Abstract
Renal disease is a major public health challenge since its prevalence has continuously increased over the last decades. At the end stage, extrarenal replacement therapy and transplantation remain the only treatments currently available. To understand how the disease progresses, further knowledge of its pathophysiology is needed. For this purpose, experimental models, using mainly rodents, have been developed to unravel the mechanisms involved in the initiation and progression of renal disease, as well as to identify potential targets for therapy. The gap junction protein connexin 43 has recently been identified as a novel player in the development of kidney disease. Its expression has been found to be altered in many types of human renal pathologies, as well as in different animal models, contributing to the activation of inflammatory and fibrotic processes that lead to renal damage. Furthermore, Cx43 genetic, pharmacogenetic, or pharmacological inhibition preserved renal function and structure. This review summarizes the existing advances on the role of this protein in renal diseases, based mainly on different in vivo animal models of acute and chronic renal diseases.
1. Introduction
Renal disease refers to a group of pathologies that affect the kidney’s structure and function. Acute and chronic kidney diseases are both common in adults and are associated with a high risk of morbidity and mortality [1].
Acute kidney injury (AKI) is characterized by a rapid decline in renal function. It occurs in about 10 to 15% of patients admitted to the hospital and affects 1 in 5 hospitalized patients worldwide [2]. AKI is more common in patients over 65 years old with co-morbidities such as cardiovascular disease, diabetes, liver disease, and pre-existing renal disease [1,3]. According to the KDIGO guidelines, chronic kidney disease (CKD) may occur after AKI and is defined by a decrease in the glomerular filtration rate of <60 mL/min/1.73 m2 for 3 months or more [1,4]. Some of the major risk factors for CKD include diabetes, high blood pressure, autoimmune disease, aging, and genetics [5]. CKD affects approximately 8 to 10% of the world population [5]. Patients with renal failure require individualized care based on their individual medical histories. Currently, AKI is treated according to the initial cause of the renal damage (i.e., sepsis, acute hemorrhage, hemodynamic shock, or inflammatory disease) and with a nephroprotection assessment, which promotes the rapid recovery of injured kidneys [6]. Concerning CKD, treatments rely mainly on limiting the disease progression and/or replacement therapies, such as extra-renal purification or renal transplantation at late stages [5,7].
Consequently, renal disease represents a real public health burden, with the current treatments, which are costly and sometimes not very effective, significantly altering the quality of life of patients. Although our understanding of the mechanisms involved in the development of kidney disease has improved in recent years, we are still searching for early prognostic markers and novel therapeutic options to prevent or reverse the progression of kidney disease. In this regard, the gap junction protein connexin 43 may be an attractive option.
2. Gap Junctions and Connexins
Cell–cell communication is essential for maintaining tissue homeostasis and structural integrity in multicellular organisms. Gap junctions are membrane channels that provide this type of communication in various organic systems, as they have been highly conserved through evolution [8]. These channels allow for the direct cytoplasmic exchange of ions and low-molecular weight metabolites (<1 kDa) between adjacent cells [9,10]. In vertebrates, this direct communication is mediated by the docking of two hemichannels, called connexons (Figure 1). The connexons are located at the plasma membrane of two adjacent cells and form a hydrophilic central pore that allows for the exchange of ions, including Na, K+, and Ca2+, small metabolites, and second messengers (cAMP, IP3, ADP, ATP, etc.) [8,11,12]. Uncoupled connexons also allow for communication between the cytoplasm and the extracellular environment [13]. Each connexon is composed of six homo- or hetero-hexameric connexin (Cx) subunits [11]. Connexins are a group of proteins named according to their approximate molecular weight (kDa). Extensive studies have identified 21 different isoforms in humans and 20 in mice. Most tissues and cells express these proteins, except for erythrocytes, sperm cells, and striated muscle cells [8]. Regarding their structure, Cxs cross the plasma membrane four times, producing two extracellular loops and one intracellular loop and exposing their N- and C-terminal tails in the cytosol [8]. This structure is highly conserved in the Cx family members. However, the intracellular loop and the N- and C-terminal tails have a large variation in their length and amino acids. Due to their structure and function in the intracellular exchange of metabolites and other second messengers, gap junctions participate in many physiological processes, such as cell proliferation, growth, or differentiation [14,15]. It is also evident that they are involved in angiogenesis, glandular secretion, and neuronal migration [16,17,18].
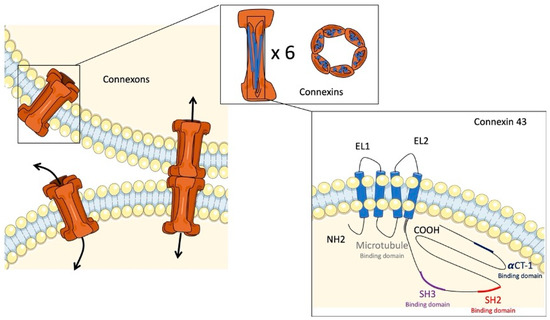
Figure 1.
Gap junctions and connexin 43. Illustration of connexons binding together to form a gap junction channel with a central pore. The connexons are represented as cones composed of six connexin subunits. Connexin 43 is represented by four transmembrane domains, two extracellular loops (EL1 and EL2), one intracellular loop, and N-terminal (NH2) and C-terminal (COOH) tails.
In recent years, several studies have suggested that Cxs are involved in renal pathophysiology. Despite being well documented, these proteins are still a challenge, both in terms of their involvement as novel therapeutic targets and in their pathophysiological mechanisms. Previous studies have shown that certain isoforms are expressed in the kidneys, including Cx26, Cx30.3, Cx31, Cx32, Cx37, Cx40, Cx43, Cx45, and Cx46 [19,20]. Many studies have focused on their function and localization, but these data are still controversial. In this review, we focus mainly on the role of Cx43 in renal disease.
3. Connexin 43
Almost ubiquitously expressed, Cx43 is the most documented constitutive gap junction protein. The gene encoding for Cx43 is the gap junction alpha-1 protein (GJA1) [21]. Its C-terminal part differs from other Cxs by the presence of about twenty phosphorylation sites and protein interaction domains, allowing it to play different physiological roles; in addition to its role in cell–cell communication, Cx43 can mediate gene transcription, cytoskeleton dynamics, ATP exocytosis, vesicle release, and cellular stress (Figure 1) [13,22]. These functions are due to the unique structure of Cx43, which is composed of an untranslated exon 1 separated by an intron from exon 2, containing an uninterrupted coding region and a 3′ untranslated region (3′-UTR) [21]. Moreover, Cx43 is abundantly expressed in ventricular cardiomyocytes and, more precisely, in intercalated discs, allowing it to electronically couple adjacent myocytes and synchronize the cardiac action potential [23,24]. Impaired trafficking can lead to arrhythmias and sudden cardiac death [25]. Many authors have investigated its role in different types of heart disease, notably atherosclerosis, where an increase in Cx43 expression has been found with a modification of its localization [26]. However, an inhibition of its expression in mice susceptible to atherosclerosis was protective, probably by limiting inflammation [22]. Other cell types express Cx43, notably astrocytes. Studies on human post mortem brains suffering from Alzheimer’s disease, and equivalent models in mice, have also shown an increased expression of Cx43 leading to neurotoxic effects through the excessive release of ATP and glutamate [27]. Studies on experimental animal models have also shown a role for Cx43 in retinal diseases. Indeed, a high expression of this Cx has been found in a diseased human eye (diabetic retinopathy, epithelioid and mixoid melanoma, etc.), playing a role in the establishment of the inflammatory process and the proliferation of tumor cells [28].
The implication of Cx43 in the inflammatory process has also been demonstrated in acute lung injury, contributing to pulmonary oedema [29]. In view of its involvement in a variety of pathological processes, the inhibition of Cx43 has shown promising results. Indeed, the use of Gap27, a pharmacological inhibitor of Cx43, in a mouse model of Parkinson’s disease was able to reduce the damage of dopaminergic neurons [30]. In addition, pharmacogenetic inhibition using antisense oligonucleotides limited inflammation and promoted wound healing [31]. While previous studies using Cx43 channel blockers reported improved outcomes, some studies in knockout animals showed the opposite results. Indeed, in a diabetic mouse model, a decrease in Cx43 expression was associated with the development and progression of diabetic retinopathy [32]. Finally, Cx43 is expressed in various cancers, including colorectal, breast, and cervical cancers [14,33,34]. However, the current studies remain controversial, showing that its expression is both protective and pro-metastatic, depending on the type of tumor [35].
4. Connexin 43 in the Kidney
The research on connexins has been an important field of biomedical research since their discovery in the mid-1970s, but Beyer et al. were the first to demonstrate Cx43 expression in rat kidneys in 1989 [36]. Later studies determined the localization of this protein in mouse models in the renal vasculature (endothelial cells), the glomerulus (podocytes and mesangial cells), and in the collecting duct [37,38]. Numerous studies have since focused on the role and molecular mechanisms mediated by Cx43 in different renal compartments where it is expressed and on its function in renal injury. This review focuses on experimental murine models of AKI and CKD that have helped us understand the role of this Cx in these diseases.
4.1. Connexin 43 in Experimental AKI
A few studies have been performed on biopsies from patients showing a variation in Cx43 expression in the kidneys [39,40]. Since then, it has become important to understand the role of this Cx in the functional and structural adaptations of the kidney in response to acute injury. The rodent models of AKI, which were developed for this purpose, will be discussed.
4.1.1. Uranyl Acetate
Hishida et al. were the first to explore the expression of Cx43 in an animal model of AKI induced by a uranyl acetate (UA) injection. UA is a derivative of uranium, a radioactive heavy metal whose acute exposures are chemically toxic to the kidneys, causing various reversible disorders, mainly characterized by acute damage to the proximal tubule [41].
To determine the mechanisms of differentiation of myofibroblasts from interstitial fibroblastic cells during AKI, Hishida et al. induced acute renal failure in Sprague–Dawley rats by intravenous injection of UA and explored its consequences 2 and 21 days after the injection. In healthy kidneys, rare immunofluorescent spots for Cx43 in the interstitium were observed. However, from day 2 onwards, more extensive staining was seen in the peritubular region, which is the main affected area following the induction of AKI by UA. The authors speculated that the upregulation of Cx43 could promote AKI by participating in the communication between myofibroblasts located around the damaged proximal tubules. However, since UA is radioactive and due to increasing safety concerns and restrictions imposed by government agencies, many authors have focused on more suitable alternatives [42].
4.1.2. Lipopolysaccharide
Lipopolysaccharide (LPS) is an endotoxin derived from the outer leaflet of the outer membrane of Gram-negative bacteria, such as Escherichia coli, that has been used by many researchers to produce experimental pathology in animals [43,44]. In the murine kidney, the injection of LPS results in an elevation in plasma urea and creatinine, as well as an increase in certain inflammatory cytokines, reversibly modifying renal function [45,46].
De Maio et al. were the first to describe a model using this endotoxin in the kidney to study Cx43 expression in adult male rats. The LPS injection induced rapid and reversible acute inflammation, which allowed for the observation of the increase in Cx43 expression only 2 h after the injection; it reached a maximum at 4 h before returning to a basal state after 12 h. The authors suggested that the Cx43-increased expression was part of an inflammatory response induced by the LPS administration [47]. Qin et al. then studied the evolution of Cx43 expression, but also the effect of its pharmacological inhibition after an LPS injection. The study was performed in adult mice to determine if Fangjifuling (FF), a traditional anti-inflammatory medicinal plant used in China, could limit the AKI induced by LPS. Although the Cx43 expression was insignificant in the renal cortex of the control mice, it was abnormally elevated around the proximal and distal tubules after the LPS injection, as previously reported by Hishida et al. In addition, its inhibition promoted the nephroprotective effect of FF against LPS-induced inflammatory and apoptotic responses; thus, confirming the involvement of Cx43 in inflammatory and apoptotic processes [48].
The above-mentioned studies were mainly interested in the expression and localization of Cx43 during an LPS-induced AKI. This model was later used to understand the role of Cx43 in inflammatory responses via inflammasome activation by Yao et al. The activation of the NLRP3 inflammasome is a major cellular event contributing to the production of inflammatory mediators. In this study, adult WT (Cx43+/+) and heterozygous (Cx43+/−) mice, in which the connexin 43 expression was genetically reduced by half, were injected intraperitoneally with LPS at different time points. As expected, the LPS administration induced renal inflammation and tubular damage, as well as it increased the levels of IL-1B, blood urea nitrogen (BUN), and proteinuria. This renal dysfunction was associated with an increased expression of Cx43 and an increase in the production of reactive oxygen species (ROS). Thus, the authors suggested that Cx43 contributes to the activation of the inflammasome through the modulation of the intracellular redox status. This process was regulated by Cx43 via Ca2+ signaling, a crucial molecule involved in ROS induction, but also via ROS propagation between adjacent cells. Thus, using this animal model allowed for the partial explanation of the role of Cx43 in inflammatory responses and inflammasome regulation [49].
4.1.3. Cisplatin
Cis-diamminedichloroplatinum (II) (cisplatin) is a drug used to treat several types of cancer. This chemotherapeutic agent contains platinum and forms complexes when it binds to DNA, causing the DNA strands to cross-link, leading to programmed cell death. Although it is an effective treatment for slowing down the growth of cancers, it is known to cause nephrotoxicity. Indeed, cisplatin-induced AKI in mice mimics manifestations of clinical AKI, such as decreased renal function and increased expression of tubular injury biomarkers [50]. Based on these observations, this model has been used to improve our knowledge of the physiology and pathophysiology of Cx43 in AKI.
Yu et al. focused on the regulatory effect of Cx43 on cisplatin-induced ferroptosis in mice. Indeed, ferroptosis is one of the main causes of cisplatin-induced AKI. Basically, it results from excessive iron-dependent lipid peroxidation that induces cell death. The expression level of Cx43 was significantly increased in cisplatin-injected mice, followed by an increase in tubular injury biomarkers, such as neutrophil gelatinase-associated lipocalin (N-GAL), kidney injury molecule-1 (KIM-1), and plasma urea [51]. Interestingly, the pharmacological inhibition of Cx43 by the Gap27 blocking peptide reduced the Cx43 levels and significantly decreased the ferroptosis markers. Moreover, in this study, human kidney proximal tubular cells (HK2 cells) were used and the Cx43 expression increased in a dose-dependent manner, while its downregulation decreased apoptosis in these cells. In addition, the Cx43 decrease resulted in an increase in SLC7A11 and GSH, which are involved in the inhibition of ferroptosis. In conclusion, the downregulation of Cx43 expression reduced AKI in this mouse model by inhibiting cisplatin-induced ferroptosis. However, further research into this mechanism is needed to provide more information on the treatment of AKI [52].
4.2. Connexin 43 in Experimental CKD
Previous studies have shown that Cx43 is expressed in the renal parenchyma and vasculature under resting and/or pathological conditions in animals and humans [36,39]. The importance of Cx43 in cell signaling and communication led to its investigation in renal pathology, particularly CKD. Regardless of the initial damage type, glomerular, endothelial, or tubulointerstitial, the main features of CKD are the recruitment of inflammatory cells and renal fibrosis [53]. Consequently, this review focuses on the role of Cx43 in these major CKD effects in different models of experimental nephropathy.
4.2.1. Cx43 and Glomerular Damage
The role of Cx43 using models of experimental glomerulopathies has been reported mainly in rodents and targeted cells, particularly podocytes, specialized in the maintenance of the glomerular filtration barrier. Glomerulonephritis (GN) represents a variety of pathologies affecting the kidney and, more precisely, the glomerulus. It is the second most common cause of CKD, often progressing to end-stage renal failure. Several causes are at the origin of this pathology, such as infections, genetics, and hereditary or autoimmune diseases. In addition, some studies reported various information, which allowed for the highlighting of glomerular lesions that can subsequently affect other renal compartments [54].
There are different types of GN; the most severe and rapidly progressive being characterized by crescent formation in the human kidney, affecting less than 50% of the total glomeruli [55]. The progression of GN is characterized by detachment, followed by loss and/or migration of podocytes to the urinary chamber; however, the underlying molecular and cellular mechanisms remain unclear.
Induction of Glomerulonephritis by Injection of Nephrotoxic Serum
In our group, GN is induced by the injection of a nephrotoxic serum (NTS), according to a protocol popularized by Salant [56]. The NTS can cause crescents and renal fibrosis, along with proteinuria and glomerular damage. We have demonstrated an abnormal expression of Cx43 in injured glomeruli in mice after an NTS injection [57]. Interestingly, Cx43 expression was mainly detected in the glomerular endothelial tuft and expressed de novo in podocytes. This increase promoted glomerular damage, contributing to disease progression. Furthermore, the inhibition of this protein using Cx43+/− mice, or the specific blockade with Cx43 antisense, was able to preserve the structure and function of the kidney. Finally, we proposed a mechanism by which proinflammatory and/or profibrotic cytokines activated the expression of Cx43 during the progression of the disease at the transcriptional level in podocytes, leading to de novo expression of Cx43 hemichannels, contributing to an exacerbated ATP release. ATP, in turn, could mediate via purinergic signaling podocyte damage, illustrated by either foot process effacement or cell death [58].
Induction of Glomerulonephritis by Anti-Thymocyte Serum (ATS) Injection
Morioka et al. were interested in another type of GN induced by an anti-thymocyte serum (ATS) injection. Mesangioproliferative GN was induced in rats by the injection of antibodies against the Thy1-1 antigen, present only on the surface of the mesangial cells of the rats, thus, allowing the establishment of mesangiolysis, followed by an expansion of the mesangial extracellular matrix. Mesangial cells and podocytes expressing Cx43 showed no change in its expression during the progression of this GN model [59].
Puromycin Aminonucleoside
Puromycin aminonucleoside (PAN) is a widely used model of nephrotic syndrome progressing to focal segmental glomerulosclerosis. PAN is known to cause a nephrotic syndrome characterized by glomerular morphological changes and severe proteinuria, but the extent of damage varies with the dose of PAN administration [60,61].
Previous studies performed by Yamamoto et al. demonstrated an increased Cx43 expression along the glomerular capillary wall in rats at early stages of PAN-induced disease. Further studies using this model confirmed that the increased expression of Cx43 was one of the earliest responses to podocyte injury ever observed. Furthermore, microinjections of the fluorescent dye Lucifer yellow in cultured podocytes suggested that Cx43-mediated gap junctional intercellular communication was present between podocytes. The authors proposed that these cells may behave as an integrated epithelium within the glomerulus, rather than individually as separate cells in response to injury [62].
Phosphate Overload-Induced Glomerulonephritis
Sekiguchi et al. studied Cx43 expression in a mouse model of phosphate overload leading to GN. Indeed, transgenic rats with overexpression of Pit-1, a type III Na-dependent phosphate transporter, were used to establish this animal model as a model for the study of podocyte damage. The uptake of phosphate at the cell membrane is critical for cell viability, but a phosphate overload may also damage the cells. Consequently, this model resulted in severe proteinuria due to the phosphate-dependent podocyte injury and damage to the glomerular barrier, leading to the development of glomerulosclerosis. An interesting finding in this model was the appearance of enhanced immunofluorescence for Cx43 in podocytes prior to any renal dysfunction being observed. Thus, the authors considered Cx43 increased expression as an early signal of podocyte damage in transgenic rats [63].
Podocyte Injury via Aldosterone Injection
Aldosterone is originally a mineralocorticoid hormone secreted in the zona glomerulosa of the adrenal cortex in response to angiotensin-2 stimulation or elevated blood potassium and is associated with the development of podocyte injury through oxidative stress induction. This is associated with albuminuria, hypertension, and the development of glomerulosclerosis. Based on these observations, Yang et al. performed aldosterone injections in mice to study the effects of Cx43 on podocyte injury and explore the possible molecular mechanism behind this effect. They demonstrated that the upregulation of Cx43 was mediated by aldosterone, which could facilitate the propagation of apoptotic signals with an increase in ROS production and the Bax/Bcl-2 ratio [64].
High-Fat Diet
Obesity has been widely recognized for several years as a result of kidney damage. This disease is characterized by an excess of body fat gained from excessive dietary energy intake. Obesity contributes to the increasing prevalence of glomerulopathy and accelerates the progression of CKD. Altered fatty acid and cholesterol metabolisms are emerging as key mediators, contributing to this pathology with renal lipid accumulation, inflammation, oxidative stress, and fibrosis.
In a study performed by Zhao et al., the expression of Cx43 was investigated in a high-fat diet model in rats for a long period of time. The hypercaloric intake caused glomerular hypertrophy and podocyte foot effacement, as well as an elevation in the podocyte injury markers. The Cx43 expression was also found to be increased within the injured glomeruli and was correlated with obesity-related inflammation, suggesting that this Cx may be a potential target in the development of obesity-related glomerulopathy [65].
4.2.2. Cx43 in Obstructive Nephropathy: The Unilateral Ureteral Obstruction Model
The unilateral ureteral obstruction model (UUO) has been currently used in rodents. The UUO consists of a ligation of the left ureter with the unobstructed right kidney serving as a control. This model presents fundamental pathogenic mechanisms that characterize some forms of CKD, in addition to tubular injuries resulting from obstructed urine flow, allowing, in the long-term, for severe damage to the renal structure and further renal fibrosis.
Sommer et al. first observed that Cx43 mRNA expression was strongly increased in ureteral ligated kidneys in rats 5 days after the UUO, indicating a possible function of this protein in the remodeling of the renal tissue after tubular damage and fibrosis [66].
By using the same model, our group confirmed an increased Cx43 expression in the early stages of renal disease in mice. Moreover, a decrease in its expression via the use of Cx43-specific antisense or in Cx43+/− mice considerably limited the recruitment of inflammatory cells and cell adhesion molecules. Regarding the fibrotic process, which occurs in response to inflammation, the decreased Cx43 expression limited the extracellular matrix remodeling, probably by inhibiting the activation of the TGFB1/ERK pathway. Consequently, Cx43 seems to play a crucial role in the inflammatory process, as also reported in AKI [48,49], but also in the progression of renal fibrosis [67]. More recently, Price et al. showed that the increased expression of Cx43 after an UUO was accompanied by a reduction in the expression of E-cadherin and ZO-1, markers commonly associated with epithelial phenotypes, and an increase in N-cadherin and β-catenin, mainly associated with mesenchymal phenotypes, reflecting an epithelial–mesenchymal transdifferentiation. This transition was blunted in Cx43+/− mice. Moreover, an augmentation of inflammatory markers, such as NLRP3, was also reported in the same mice, as well as an increase in the ATP release mediated by Cx43 hemichannels, which may promote tubular lesions and the establishment of the inflammatory process [68]. Indeed, additional studies have shown that the release of ATP activated purinergic receptors, such as P2 × 7, which contribute to fibrosis in several types of diseases [69].
4.2.3. Cx43 and Hypertensive Nephropathy
High blood pressure is one of the main causes of CKD. This hypertension can be primary or secondary to the development of the disease. It is widely recognized that the kidney plays a crucial role in the regulation of blood pressure. In this section, we report the main hypertensive animal models used in studies to determine Cx43 functions in this pathological condition.
Stimulation of Renin Secretion
Haefliger et al. used a hypertensive rat model to study the distribution of Cx43 in the kidney by clipping the renal artery to stimulate renin secretion. In this context, Cx43 was expressed in renal vessel endothelial cells at moderate levels [70]. Following this observation and the potential expression of Cx43 in endothelial cells, Garcia-Pinto et al. also used this model to study its distribution more precisely. A Cx43 expression was observed in the interdigitations and in the cytoplasm of endothelial and tubular cells. However, in spontaneously hypertensive rats, Cx43 expression was found to be decreased [71]. Over time, several authors showed contradictory data. To study the role of Cx43 in hypertension-induced CKD, RenTg mice, a model of hypertension-induced CKD, where renin was expressed ectopically in the liver, were crossed with Cx43+/− mice. Interestingly, the upregulation of Cx43 contributed to structural damage in the renal cortex in the RenTg mice, and the RenTgCx43+/− mice showed improved tissue structure and function. Accordingly, Cx43 expression increased as renal disease progressed, whereas a Cx43 decrease in expression was beneficial in chronic inflammation [67].
Renal Injury via Angiotensin II
The renin–angiotensin system (RAS) is an endocrine network composed of renin and angiotensin II (AngII) that acts on the kidneys and adrenal glands, regulating blood pressure, intravascular volume, and electrolyte balance. Moreover, it is well established in the literature that AngII contributes to the rise in blood pressure, which causes progressive damage to nephrons, generating chronic lesions and further end-stage renal disease. The infusion of AngII in rats showed a possible correlation between the activation of the RhoA/ROCK pathway and the expression of Cx43, leading to an increase in pro-inflammatory cytokines, such as the tumor necrosis factor α (TNF-α) and interleukine-1β (IL-1β), and further kidney damage [72].
4.2.4. Cx43 and Diabetic Nephropathy
Chronic hyperglycemia and high blood pressure are the main risk factors for the development of diabetic nephropathy (DN), which represents a major cause of CKD worldwide. There are many pathways and mediators involved in the development and progression of DN, but they still need to be further explored. Renal fibrosis is the main outcome of DN, which is characterized by glomerular sclerosis and tubulointerstitial fibrosis.
Different experimental models of DN have been used to study the expression and role of Cx43 in this pathology. Satriano et al. studied this protein in rats with type 1 diabetes induced by a streptozotocin (STZ) treatment, a cytotoxic substance on the beta cells of the islets of Langherans. They observed a decreased expression of Cx43 after the induction of DN [73]. Similar data were reported by Hu et al. and Zhang et al. [74,75]. The use of db/db mice with type 2 diabetes and leptin receptor deficiency also reported that elevated glucose levels reduced Cx43 expression. In addition, an increase in Cx43 expression improved the activation of the Nrf2/ARE pathway, a crucial cellular defense mechanism against oxidative stress, ultimately attenuating renal fibrosis in diabetic mice [76]. Additional studies performed in db/db mice also showed that Cx43 activation reduced the development of diabetic renal fibrosis by regulating EMT via the SIRT-1 HIF signaling pathway, involved in metabolism and inflammation. In the same study, overexpression of Cx43 inhibited EMT and reduced the expression of ECM components, such as fibronectin (FN), collagen I, and collagen IV, in NRK-52E (rat kidney proximal tubular epithelial cell line) cells induced by high glucose [77]. Finally, the use of boldine, a well-known anti-inflammatory and hypoglycemic alkaloid, in mesangial cells prevented the increase in oxidative stress, the decline in gap junction intercellular communication, and the increase in cell permeability due to Cx43 hemichannel activity resulting from high glucose and proinflammatory cytokines [78].
All of the above-mentioned observations on Cx43 expression and the proposed functions in different models of experimental nephropathy are listed in Table 1.

Table 1.
Physiopathological functions of Cx43 in different in vivo models of renal disease.
5. Connexin 43 and Human Nephropathy
The difficulty of obtaining samples reduces the number of human studies. Consequently, existing data are mainly based on the in vitro culture of human cells and a few renal biopsies of patients with different types of nephropathies.
Cx43 was first detected in human kidneys in 1997 by Hillis et al. The authors studied the expression of this protein in biopsies from patients with various inflammatory glomerulopathies and found increased expression of Cx43 in infiltrating and damaged tubular epithelial cells and de novo expression in interstitial injured sites [39]. The pattern of expression of Cx43 was closely paralleled by that of cell adhesion markers, and the authors speculated that this protein was implicated in tubulointerstitial inflammation.
Later, Sawai et al. reported that the altered expression of Cx43 was associated with progressive diabetic type 2 nephropathy in human biopsies. Similar data were reported in in vivo models [79]. Furthermore, Guo et al. found that diabetic patients showed decreased expression of Cx43 in the glomerulus when the MAPK/mTOR pathways were activated [80].
As previously shown in rodents, Kurtz et al. reported Cx43 expression in human kidneys, mainly in the endothelium of the arteries/arterioles and the juxtaglomerular apparatus [81]. Our research group reported that immunofluorescence for Cx43 was highly induced in biopsies of patients suffering from different types of nephropathies, such as IgA and C3 glomerulonephritis, nephroangiosclerosis, and obstructive nephritis [58,67].
Finally, a mechanism has been recently proposed based both on biopsies of CKD patients and in vivo and in vitro models of tubulopathy, underlying aberrant Cx43 hemichannel activity via ATP release, supporting purinergic receptors and inflammasome activation, thus, promoting the progression of the disease [82].
6. Conclusions
Chronic kidney disease is a major global health burden due to the increasing prevalence of its associated risk factors. Despite recent medical advances, end-stage renal disease remains a major challenge. Therefore, finding new therapeutic targets to detect renal disease earlier and improve patient outcomes is a medical priority. Among some candidates, the gap junction protein Cx43 has emerged as a promising therapeutic target. Indeed, even though its expression depends on the type of nephropathy, it appears crucial to the progression of inflammation and renal fibrosis. Furthermore, Cx43 inhibition, whether pharmacological, genetic, or pharmacogenetic, was able to limit renal damage and improve renal function in animal models of experimental nephropathy. Hence, new therapies targeting Cx43 could be developed to delay kidney disease progression in the near future.
Author Contributions
Conceptualization, E.R., L.B. and C.E.C.; writing—original draft preparation, E.R. and C.E.C.; writing—review and editing, E.R., L.B. and C.E.C.; supervision, C.E.C.; project administration, C.E.C.; funding acquisition, C.E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the French National Research Agency (ANR ConTarKid grant to C.E.C., ANR-15-CE14-0024), the French National Institute of Health and Medical Research (Inserm) and the Sorbonne University. E.R. is a doctoral fellow of the French Ministry of National Education (“Ecole Doctorale de Physiologie and Physiopathologie”, ED 394). L.B. is a fellow of the SFAR (“Société Française d’Anesthésie et de Réanimation”).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The figures were designed with Servier Medical Art by Servier, which is licensed under the Creative Commons Attribution 3.0 Unported License.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bao, Y.-W.; Yuan, Y.; Chen, J.-H.; Lin, W.-Q. Kidney disease models: Tools to identify mechanisms and potential therapeutic targets. Zool. Res. 2018, 39, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World Incidence of AKI: A Meta-Analysis. Clin. J. Am. Soc. Nephrol. CJASN 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef]
- Harty, J. Prevention and Management of Acute Kidney Injury. Ulst. Med. J. 2014, 83, 149–157. [Google Scholar]
- Levey, A.S.; Becker, C.; Inker, L.A. Glomerular Filtration Rate and Albuminuria for Detection and Staging of Acute and Chronic Kidney Disease in Adults: A systematic review. JAMA 2015, 313, 837–846. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Axelsen, L.N.; Sorgen, P.L.; Verma, V.; Delmar, M.; Holstein-Rathlou, N.-H. Gap Junctions. Compr. Physiol. 2012, 2, a002576. [Google Scholar] [CrossRef]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef]
- Zhou, J.Z.; Jiang, J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions--an update. FEBS Lett. 2014, 588, 1186–1192. [Google Scholar] [CrossRef]
- Leybaert, L.; Lampe, P.D.; Dhein, S.; Kwak, B.; Ferdinandy, P.; Beyer, E.; Laird, D.W.; Naus, C.C.; Green, C.R.; Schulz, R. Connexins in Cardiovascular and Neurovascular Health and Disease: Pharmacological Implications. Pharmacol. Rev. 2017, 69, 396–478. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Sánchez, H.A.; Lee, S.C.; Altenberg, G.A.; Nathanson, M.H.; Sáez, J.C. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol. 2010, 299, C1504–C1515. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta Biomembr. 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Nalewajska, M.; Marchelek-Myśliwiec, M.; Opara-Bajerowicz, M.; Dziedziejko, V.; Pawlik, A. Connexins—Therapeutic Targets in Cancers. Int. J. Mol. Sci. 2020, 21, 9119. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Meda, P.; Chanson, M.; Pepper, M.; Giordano, E.; Bosco, D.; Traub, O.; Willecke, K.; El Aoumari, A.; Gros, D.; Beyer, E.C.; et al. In vivo modulation of connexin 43 gene expression and junctional coupling of pancreatic B-cells. Exp. Cell Res. 1991, 192, 469–480. [Google Scholar] [CrossRef]
- Fushiki, S.; Velazquez, J.L.P.; Zhang, L.; Bechberger, J.F.; Carlen, P.; Naus, C.C.G. Changes in Neuronal Migration in Neocortex of Connexin43 Null Mutant Mice. J. Neuropathol. Exp. Neurol. 2003, 62, 304–314. [Google Scholar] [CrossRef]
- Hanner, F.; Sorensen, C.M.; Holstein-Rathlou, N.-H.; Peti-Peterdi, J. Connexins and the kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1143–R1155. [Google Scholar] [CrossRef]
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef]
- Söhl, G.; Willecke, K. Gap junctions and the connexin protein family. Cardiovasc. Res. 2004, 62, 228–232. [Google Scholar] [CrossRef]
- Prakoura, N.; Kavvadas, P.; Chadjichristos, C.E. Connexin 43: A New Therapeutic Target Against Chronic Kidney Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I. Connexins and the heart. Trends Cardiovasc. Med. 1992, 2, 50–55. [Google Scholar] [CrossRef]
- Boengler, K.; Schulz, R. Connexin 43 and Mitochondria in Cardiovascular Health and Disease. In Mitochondrial Dynamics in Cardiovascular Medicine; Santulli, G., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 227–246. [Google Scholar] [CrossRef]
- Epifantseva, I.; Shaw, R.M. Intracellular trafficking pathways of Cx43 gap junction channels. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Burger, F.; Pelli, G.; Mach, F.; Kwak, B.R. Dual Benefit of Reduced Cx43 on Atherosclerosis in LDL Receptor-Deficient Mice. Cell Commun. Adhes. 2003, 10, 395–400. [Google Scholar] [CrossRef]
- Yi, C.; Mei, X.; Ezan, P.; Mato, S.; Matias, I.; Giaume, C.; Koulakoff, A. Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 2016, 23, 1691–1701. [Google Scholar] [CrossRef]
- Žužul, M.; Lozić, M.; Filipović, N.; Čanović, S.; Pavičić, A.D.; Petričević, J.; Kunac, N.; Šoljić, V.; Saraga-Babić, M.; Konjevoda, S.; et al. The Expression of Connexin 37, 40, 43, 45 and Pannexin 1 in the Early Human Retina and Choroid Development and Tumorigenesis. Int. J. Mol. Sci. 2022, 23, 5918. [Google Scholar] [CrossRef]
- Parthasarathi, K.; Ichimura, H.; Monma, E.; Lindert, J.; Quadri, S.; Issekutz, A.; Bhattacharya, J. Connexin 43 mediates spread of Ca2+-dependent proinflammatory responses in lung capillaries. J. Clin. Investig. 2006, 116, 2193–2200. [Google Scholar] [CrossRef]
- Quan, H.H.; Xu, W.X.; Qi, Y.Z.; Li, Q.R.; Zhou, H.; Huang, J. Inhibition connexin 43 by mimetic peptide Gap27 mediates protective effects on 6-hydroxydopamine induced Parkinson’s disease mouse model. Beijing Da Xue Xue Bao 2022, 54, 421–426. [Google Scholar]
- Qiu, C.; Coutinho, P.; Frank, S.; Franke, S.; Law, L.-Y.; Martin, P.; Green, C.R.; Becker, D.L. Targeting Connexin43 Expression Accelerates the Rate of Wound Repair. Curr. Biol. 2003, 13, 1697–1703. [Google Scholar] [CrossRef]
- Bobbie, M.W.; Roy, S.; Trudeau, K.; Munger, S.J.; Simon, A.M.; Roy, S. Reduced Connexin 43 Expression and Its Effect on the Development of Vascular Lesions in Retinas of Diabetic Mice. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Dong, L.; MacDonald, A.I.; Akbari, S.; Edward, M.; Hodgins, M.B.; Johnstone, S.R.; Graham, S.V. HPV16 E6 Controls the Gap Junction Protein Cx43 in Cervical Tumour Cells. Viruses 2015, 7, 5243–5256. [Google Scholar] [CrossRef] [PubMed]
- Phillips, S.L.; Williams, C.B.; Zambrano, J.N.; Williams, C.J.; Yeh, E.S. Connexin 43 in the development and progression of breast cancer: What’s the connection? (Review). Int. J. Oncol. 2017, 51, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Bonacquisti, E.E.; Nguyen, J. Connexin 43 (Cx43) in cancer: Implications for therapeutic approaches via gap junctions. Cancer Lett. 2019, 442, 439–444. [Google Scholar] [CrossRef]
- Barajas, L.; Liu, L.; Tucker, M. Localization of connexin43 in rat kidney. Kidney Int. 1994, 46, 621–626. [Google Scholar] [CrossRef]
- Arensbak, B.; Mikkelsen, H.B.; Gustafsson, F.; Christensen, T.; Holstein-Rathlou, N.-H. Expression of connexin 37, 40, and 43 mRNA and protein in renal preglomerular arterioles. Histochem. Cell Biol. 2001, 115, 479–487. [Google Scholar] [CrossRef]
- Guo, R.; Liu, L.; Barajas, L. RT-PCR study of the distribution of connexin 43 mRNA in the glomerulus and renal tubular segments. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1998, 275, R439–R447. [Google Scholar] [CrossRef]
- Hillis, G.S.; Duthie, L.A.; Mlynski, R.; McKay, N.G.; Mistry, S.; MacLeod, A.M.; Simpson, J.G.; Haites, N.E. The Expression of Connexin 43 in Human Kidney and Cultured Renal Cells. Nephron 1997, 75, 458–463. [Google Scholar] [CrossRef]
- Hillis, G.S.; Duthie, L.A.; Brown, P.A.; Simpson, J.G.; MacLeod, A.M.; Haites, N.E. Upregulation and co-localization of connexin43 and cellular adhesion molecules in inflammatory renal disease. J. Pathol. 1997, 182, 373–379. [Google Scholar] [CrossRef]
- Homma-Takeda, S.; Numako, C.; Kitahara, K.; Yoshida, T.; Oikawa, M.; Terada, Y.; Kokubo, T.; Shimada, Y. Phosphorus Localization and Its Involvement in the Formation of Concentrated Uranium in the Renal Proximal Tubules of Rats Exposed to Uranyl Acetate. Int. J. Mol. Sci. 2019, 20, 4677. [Google Scholar] [CrossRef]
- Fujigaki, Y.; Muranaka, Y.; Sun, D.; Goto, T.; Zhou, H.; Sakakima, M.; Fukasawa, H.; Yonemura, K.; Yamamoto, T.; Hishida, A. Transient myofibroblast differentiation of interstitial fibroblastic cells relevant to tubular dilatation in uranyl acetate-induced acute renal failure in rats. Virchows Arch. Int. J. Pathol. 2004, 446, 164–176. [Google Scholar] [CrossRef]
- Elmas, M.; Yazar, E.; Uney, K.; Karabacak, A.E. Influence of Escherichia coli Endotoxin-Induced Endotoxaemia on the Pharmacokinetics of Enrofloxacin after Intravenous Administration in Rabbits. J. Vet. Med. A Physiol. Pathol. Clin. Med. 2006, 53, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.; Toelboell, T.; Andersen, P.H. Dose dependency and individual variability in selected clinical, haematological and blood biochemical responses after systemic lipopolysaccharide challenge in cattle. Vet. Res. 2005, 36, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-L.; Feng, Y.; Wu, M.; Wang, B.; Li, Z.-L.; Zhong, X.; Wu, W.-J.; Chen, J.; Ni, H.-F.; Tang, T.-T.; et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differ. 2019, 27, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cobo, M. Expression of the connexin 43 gene is increased int he kidneys and the lungs of rats injectied with bacterial lipopolysaccharide. Shock 1998, 10, 97–102. [Google Scholar] [CrossRef]
- Su, Z.; Yu, P.; Sheng, L.; Ye, J.; Qin, Z. Fangjifuling Ameliorates Lipopolysaccharide-Induced Renal Injury via Inhibition of Inflammatory and Apoptotic Response in Mice. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 49, 2124–2137. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, Z.; Zhang, Z.; Obata, F.; Yang, X.; Zhang, X.; Huang, Y.; Mitsui, T.; Fan, J.; Takeda, M.; et al. Connexin43 Contributes to Inflammasome Activation and Lipopolysaccharide-Initiated Acute Renal Injury via Modulation of Intracellular Oxidative Status. Antioxid. Redox Signal. 2019, 31, 1194–1212. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Lombardi, A.M.; Nguyen, K.N.; Edelstein, C.L. Recent Advances in Models, Mechanisms, Biomarkers, and Interventions in Cisplatin-Induced Acute Kidney Injury. Int. J. Mol. Sci. 2019, 20, 3011. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Hyland, C.; Falk, M.M.; Iovine, M.K. Connexin 43 gap junctional intercellular communication inhibits evx1 expression and joint formation in regenerating fins. Development 2020, 147, dev190512. [Google Scholar] [CrossRef]
- Yu, M.; Lin, Z.; Tian, X.; Chen, S.; Liang, X.; Qin, M.; Zhu, Q.; Wu, Y.; Zhong, S. Downregulation of Cx43 reduces cisplatin-induced acute renal injury by inhibiting ferroptosis. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2021, 158, 112672. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Chadban, S.; Atkins, R. Glomerulonephritis. Lancet 2005, 365, 1797–1806. [Google Scholar] [CrossRef]
- Thorner, P.S.; Ho, M.; Eremina, V.; Sado, Y.; Quaggin, S. Podocytes Contribute to the Formation of Glomerular Crescents. J. Am. Soc. Nephrol. JASN 2008, 19, 495–502. [Google Scholar] [CrossRef]
- Salant, D.J.; Cybulsky, A.V. Experimental glomerulonephritis. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1988; Volume 162, pp. 421–461. [Google Scholar] [CrossRef]
- Toubas, J.; Beck, S.; Pageaud, A.-L.; Huby, A.-C.; Mael-Ainin, M.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Alteration of connexin expression is an early signal for chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F24–F32. [Google Scholar] [CrossRef] [PubMed]
- Kavvadas, P.; Abed, A.; Poulain, C.; Authier, F.; Labéjof, L.-P.; Calmont, A.; Afieri, C.; Prakoura, N.; Dussaule, J.-C.; Chatziantoniou, C.; et al. Decreased Expression of Connexin 43 Blunts the Progression of Experimental GN. J. Am. Soc. Nephrol. JASN 2017, 28, 2915–2930. [Google Scholar] [CrossRef]
- Morioka, T.; Okada, S.; Nameta, M.; Kamal, F.; Yanakieva-Georgieva, N.T.; Yao, J.; Sato, A.; Piao, H.; Oite, T. Glomerular expression of connexin 40 and connexin 43 in rat experimental glomerulonephritis. Clin. Exp. Nephrol. 2012, 17, 191–204. [Google Scholar] [CrossRef]
- Diamond, J.R.; Karnovsky, M.J. Focal and segmental glomerulosclerosis following a single intravenous dose of puromycin aminonucleoside. Am. J. Pathol. 1986, 122, 481–487. [Google Scholar] [PubMed]
- Zhou, Y.; Kim, C.; Pablo, J.L.B.; Zhang, F.; Jung, J.Y.; Xiao, L.; Bazua-Valenti, S.; Emani, M.; Hopkins, C.R.; Weins, A.; et al. TRPC5 Channel Inhibition Protects Podocytes in Puromycin-Aminonucleoside Induced Nephrosis Models. Front. Med. 2021, 8, 721865. [Google Scholar] [CrossRef]
- Yaoita, E.; Yao, J.; Yoshida, Y.; Morioka, T.; Nameta, M.; Takata, T.; Kamiie, J.-I.; Fujinaka, H.; Oite, T.; Yamamoto, T. Up-Regulation of Connexin43 in Glomerular Podocytes in Response to Injury. Am. J. Pathol. 2002, 161, 1597–1606. [Google Scholar] [CrossRef]
- Sekiguchi, S.; Suzuki, A.; Asano, S.; Nishiwaki-Yasuda, K.; Shibata, M.; Nagao, S.; Yamamoto, N.; Matsuyama, M.; Sato, Y.; Yan, K.; et al. Phosphate overload induces podocyte injury via type III Na-dependent phosphate transporter. Am. J. Physiol. Ren. Physiol. 2011, 300, F848–F856. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Wang, B.; Li, M.; Jiang, B. Connexin 43 is Involved in Aldosterone-Induced Podocyte Injury. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2014, 34, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, G.; Wang, Y.; Liu, Z. Alteration of Connexin43 expression in a rat model of obesity-related glomerulopathy. Exp. Mol. Pathol. 2018, 104, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.; Eismann, U.; Deuther-Conrad, W.; Wendt, T.; Mohorn, T.; Fünfstück, R.; Stein, G. Time Course of Cytokine mRNA Expression in Kidneys of Rats with Unilateral Ureteral Obstruction. Nephron 2000, 84, 49–57. [Google Scholar] [CrossRef]
- Abed, A.; Toubas, J.; Kavvadas, P.; Authier, F.; Cathelin, D.; Alfieri, C.; Boffa, J.-J.; Dussaule, J.-C.; Chatziantoniou, C.; Chadjichristos, C.E. Targeting connexin 43 protects against the progression of experimental chronic kidney disease in mice. Kidney Int. 2014, 86, 768–779. [Google Scholar] [CrossRef]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2017, 10, a029348. [Google Scholar] [CrossRef]
- Xu, H.; Wang, M.; Li, Y.; Shi, M.; Wang, Z.; Cao, C.; Hong, Y.; Bin Hu, B.; Zhu, H.; Zhao, Z.; et al. Blocking connexin 43 and its promotion of ATP release from renal tubular epithelial cells ameliorates renal fibrosis. Cell Death Dis. 2022, 13, 511. [Google Scholar] [CrossRef]
- Haefliger, J.-A.; Demotz, S.; Braissant, O.; Suter, E.; Waeber, B.; Nicod, P.; Meda, P. Connexins 40 and 43 are differentially regulated within the kidneys of rats with renovascular hypertension. Kidney Int. 2001, 60, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pinto, A.B.; de Matos, V.S.; Rocha, V.; Moraes-Teixeira, J.; Carvalho, J.J. Low-Intensity physical activity beneficially alters the ultrastructural renal morphology of spontaneously hypertensive rats. Clinics 2011, 66, 855–863. [Google Scholar] [CrossRef]
- Gómez, G.I.; Velarde, V.; Sáez, J.C. Role of a RhoA/ROCK-Dependent Pathway on Renal Connexin43 Regulation in the Angiotensin II-Induced Renal Damage. Int. J. Mol. Sci. 2019, 20, 4408. [Google Scholar] [CrossRef]
- Satriano, J.; Mansoury, H.; Deng, A.; Sharma, K.; Vallon, V.; Blantz, R.C.; Thomson, S.C. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am. J. Physiol. Cell Physiol. 2010, 299, C374–C380. [Google Scholar] [CrossRef]
- Hu, C.; Cong, X.D.; Dai, D.-Z.; Zhang, Y.; Zhang, G.L.; Dai, Y. Argirein alleviates diabetic nephropathy through attenuating NADPH oxidase, Cx43, and PERK in renal tissue. Naunyn-Schmiedebergs Arch. Pharmakol. 2011, 383, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, Y.; Ma, Z.; Li, Y.; Guo, J.; Meng, Q.; Bian, H. A novel formula from mulberry leaf ameliorates diabetic nephropathy in rats via inhibiting the TGF-β1 pathway. Food Funct. 2015, 6, 3307–3315. [Google Scholar] [CrossRef]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-β1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2016, 102, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Huang, K.; Haiming, X.; Lin, Z.; Yang, Y.; Zhang, M.; Liu, P.; Huang, H. Connexin 43 prevents the progression of diabetic renal tubulointerstitial fibrosis by regulating the SIRT1-HIF-1α signaling pathway. Clin. Sci. 2020, 134, 1573–1592. [Google Scholar] [CrossRef]
- Hernández-Salinas, R.; Vielma, A.Z.; Arismendi, M.N.; Boric, M.P.; Sáez, J.C.; Velarde, V. Boldine Prevents Renal Alterations in Diabetic Rats. J. Diabetes Res. 2013, 2013, 593672. [Google Scholar] [CrossRef]
- Sawai, K.; Mukoyama, M.; Mori, K.; Yokoi, H.; Koshikawa, M.; Yoshioka, T.; Takeda, R.; Sugawara, A.; Kuwahara, T.; Saleem, M.A.; et al. Redistribution of connexin43 expression in glomerular podocytes predicts poor renal prognosis in patients with type 2 diabetes and overt nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.–Eur. Ren. Assoc. 2006, 21, 2472–2477. [Google Scholar] [CrossRef]
- Guo, Y.-N.; Wang, J.-C.; Cai, G.-Y.; Hu, X.; Cui, S.-Y.; Lv, Y.; Yin, Z.; Fu, B.; Hong, Q.; Chen, X.-M. AMPK-mediated downregulation of connexin43 and premature senescence of mesangial cells under high-glucose conditions. Exp. Gerontol. 2014, 51, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, L.; Madsen, K.; Kurt, B.; Jensen, B.L.; Walter, S.; Banas, B.; Wagner, C.; Kurtz, A. High-Level Connexin Expression in the Human Juxtaglomerular Apparatus. Nephron Physiol. 2010, 116, p1–p8. [Google Scholar] [CrossRef]
- Potter, J.; Price, G.; Cliff, C.; Green, C.; Squires, P.; Hills, C. Collagen I Modifies Connexin-43 Hemichannel Activity via Integrin α2β1 Binding in TGFβ1-Evoked Renal Tubular Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 3644. [Google Scholar] [CrossRef] [PubMed]































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).