetes 2021, 70, db210270. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Sardu, C.; Trotta, M.; Scisciola, L.; Turriziani, F.; Ferreraccio, F.; Panarese, I.; Petrella, L.; Fanelli, M.; Modugno, P.; et al. Sodium-glucose co-transporter2 expression and inflammatory activity in diabetic atherosclerotic plaques: Effects of sodium-glucose co-transporter2 inhibitor treatment. Mol. Metab. 2021, 54, 101337. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kitada, M.; Ogura, Y.; Liu, H.; Koya, D. Dapagliflozin Restores Impaired Autophagy and Suppresses Inflammation in High Glucose-Treated HK-2 Cells. Cells 2021, 10, 1457. [Google Scholar] [CrossRef]
- Madonna, R.; Doria, V.; Minnucci, I.; Pucci, A.; Pierdomenico, D.S.; de Caterina, R. Empagliflozin reduces the senescence of cardiac stromal cells and improves cardiac function in a murine model of diabetes. J. Cell. Mol. Med. 2020, 24, 12331–12340. [Google Scholar] [CrossRef]
- Musso, G.; Saba, F.; Cassader, M.; Gambino, R. Diabetic ketoacidosis with SGLT2 inhibitors. BMJ 2020, 371, m4147. [Google Scholar] [CrossRef]
- Lin, C.; Zhu, X.; Cai, X.; Yang, W.; Lv, F.; Nie, L.; Ji, L. SGLT2 inhibitors and lower limb complications: An updated meta-analysis. Cardiovasc. Diabetol. 2021, 20, 1–12. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Kalra, S.; Purandare, V.; Vasnawala, H. Genital infections with sodium glucose cotransporter-2 inhibitors: Occurrence and management in patients with type 2 diabetes mellitus. Indian J. Endocrinol. Metab. 2018, 22, 837–842. [Google Scholar] [CrossRef]
- Lissoni, A.; Wang, N.; Nezlobinskii, T.; de Smet, M.; Panfilov, A.V.; Vandersickel, N.; Leybaert, L.; Witschas, K. Gap19, a Cx43 hemichannel inhibitor, acts as a gating modifier that decreases main state opening while increasing substrate gating. Int. J. Mol. Sci. 2020, 21, 7340. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, T.; Smet, M.; Verwaerde, S.; Decrock, E.; Czekaj, A.; Wandenbroucke, R.E.; Lemeire, K.; Gonçalves, A.; Declercq, W.; Vandenabeele, P.; et al. Blocking connexin43 hemichannels protects mice against tumour necrosis factor-induced inflammatory shock. Sci. Rep. 2019, 9, 16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Griffith, J.; Nor, M.; Zhang, J.; Freestnoe, P.S.; Danesh-Meyer, H.V.; Rupenthal, I.D.; Acosta, M.; Nicholson, L.F.B.; O’Carroll, S.J.; et al. Tonabersat Prevents Inflammatory Damage in the Central Nervous System by Blocking Connexin43 Hemichannels. Neurother. J. Am. Soc. Exp. Neurother. 2017, 14, 1148–1165. [Google Scholar] [CrossRef] [Green Version]
- King, D.; Sedovy, M.; Leng, X.; Xue, J.; Lamouille, S.; Koval, M.; Isakson, B.E.; Johnstone, S.R. Mechanisms of connexin mimetic peptides. Int. J. Mol. Sci. 2021, 22, 10186. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.; Green, C.; Kho, D.; Zhang, J.; Graham, E.S.; Acosta, M.L.; Rupenthal, I.D. The inflammasome pathway is amplified and perpetuated in an autocrine manner through connexin43 hemichannel mediated ATP release. Biochim. Biophys. Acta 2018, 1862, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, S.; Park, H.; Lee, Y.J.; Park, S.H.; Lee, K.J.; Lee, D.G.; Kang, H.; Kim, J.E. Contribution of autophagy-notch1-mediated nlrp3 inflammasome activation to chronic inflammation and fibrosis in keloid fibroblasts. Int. J. Mol. Sci. 2020, 21, 8050. [Google Scholar] [CrossRef]
- Van Campenhout, R.; Gomes, A.; Groof, T.; Muyldermans, S.; Devoogdt, N.; Vinken, M. Mechanisms underlying connexin hemichannel activation in disease. Int. J. Mol. Sci. 2021, 22, 3503. [Google Scholar] [CrossRef]
- Price, G.W.; Chadjichristos, C.; Kavvadas, P.; Tang, S.C.W.; Yiu, W.H.; Green, C.R.; Potter, J.A.; Siamantouras, E.; Squires, P.E.; Hills, C.E. Blocking Connexin-43 mediated hemichannel activity protects against early tubular injury in experimental chronic kidney disease. Cell Commun. Signal. 2020, 18, 1–17. [Google Scholar] [CrossRef]
- Laird, D.; Lampe, P. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905. [Google Scholar] [CrossRef]
- Rhett, J.M.; Yeh, E.S. Molecular Sciences the Potential for Connexin Hemichannels to Drive Breast Cancer Progression through Regulation of the Inflammatory Response. Int. J. Mol. Sci. 2018, 19, 1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, Á.; Magyar, C.; Héja, L.; Kardos, J. Peptide binding sites of connexin proteins. Chemistry 2020, 2, 662–673. [Google Scholar] [CrossRef]
- Wang, N.; Bock, M.; Antoons, G.; Gadicharla, A.K.; Bol, M.; Decrock, E.; Evans, W.H.; Sipido, K.R.; Bukauskas, F.F.; Leybaert, L. Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res. Cardiol. 2012, 107, 304. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Ghatnekar, G.; Grek, C.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [Green Version]
- Evans, W.; Leybaert, L. Mimetic Peptides as Blockers of Connexin Channel-Facilitated Intercellular Communication. Cell Commun. Adhes. 2007, 14, 265–273. [Google Scholar] [CrossRef]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; de Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [Green Version]
- Coutinho, F.; Green, C.; Acosta, M.; Rupenthal, I.D. Xentry-Gap19 inhibits Connexin43 hemichannel opening especially during hypoxic injury. Drug Deliv. Transl. Res. 2020, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; de Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Yang, L.; Chen, J.; Chen, Y.; Zhang, L.; Wang, L.; Li, X.; Li, Y.; Yu, H. Inhibition of Connexin43 hemichannels with Gap19 protects cerebral ischemia/reperfusion injury via the JAK2/STAT3 pathway in mice. Brain Res. Bull. 2019, 146, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Yanguas, S.C.; Willebrords, J.; Weemhoff, J.L. Connexin hemichannel inhibition reduces acetaminophen-induced liver injury in mice. Toxicol. Lett. 2017, 278, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Tarzemany, R.; Jiang, G.; Jiang, J.; Larjava, H.; Häkkinen, L. Connexin 43 Hemichannels Regulate the Expression of Wound Healing-Associated Genes in Human Gingival Fibroblasts. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Warner, A.; Clements, D.; Parikh, S.; Evans, W.H.; DeHaan, R.L. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 1995, 488, 721–728. [Google Scholar] [CrossRef]
- Orellana, J.; Shoji, K.; Abudara, V.; Ezan, P.; Amigou, E.; Sáez, P.J.; Jiang, J.X.; Naus, C.C.; Sáez, J.C.; Giaume, C. Amyloid β-induced death in neurons involves glial and neuronal hemichannels. J. Neurosci. 2011, 31, 4962–4977. [Google Scholar] [CrossRef] [PubMed]
- Elbadawy, H.M.; Mirabelli, P.; Xeroudaki, M.; Parekh, M.; Bertolin, M.; Breda, C.; Cagini, C.; Ponzin, D.; Lagali, N.; Ferrari, S. Effect of connexin 43 inhibition by the mimetic peptide Gap27 on corneal wound healing, inflammation and neovascularization. Br. J. Pharmacol. 2016, 173, 2880–2893. [Google Scholar] [CrossRef] [PubMed]
- Faniku, C.; O’Shaughnessy, E.; Lorraine, C.; Johnstone, S.R.; Graham, A.; Greenhough, S.; Martin, P.E.M. The Connexin Mimetic Peptide Gap27 and Cx43-Knockdown Reveal Differential Roles for Connexin43 in Wound Closure Events in Skin Model Systems. Int. J. Mol. Sci. 2018, 19, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.; Green, C.; Rupenthal, I.; Mugisho, O.O. Connexin43 hemichannel block protects against retinal pigment epithelial cell barrier breakdown. Acta Diabetol. 2020, 57, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Nor, M.; Danesh-Meyer, H.; Vessey, K.A.; Fletcher, E.L.; O’Carroll, S.J.; Acosta, M.L.; Green, C.R. Connexin43 Mimetic Peptide Improves Retinal Function and Reduces Inflammation in a Light-Damaged Albino Rat Model. Investig. Opth. Vis. Sci. 2016, 57, 3961–3973. [Google Scholar] [CrossRef] [Green Version]
- Nor, M.N.M.; Rupenthal, I.D.; Green, C.R.; Acosta, M.L. Connexin hemichannel block using orally delivered Tonabersat improves outcomes in animal models of retinal disease. Neurotherapeutics 2020, 17, 371–387. [Google Scholar] [CrossRef]
- Jiang, J.; Hoagland, D.; Palatinus, J.; He, H.; Iyyathurai, J.; Jourdan, J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of α Carboxyl Terminus 1 Peptide with the Connexin 43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A peptide mimetic of the connexin43 carboxyl terminus reduces gap junction remodeling and induced arrhythmia following ventricular injury. Circ Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Bryant, Z.; Ghatnekar, G.; Singh, U.P.; Gourdie, R.G.; Potts, J.D. A Synthetic Connexin 43 Mimetic Peptide Augments Corneal Wound Healing. Exp. Eye Res. 2013, 115, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A Multicenter Randomized Controlled Trial Evaluating a Cx43-Mimetic Peptide in Cutaneous Scarring. J. Investig. Dermatol. 2017, 137, 620. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, J.; Richardson, W.; Marsh, S.; Rhett, M.; Bustos, F.; Degen, K.; Ghatnekar, G.S.; Grek, C.L.; Jourdan, L.J.; Holmes, J.W.; et al. The connexin 43 carboxyl terminal mimetic peptide αCT1 prompts differentiation of a collagen scar matrix in humans resembling unwounded skin. FASEB J. 2020, 35, e21762. [Google Scholar] [CrossRef]
- Butera, J.; Larsen, B.; Herman, J.; Kerns, E.; Di, L.; Alimardanov, A.; Swillo, R.E.; Morgan, G.A.; Liu, K.; Wang, Q.; et al. Discovery of (2S,4R)-1-(2-aminoacetyl)-4-benzamidopyrrolidine-2-carboxylic acid hydrochloride (GAP-134)13, an orally active small molecule gap-junction modifier for the treatment of atrial fibrillation. J. Med. Chem. 2009, 52, 908–911. [Google Scholar] [CrossRef]
- Kim, D.; Mouritzen, U.; Larsen, B.D.; Roy, S. Inhibition of Cx43 gap junction uncoupling prevents high glucose-induced apoptosis and reduces excess cell monolayer permeability in retinal vascular endothelial cells. Exp. Eye Res. 2018, 173, 85–90. [Google Scholar] [CrossRef]
- Squires, P.; Price, G.W.; Mouritzen, U.; Potter, J.A.; Williams, B.M.; Hills, C.E. Danegaptide Prevents TGFβ1-Induced Damage in Human Proximal Tubule Epithelial Cells of the Kidney. Int. J. Mol. Sci. 2021, 22, 2809. [Google Scholar] [CrossRef]
- Skyschally, A.; Walter, B.; Hansen, R.; Heusch, G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn. Schmiedebergs Arch. Pharmacol. 2013, 386, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Rossman, E.I.; Kantrowitz, J.; Butera, J.; Peterson, J.S.; Gardell, S.J.; Vlasuk, G.P. GAP-134 ([2S,4R]-1-[2-Aminoacetyl]4-Bensamidopyrrolidine-2-Carboxylic Acid] prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J. Cardiovasc. Pharmacol. Ther. 2009, 14, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Howard, L.P.; Mangat, I.; Moe, G.W.; Hu, X.; So, P.P.; Tarulli, E.; Ramadeen, A.; Rossman, E.I.; Hennan, J.K.; et al. Effects of chronic gap junction conduction-enhancing antiarrhythmic peptide GAP-134 administration on experimental atrial fibrillation in dogs. Circ. Arrhythmia Electrophysiol. 2009, 2, 171–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossman, E.I.; Liu, K.; Morgan, G.A.; Swillo, R.E.; Krueger, J.A.; Gardell, S.J.; Butera, J.; Gruver, M.; Kantrowitz, J.; Feldman, H.S.; et al. The gap junction modifier, GAP-134 [(2S,4R]-1-[2-Aminoacetyl]4-Bensamidopyrrolidine-2-Carboxylic Acid], improves conduction and reduces atrial fibrillation/flutter in the canine sterile pericarditis model. J. Pharmacol. Exp. Ther. 2009, 329, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, Z.; Chaiban, J.T.; Krikorian, A. Novel insights into the pathophysiology and clinical aspects of diabetic nephropathy. Rev. Endocr. Metab. Disord. 2017, 18, 21–28. [Google Scholar] [CrossRef]
- Kravets, I.; Mallipattu, S.K. The Role of Podocytes and Podocyte-Associated Biomarkers in Diagnosis and Treatment of Diabetic Kidney Disease. J. Endocr. Soc. 2020, 4, bvaa029. [Google Scholar] [CrossRef] [Green Version]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, S.; Alves, A.; Preguiça, I.; Barbosa, A.; Vieira, P.; Mendes, F.; Martins, D.; Viana, S.D.; Reis, F. Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet. Nutrients 2020, 12, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Huang, H.; Gao, R.; Liu, Y. Dynamic Phenotypes and Molecular Mechanisms to Understand the Pathogenesis of Diabetic Nephropathy in Two Widely Used Animal Models of Type 2 Diabetes Mellitus. Front. Cell Dev. Biol. 2020, 8, 172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Y.; Zhao, Y. Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS ONE 2019, 14, e0221991. [Google Scholar] [CrossRef]
- Xiong, J.; Wang, Y.; Shao, N.; Gao, P.; Tang, H.; Su, H.; Zhang, C.; Meng, X.F. The Expression and Significance of NLRP3 Inflammasome in Patients with Primary Glomerular Diseases. Kidney Blood Press. Res. 2015, 40, 344–354. [Google Scholar] [CrossRef]
- Kavvadas, P.; Abed, A.; Poulain, C.; Authier, F.; Labéjof, L.P.; Calmont, A.; Afieri, C.; Prakoura, N.; Dussaule, J.C.; Chatziantoniou, C.; et al. Decreased expression of connexin 43 blunts the progression of experimental GN. J. Am. Soc. Nephrol. 2017, 28, 2915–2930. [Google Scholar] [CrossRef]
- Ougaard, M.K.E.; Kvist, P.H.; Jensen, H.E.; Hess, C.; Rune, I.; Søndergaard, H. Murine Nephrotoxic Nephritis as a Model of Chronic Kidney Disease. Int. J. Nephrol. 2018, 2018, 8424502. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Zhao, Y.; Na, C.; Yang, M.; Zhu, X.; Shi, H.; Gan, W.; Zhang, A. Connexin 43-autophagy loop in the podocyte injury of diabetic nephropathy. Int. J. Mol. Med. 2019, 44, 1781–1788. [Google Scholar] [CrossRef]
- Liu, B.C.; Tang, T.T.; Lv, L.L.; Lan, H.Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579. [Google Scholar] [CrossRef]
- Hills, C.; Price, G.W.; Wall, M.J.; Kaufmann, T.J.; Chi-Wai Tang, S.; Yiu, W.H.; Squires, P.E. Transforming Growth Factor Beta 1 Drives a Switch in Connexin Mediated Cell-to-Cell Communication in Tubular Cells of the Diabetic Kidney. Cell. Physiol. Biochem. 2018, 45, 2369–2388. [Google Scholar] [CrossRef] [Green Version]
- Potter, J.A.; Price, G.W.; Cliff, C.L.; Green, C.R.; Squires, P.E.; Hills, C.E. Collagen I modifies connexin-43 hemichannel activity via integrin α2β1 binding in TGFβ1-evoked renal tubular epithelial cells. Int. J. Mol. Sci. 2021, 22, 3644. [Google Scholar] [CrossRef]
- Hills, C.E.; Price, G.W.; Squires, P.E. Mind the gap: Connexins and cell–cell communication in the diabetic kidney. Diabetologia 2015, 58, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Siamantouras, E.; Hills, C.E.; Liu, K.K.; Squires, P.E. Examining Cell-Cell Interactions in the Kidney Using AFM Single-Cell Force Spectroscopy. Methods Mol. Biol. 2020, 2067, 189–201. [Google Scholar] [CrossRef]
- Siamantouras, E.; Hills, C.E.; Squires, P.E.; Liu, K.K. Quantifying cellular mechanics and adhesion in renal tubular injury using single cell force spectroscopy. Nanomedicine 2016, 12, 1013–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siamantouras, E.; Price, G.W.; Potter, J.A.; Hills, C.E.; Squires, P.E. Purinergic receptor (P2X7) activation reduces cell-cell adhesion between tubular epithelial cells of the proximal kidney. Nanomed. Nanotechnol. Biol. Med. 2019, 22, 102108. [Google Scholar] [CrossRef] [PubMed]
- Menzies, R.I.; Booth, J.W.R.; Mullins, J.J.; Bailey, M.A.; Tam, F.W.K.; Norman, J.T.; Unwin, R.J. Hyperglycemia-induced Renal P2X7 Receptor Activation Enhances Diabetes-related Injury. EBioMedicine 2017, 19, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.-W.; Yuan, Y.; Chen, J.-H.; Lin, W.-Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Sci. Press Zool. Res. 2018, 39, 72–86. [Google Scholar] [CrossRef]
- McHugh, S.M.; Roman, S.; Davis, B.; Koch, A.; Pickett, A.M.; Richardson, J.C.; Miller, S.R.; Wetten, S.; Cox, C.J.; Karpe, F.; et al. Effects of genetic variation in the P2RX7 gene on pharmacodynamics of a P2X7 receptor antagonist: A prospective genotyping approach. Br. J. Clin. Pharmacol. 2012, 74, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-h.; Xiao, H.-m.; Zhang, M.; Lin, Z.-y.; Yang, Y.; Chen, R.; Liu, P.-q.; Huang, K.-p.; Huang, H.-q. USP9X deubiquitinates connexin43 to prevent high glucose-induced epithelial-to-mesenchymal transition in NRK-52E cells. Biochem. Pharmacol. 2021, 188, 114562. [Google Scholar] [CrossRef]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.S.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef]
- Hills, C.E.; Squires, P.E. TGF-β1-Induced Epithelial-to-Mesenchymal Transition and Therapeutic Intervention in Diabetic Nephropathy. Am. J. Nephrol. 2010, 31, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Davidson, J.O.; Green, C.R.; Louise, L.F.; O’Carroll, S.J.; Fraser, M.; Bennet, L.; Jan Gunn, A. Connexin hemichannel blockade improves outcomes in a model of fetal ischemia. Ann. Neurol. 2012, 71, 121–132. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.B.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Hills, C.E.; Squires, P.E. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011, 22, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abed, A.; Toubas, J.; Kavvadas, P.; Authier, F.; Cathelin, D.; Alfieri, C.; Boffa, J.-J.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Targeting connexin 43 protects against the progression of experimental chronic kidney disease in mice. Kidney Int. 2014, 86, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Q.; Fu, Y.-x.; Shu, A.-m.; Lv, X.; Chen, Y.-p.; Gao, Y.-y.; Chen, J.; Wang, W.; Lv, G.-h.; Lu, J.-f.; et al. Loganin alleviates macrophage infiltration and activation by inhibiting the MCP-1/CCR2 axis in diabetic nephropathy. Life Sci. 2021, 272, 118808. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Jiang, H.; Wang, B.; Sui, W.; Zhou, H.; Zou, G. Expression and Significance of RANTES and MCP-1 in Renal Tissue with Chronic Renal Allograft Dysfunction. Transplant. Proc. 2016, 48, 2034–2039. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Lin, S.C.; Chen, G.; He, L.; Hu, Z.; Chan, L.; Trial, J.A.; Entman, M.L.; Wang, Y. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1644–1659. [Google Scholar] [CrossRef] [Green Version]
- National Diabetes Statistics Report. 2020. Available online: https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf (accessed on 24 November 2021).
- Mathebula, S.D. Biochemical changes in diabetic retinopathy triggered by hyperglycaemia: A review. Afr. Vis. Eye Health 2018, 77, 1–7. [Google Scholar] [CrossRef]
- Maugeri, G.; Bucolo, C.; Drago, F.; Rossi, S.; di Rosa, M.; Imbesi, R.; D’Agata, V.; Giunta, S. Attenuation of High Glucose-Induced Damage in RPE Cells through p38 MAPK Signaling Pathway Inhibition. Front. Pharmacol. 2021, 12, 1125. [Google Scholar] [CrossRef]
- Mehanna, C.J.; Abdul Fattah, M.; Haddad, S.; Tamim, H.; Ghazi, N.; Salti, H. Anti-VEGF Therapy for Persistent Neovascularization after Complete Panretinal Photocoagulation in Proliferative Diabetic Retinopathy. Ophthalmol. Retin. 2019, 3, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Tham, Y.C.; Yu, M.; Chee, M.L.; Rim, T.H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef]
- Everett, L.A.; Paulus, Y.M. Laser Therapy in the Treatment of Diabetic Retinopathy and Diabetic Macular Edema. Curr. Diab. Rep. 2021, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Singh, R.P. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context 2018, 7, 212532. [Google Scholar] [CrossRef]
- Che, D.; Zhou, T.; Lan, Y.; Xie, J.; Gong, H.; Li, C.; Feng, J.; Hong, H.; Qi, W.; Ma, C.; et al. High glucose-induced epithelial-mesenchymal transition contributes to the upregulation of fibrogenic factors in retinal pigment epithelial cells. Int. J. Mol. Med. 2016, 38, 1815–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, H.; Yin, N.; Rupenthal, I.D.; Green, C.R.; Odunayo, I.; Mugisho, O. Blocking connexin43 hemichannels prevents TGF-β2 upregulation and epithelial–mesenchymal transition in retinal pigment epithelial cells. Cell Biol. Int. 2021. [Google Scholar] [CrossRef]
- Mugisho, O.O.; Green, C.R.; Squirrell, D.M.; Bould, S.; Danesh-Meyer, H.V.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. J. Mol. Med. 2019, 97, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Mugisho, O.O.; Green, C.R.; Zhang, J.; Binz, N.; Acosta, M.L.; Rakoczy, E.; Rupenthal, I.D. Immunohistochemical Characterization of Connexin43 Expression in a Mouse Model of Diabetic Retinopathy and in Human Donor Retinas. Int. J. Mol. Sci. 2017, 18, 2567. [Google Scholar] [CrossRef] [Green Version]
- Mugisho, O.O.; Rupenthal, I.D.; Squirrell, D.M.; Bould, S.J.; Danesh-Meyer, H.V.; Zhang, J.; Green, C.R.; Acosta, M.L. Intravitreal pro-inflammatory cytokines in non-obese diabetic mice: Modelling signs of diabetic retinopathy. PLoS ONE 2018, 13, e0202156. [Google Scholar] [CrossRef] [PubMed]
- Louie, H.H.; Shome, A.; Kuo, C.Y.; Rupenthal, I.D.; Green, C.R.; Mugisho, O.O. Connexin43 hemichannel block inhibits NLRP3 inflammasome activation in a human retinal explant model of diabetic retinopathy. Exp. Eye Res. 2021, 202, 108384. [Google Scholar] [CrossRef] [PubMed]
- Green, C.R.; Mat Nor, M.N.; Mugisho, O.O.; Rupenthal, I.D.; Squirrell, D.M.; Acosta, M.L. Connexin hemichannel block shuts down inflammation in an animal model of chronic diabetic retinopathy to improve structural and functional outcomes | IOVS | ARVO Journals. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2784. [Google Scholar]
- Tien, T.; Muto, T.; Zhang, J.; Sohn, E.H.; Mullins, R.F.; Roy, S. Association of reduced Connexin 43 expression with retinal vascular lesions in human diabetic retinopathy. Exp. Eye Res. 2016, 146, 103–106. [Google Scholar] [CrossRef]
- Li, A.F.; Roy, S. High glucose-induced downregulation of connexin 43 expression promotes apoptosis in microvascular endothelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Li, A.F.; Sato, T.; Haimovici, R.; Okamoto, T.; Roy, S. High Glucose Alters Connexin 43 Expression and Gap Junction Intercellular Communication Activity in Retinal Pericytes. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5376–5382. [Google Scholar] [CrossRef] [Green Version]
- European Commission (DG ECFIN) and Economic Policy Committee (Ageing Working Group). The 2015 Ageing Report Economic and Budgetary Projections for the 28 EU Member States (2013–2060). Available online: https://ec.europa.eu/economy_finance/publications/european_economy/2015/pdf/ee3_en.pdf (accessed on 24 November 2021).
- Raman, K.S.; Matsubara, J.A. Dysregulation of the NLRP3 Inflammasome in Diabetic Retinopathy and Potential Therapeutic Targets. Ocul. Immunol. Inflamm. 2020, 1–9. [Google Scholar] [CrossRef]
- Moreno, J.A.; Gomez-Guerrero, C.; Mas, S.; Sanz, A.B.; Lorenzo, O.; Ruiz-Ortega, M.; Opazo, L.; Mezzano, S.; Egido, J. Targeting inflammation in diabetic nephropathy: A tale of hope. Expert. Opin. Investig. Drugs 2018, 27, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.-y.; Tang, L.-q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 2016, 114, 251–264. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, X.; Liao, N.; Mi, L.; Peng, Y.; Liu, B.; Zhang, S.; Wen, F. Enhanced Expression of NLRP3 Inflammasome-Related Inflammation in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 978–985. [Google Scholar] [CrossRef]
- Hou, Y.; Lin, S.; Qiu, J.; Sun, W.; Dong, M.; Xiang, Y.; Wang, L.; Du, P. NLRP3 inflammasome negatively regulates podocyte autophagy in diabetic nephropathy. Biochem. Biophys. Res. Commun. 2020, 521, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Han, W.; Song, S.; Du, Y.; Liu, C.; Chen, N.; Wu, H.; Shi, Y.; Duan, H. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol. Cell. Endocrinol. 2018, 478, 115–125. [Google Scholar] [CrossRef]
- Hickson, L.T.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, S.E.; Halai, R.; Cooper, M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef] [PubMed]
- Masson, W.; Lobo, M.; Barbagelata, L.; Lavalle-Cobo, A.; Molinero, G. Effect of anti-inflammatory therapy on major cardiovascular events in patients with diabetes: A meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2021, 15, 102164. [Google Scholar] [CrossRef]
- Tittarelli, A. Connexin channels modulation in pathophysiology and treatment of immune and inflammatory disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166258. [Google Scholar] [CrossRef] [PubMed]
- Price, G.W.; Potter, J.A.; Williams, B.M.; Cliff, C.L.; Squires, P.E.; Hills, C.E. Connexin-mediated cell communication in the kidney: A potential therapeutic target for future intervention of diabetic kidney disease? Joan Mott Prize Lecture. Exp. Physiol. 2020, 105, 219–229. [Google Scholar] [CrossRef]
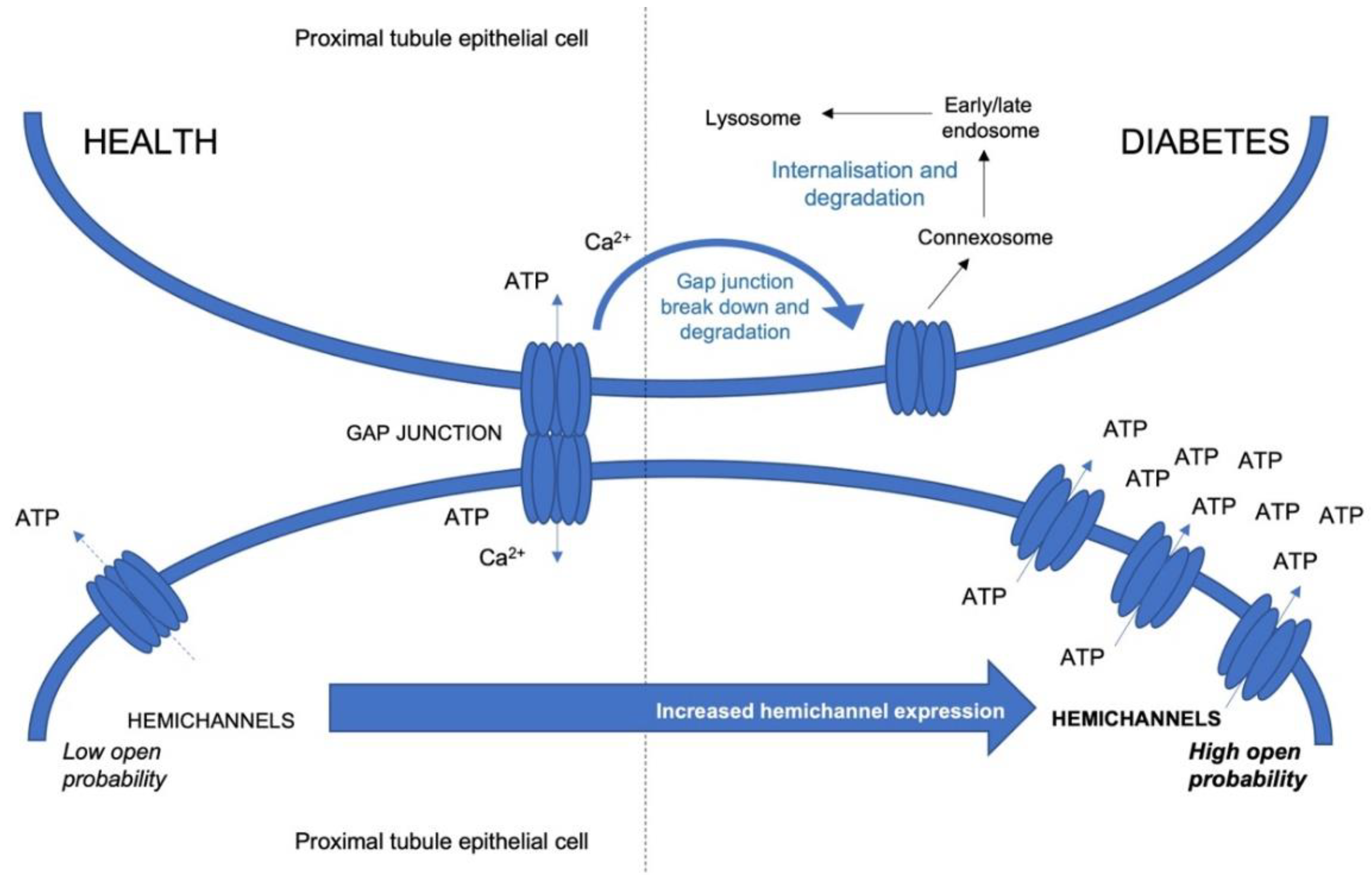

{kind=link}
| Hemichannel Blocker/ Therapeutic Agent | Sequence/ Formula | Mechanism of Action | Examples of Models Trialled in | Clinical Trials? |
|---|---|---|---|---|
| Gap19 | KQIEIKKFK Also: Transactivator of transcription (TAT)-Gap19 -YGRKKRRQRRR-KQIEIKKFK Xentry (XG19) -lclrpvGG-KQIEIKKFK | Binds to the intracellular loop of Cx43, whilst not affecting gap junction communication [115]. Exhibits low cell permeability, so is often coupled with TAT which aids transcription or Xentry which is a cell penetrating peptide [116]. | Primary mouse cardiomyocytes [117]; Cerebral ischaemia/injury in mice [118]; Primary mouse astrocytes/hippocampal slices (TAT-Gap19) [115]; Immortalised human retinal pigment epithelium cells (ARPE-19)/primary human retinal microvascular endothelial cells (hREMC) (XG19) [116]; Isolated rat hepatocytes [119]; Human gingival fibroblasts [120]. | None found. |
| Gap26 | VCYDKSFPISHVR | Originally developed to block gap junction communication [121]. Now shown to also block hemichannels, Gap26 binds to the first extracellular loop of Cx43 [112]. | Isolated pig ventricular cardiomyocytes [117]; Cultured microglia, astrocytes and neurons [122]. | None found. |
| Gap27 | SRPTEKTIFII | Originally designed for gap junction blockade [121], Gap27 can also block hemichannels by binding to the second extracellular loop of Cx43 [112]. | Isolated pig ventricular cardiomyocytes [117]; Primary human corneal epithelial cells in vitro, human corneas ex vivo rat wound healing model in vivo [123]; Adult keratinocytes, juvenile foreskin, human neonatal fibroblasts and adult dermal tissue as models of wound healing [124]. | None found. |
| Peptide 5 | VDCFLSRPTEKT | Binds to the second extracellular loop of Cx43, preventing hemichannel opening [111]. | Human primary proximal tubule epithelial cells and clonal tubular kidney epithelial cells [108]; Retinal pigment epithelial cells [105,125]; Patch-clamp inflammatory model in mice [102]; Light-damaged albino rat model [126]. | None found |
| Tonabersat (Xiflam) | C₂₀H₁₉ClFNO₄ | Able to block gap junctions (at high concentration), this small molecule, a benzopyran derivative can block Cx43 hemichannels at lower doses [50]. | Human retinal pigment epithelial cells (ARPE-19) [50]; Rat model of diabetic retinopathy [127]. | Phase II clinical trials in migraines-NCT00311662 NCT00534560 NCT00332007 |
| alpha connexin carboxyl terminus 1 (αCT1) | Ant-RPRPDDLEI | Binds to the COOH tail (cytoplasmic terminus) of Cx43 [113], mediating phosphorylation of Cx43 at serine 368 [128]. Has also been shown to affect gap junction remodelling [129]. | Rat model corneal wound [130]; Beneficial in a randomised control trial assessing cutaneous scarring [131]; Human biopsy tissue/rat and guinea pig scars [132]. | Clinical trials for diabetic foot ulcers as ‘Grannexin gel’ Phase I-NCT02652754 Phase II-NCT02652572 Terminated at phase III May 2020 (NCT02667327)–no safety concerns |
| Danegaptide (GAP-134) | C14H17N3O4 | Not fully elucidated. As a gap-junction modifier, it maintains gap junction coupling during cellular stress [133,134], and has been shown to block Cx43 hemichannels in human proximal tubule epithelial cells [135]. | Primary human proximal tubule epithelial cells [135]; Rat Retinal Endothelial cells during high glucose stress [134]; Myocardial infarct in pigs [136] and dogs [137]; Atrial fibrillation models in dogs [138,139]. | Phase II for myocardial infarction-NCT01977755 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cliff, C.L.; Williams, B.M.; Chadjichristos, C.E.; Mouritzen, U.; Squires, P.E.; Hills, C.E. Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes. Int. J. Mol. Sci. 2022, 23, 600. https://doi.org/10.3390/ijms23020600
Cliff CL, Williams BM, Chadjichristos CE, Mouritzen U, Squires PE, Hills CE. Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes. International Journal of Molecular Sciences. 2022; 23(2):600. https://doi.org/10.3390/ijms23020600
Chicago/Turabian StyleCliff, Chelsy L., Bethany M. Williams, Christos E. Chadjichristos, Ulrik Mouritzen, Paul E. Squires, and Claire E. Hills. 2022. "Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes" International Journal of Molecular Sciences 23, no. 2: 600. https://doi.org/10.3390/ijms23020600
APA StyleCliff, C. L., Williams, B. M., Chadjichristos, C. E., Mouritzen, U., Squires, P. E., & Hills, C. E. (2022). Connexin 43: A Target for the Treatment of Inflammation in Secondary Complications of the Kidney and Eye in Diabetes. International Journal of Molecular Sciences, 23(2), 600. https://doi.org/10.3390/ijms23020600