

Addressing Noise and Estimating Uncertainty in Biomedical Data through the Exploration of Chemical Space

 , and
, and 
{kind=link}
{kind=link}
Abstract
Highlights
- Uncertainty is intrinsic to any estimation problem and mainly comes from noise in data and modeling hypotheses. Uncertainty is introduced by the question that we try to elucidate.
- Noise in data always introduces errors in the predictions because it enters the cost function in a nonlinear way. Uncertainty also means ambiguity in the identification. The best way to address both issues is sampling.
- Sampling in high-dimensional spaces is a very challenging task that requires the use of dimension-reduction techniques. When the character of a problem is determined, it becomes linearly separable.
- Sampling is hampered by high computational costs to perform forward predictions. Random sampling methods are highly ineffective. Smart model parameterizations, forward surrogates, and parallel computing are sampling techniques that can overcome these bottlenecks.
1. Introduction
The Concept of Noise and Its Role on Uncertainty in Biomedical Data
- The phenotype prediction problem, which consists of identifying the altered genetic pathways that are responsible for the development of a disease, given a set of samples belonging to both disease and healthy control classes.
- The protein folding problem, i.e., protein tertiary structure prediction from its sequence.
- The single-nucleotide polymorphisms (SNPs) problem, which consists of predicting if a given mutation (or a set of mutations) in the genome correlates with disease. Mutations are deleterious if they decrease the fitness of an organism and might be causing a disease. Mutational effects can be favorable, harmful, or neutral, depending on their context or position. The deleterious mutation hypothesis assumes that sex exists to purge a species of damaging genetic mutations. The majority of deleterious mutations are marginally deleterious, and the introduction of each mutation has an increasingly considerable effect on the fitness of the organism [1]. Most non-neutral mutations are deleterious. A related problem consists of predicting how a given mutation in the DNA affects the expression of the different genes in the transcriptome.
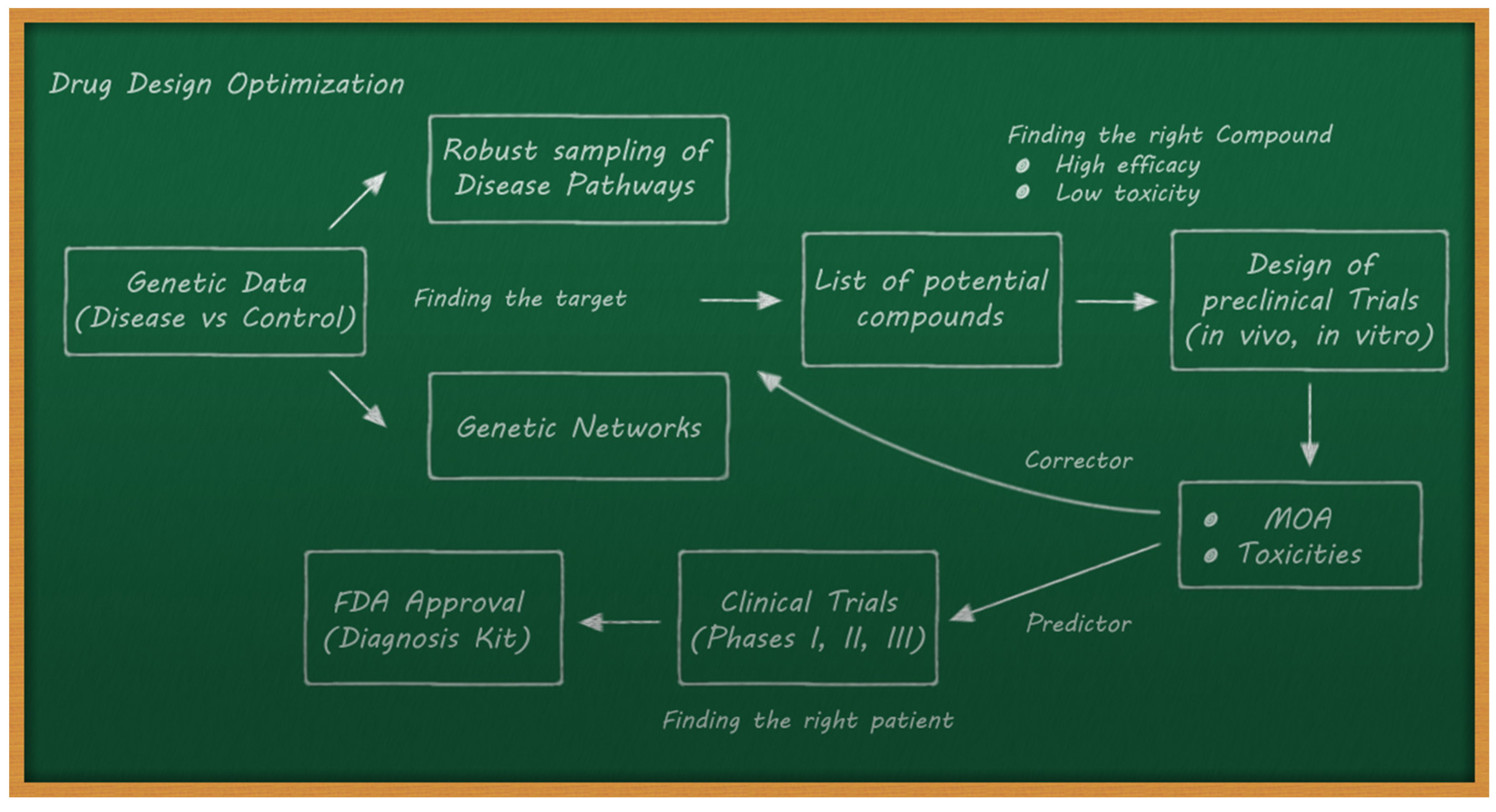
- The de novo drug design problem, which consists of optimizing the structure of new drugs with the mechanism of action designed to fight a disease, maximizing efficacy, and minimizing harmful side effects.
2. Discussion
3. Methodology
3.1. Inverse Problem
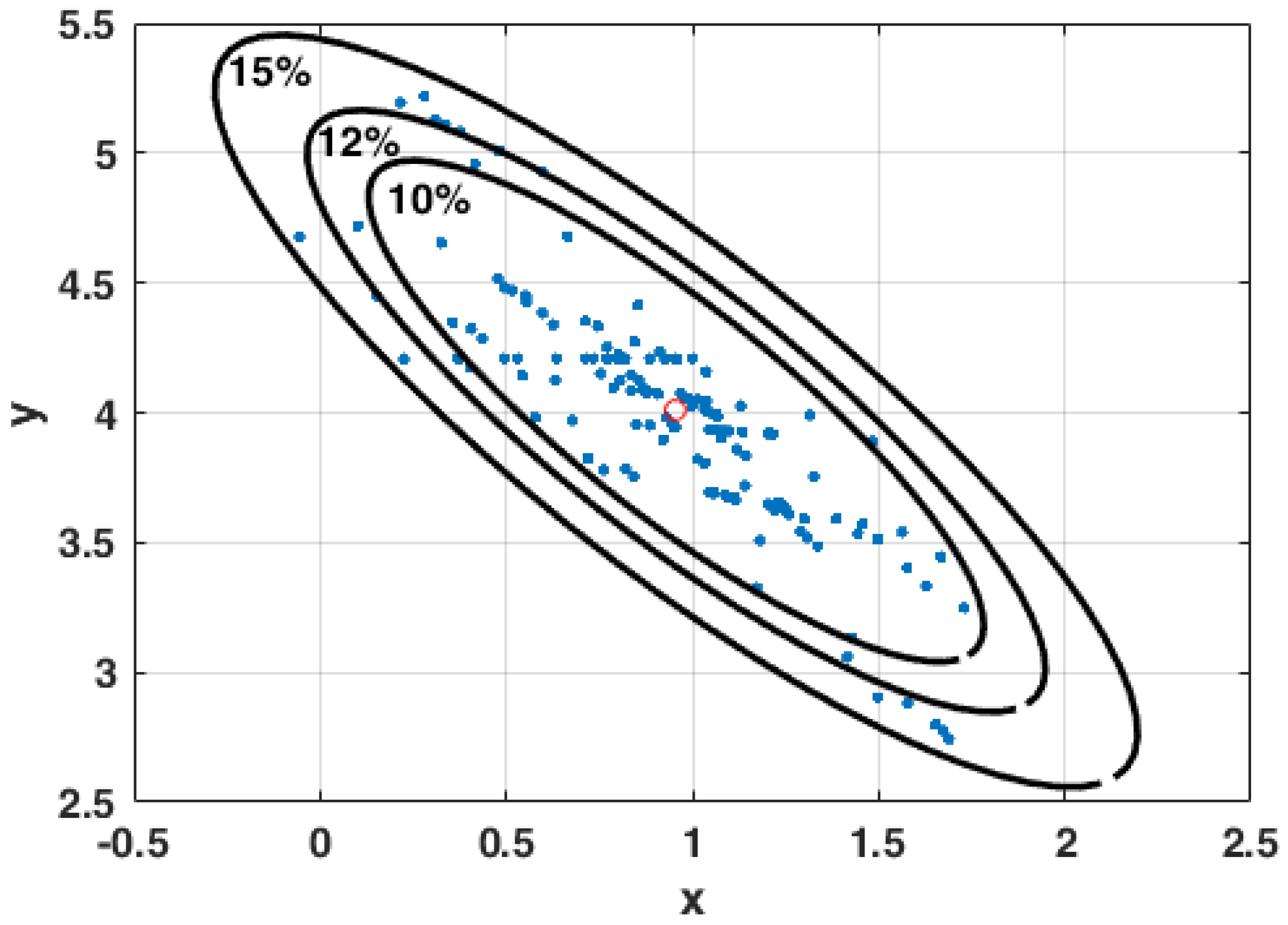
3.2. Modeling Errors
3.3. The Curse of Dimensionality
4. Specific Problems
4.1. Phenotype Prediction in Drug Design
- ✓
- The lack of a conceptual model that relates the different genes/probes to the class prediction.
- ✓
- The incomplete knowledge of genetic functions and genetic pathways, which introduces errors in the analysis.
- ✓
- ✓
- The highly underdetermined character of the phenotype inverse problem, since the number of monitored genetic probes is much greater than the number of observed samples. In fact, the genomic big data is never big because the patient dimension is always relatively small. Therefore, it is preferable to talk about little big data.
4.2. The Uncertainty Space in Phenotype Prediction
- ○
- Lack of a conceptual model explaining the disease: the lack of this model might provoke the discovery of spurious relationships.
- ○
- Highly underdetermined character: the number of patients is very reduced compared to the number of possible causes.
- ○
- Noise in data (genetic expressions and class vectors).
- ○
- The question/problem statement itself might be partially erroneous.
4.3. Drug Discovery and Drug Repositioning/Repurposing
- ○
- Performing a correct selection of the potential compounds in the preclinical trials to maximize efficacy and minimize toxicity.
- ○
- Trying to predict the outcomes of clinical trials via preclinical data and AI methods. This implies an adequate data acquisition pipeline.
- ○
- The sampling of the altered pathways, as seen in the phenotype prediction problem.
- ○
- Lack of a conceptual model relating the expression of the altered genes to the list of selected potential compounds, otherwise considered the hypothesis of achieving homeostasis via the effect of these compounds. Moreover, the CMAP data (methodology) comes from in vitro experiments and the altered pathways should be rebalanced for humans.
- ○
- Incomplete coverage of CMAP data: the perturbed genes, the compounds, the tissues, and the doses.
- ○
- Lastly, the extrapolation from preclinical analysis (in vivo and in vitro) to clinical trials (humans) with respect to the mechanisms of action (MOA) and the generation of toxicities (side effects) is not obvious.
4.4. De Novo Design: Sampling the Chemical Space
- ○
- The huge dimension of the chemical space.
- ○
- The lack of a conceptual model relating the chemical structure of one compound to its effect in transcriptomics. The idea is to train AI algorithms to learn the chemical space.
- ○
- Highly underdetermined character of the problem since there are not enough examples to train these neural networks.
- ○
- The parameterization of the compound structure is also an additional source of ambiguity.
- ○
- Noise in experimental data (mainly genetics).
- ○
- The question/problem statement itself might be partially erroneous: can the structure of the compound be related to its effect?
4.5. Uncertainty in the Prediction of Protein Mutations
- ○
- The lack of a conceptual model relating the mutation to its effect. The idea is to train AI algorithms to learn the chemical space.
- ○
- Highly underdetermined character of this problem since there are not enough examples to train these neural networks.
- ○
- The unknown effect of simultaneous mutations.
- ○
- The extrapolation of the effect of one mutation in one protein to other proteins.
- ○
- Noise in experimental data (mainly proteomics).
- ○
- The question/problem statement itself might be partially erroneous: can the structure of the compound be related to its effect?
4.6. Different Approaches to Decrease Uncertainty
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Charlesworth, B. The effects of deleterious mutations on evolution at linked sites. Genetics 2012, 190, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Fernández Martínez, J.L.; Fernandez Muniz, M.Z.; Tompkins, M.J. On the topography of the cost functional in linear and nonlinear inverse problems. Geophysics 2012, 77, W1–W15. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L. The effect of noise and Tikhonov’s regularization in inverse problems. Part I: The linear case. J. Appl. Geophys. 2014, 108, 176–185. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Pallero, J.L.G.; Fernández-Muñiz, Z.; Pedruelo-González, L.M. The effect of noise and Tikhonov’s regularization in inverse problems. Part II: The nonlinear case. J. Appl. Geophys. 2014, 108, 186–193. [Google Scholar] [CrossRef]
- Tarantola, A.; Valette, B. Inverse Problems = Quest for Information. J. Geophys. 1982, 50, 159–170. [Google Scholar]
- Tarantola, A.; Valette, B. Generalized Nonlinear Inverse Problems Solved Using the Least Squares Criterion. Rev. Geophys. 1982, 20, 219–232. [Google Scholar] [CrossRef]
- Bellman, R. Dynamic Programming and Lagrange Multipliers. Proc. Natl. Acad. Sci. USA 1956, 42, 767–769. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Fernández-Muñiz, Z. The curse of dimensionality in inverse problems. J. Comput. Appl. Math. 2020, 369, 112571. [Google Scholar] [CrossRef]
- Schmidhuber, J. Deep Learning in neural networks: An overview. Neural Netw. 2015, 61, 85–117. [Google Scholar] [CrossRef]
- Valueva, M.V.; Nagornov, N.N.; Lyakhov, P.A.; Valuev, G.V.; Chervyakov, N.I. Application of the residue number system to reduce hardware costs of the convolutional neural network implementation. Math. Comput. Simul. 2020, 177, 232–243. [Google Scholar] [CrossRef]
- de Andrés-Galiana, E.J.; Fernández-Martínez, J.L.; Sonis, S.T. Sensitivity analysis of gene ranking methods in phenotype prediction. J. Biomed. Inform. 2016, 64, 255–264. [Google Scholar] [CrossRef] [PubMed]
- de Andrés-Galiana, E.J.; Fernández-Martínez, J.L.; Saligan, L.N.; Sonis, S.T. Impact of microarray preprocessing techniques in unraveling biological pathways. J. Comput. Biol. 2016, 23, 957–968. [Google Scholar] [CrossRef]
- Luis Fernández-Martínez, J.; Álvarez, O.; De Andrés-Galiana, E.J.; De La Viña, J.F.-S.; Huergo, L.; Luis, J. Robust Sampling of Altered Pathways for Drug Repositioning Reveals Promising Novel Therapeutics for Inclusion Body Myositis. J. Rare Dis. Res. Treat. 2019, 4, 7–15. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Fernández-Muñiz, Z.; Pallero, J.L.G.; Pedruelo-González, L.M. From Thomas Bayes to Albert Tarantola. New insights to understand uncertainty in inverse problems from a deterministic point of view. J. Appl. Geophys. 2013, 98, 62–72. [Google Scholar] [CrossRef]
- de Andrés-Galiana, E.J.; Fernández-Martínez, J.L.; Sonis, S.T. Design of Biomedical Robots for Phenotype Prediction Problems. J. Comput. Biol. 2016, 23, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Martínez, J.L.; Cernea, A.; Fernández-Ovies, F.J.; Fernández-Muñiz, Z.; Alvarez-Machancoses, O.; Saligan, L.; Sonis, S.T. Sampling defective pathways in phenotype prediction problems via the Holdout sampler. Int. Conf. Bioinform. Biomed. Eng. 2018, 10814, 24–32. [Google Scholar]
- Cernea, A.; Fernández-Martínez, J.L.; deAndrés-Galiana, E.J.; Fernández-Ovies, F.J.; Fernández-Muñiz, Z.; Alvarez-Machancoses, O.; Saligan, L.; Sonis, S.T. Sampling defective pathways in phenotype prediction problems via the Fisher’s Ratio Sampler. In Bioinformatics and Biomedical Engineering. IWBBIO 2018; Lecture Notes in Computer Science; Springer: Cham, Switzerlands, 2018; Volume 10814, pp. 15–23. [Google Scholar] [CrossRef]
- Cernea, A.; Fernández-Martínez, J.L.; deAndrés-Galiana, E.J.; Fernández-Ovies, F.J.; Fernández-Muñiz, Z.; Alvarez-Machancoses, Ó.; Saligan, L.; Sonis, S.T. Comparison of Different Sampling Algorithms for Phenotype Prediction. Bioinformatics and Biomedical Engineering. IWBBIO 2018 Lecture Notes in Computer Science. 2018, 10814, 33–45. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; deAndrés-Galiana, E.J.; deAndrés-Galiana, E.J.; Cernea, A.; Fernández-Ovies, F.J.; Menéndez, M. Sampling Defective Pathways in Parkinson Disease. J. Med. Inform. Decis. Mak. 2019, 1, 37–52. [Google Scholar] [CrossRef]
- Fernández-Martínez, J.L.; Álvarez-Machancoses, Ó.; deAndrés-Galiana, E.J.; Bea, G.; Kloczkowski, A. Robust Sampling of Defective Pathways in Alzheimer’s Disease. Implications in Drug Repositioning. Int. J. Mol. Sci. 2020, 21, 3594. [Google Scholar] [CrossRef]
- deAndrés-Galiana, E.J.; Bea, G.; Fernández-Martínez, J.L.; Saligan, L.N. Analysis of defective pathways and drug repositioning in Multiple Sclerosis via machine learning approaches. Comput. Biol. Med. 2019, 115, 103492. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Martínez, J.L.; de Andrés-Galiana, E.J.; Fernández-Ovies, F.J.; Cernea, A.; Kloczkowski, A. Robust Sampling of Defective Pathways in Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 4681. [Google Scholar] [CrossRef] [PubMed]
- Cernea, A.; Fernández-Martínez, J.L.; Deandrés-Galiana, E.J.; Fernández-Ovies, F.J.; Alvarez-Machancoses, O.; Fernández-Muñiz, Z.; Saligan, L.; Sonis, S.T. Robust pathway sampling in phenotype prediction. Application to triple negative breast cancer. BMC Bioinform. 2020, 21, 89. [Google Scholar] [CrossRef]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug. Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Machancoses, Ó.; Fernández-Martínez, J.L. Using artificial intelligence methods to speed up drug discovery. Expert Opin. Drug. Discov. 2019, 14, 769–777. [Google Scholar] [CrossRef]
- Ertl, P. Cheminformatics analysis of organic substituents: Identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups. J. Chem. Inf. Comput. Sci. 2003, 43, 374–380. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning phrase representations using RNN encoder-decoder for statistical machine translation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; Association for Computational Linguistics: Doha, Qatar, 2014; pp. 1724–1734. [Google Scholar] [CrossRef]
- Deng, L.; Yu, D. Deep learning: Methods and applications. Found. Trends Signal Process. 2013, 7, 197–387. [Google Scholar] [CrossRef]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design—A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative Adversarial Nets. Commun. ACM 2020, 63, 139–144. [Google Scholar] [CrossRef]
- Kusner, M.J.; Paige, B.; Miguel Hernández-Lobato, J. Grammar Variational Autoencoder. arXiv 2017, arXiv:1703.01925. [Google Scholar]
- Dai, H.; Tian, Y.; Dai, B.; Skiena, S.; Song, L. Syntax-Directed Variational Autoencoder for Structured Data. arXiv 2018, arXiv:1802.08786. [Google Scholar]
- Segler, M.H.S.; Kogej, T.; Tyrchan, C.; Waller, M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018, 4, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Ramsundar, B.; Liu, B.; Wu, Z.; Verras, A.; Tudor, M.; Sheridan, R.P.; Pande, V. Is Multitask Deep Learning Practical for Pharma? J. Chem. Inf. Model. 2017, 57, 2068–2076. [Google Scholar] [CrossRef]
- Ramsundar, B.; Kearnes, S.; Riley, P.; Webster, D.; Konerding, D.; Pande, V. Massively Multitask Networks for Drug Discovery. arXiv 2015, arXiv:1502.02072. [Google Scholar]
- Unterthiner, T.; Mayr, A.; Unter Klambauer, G.; Steijaert, M.; Wegner, J.K.; Johnson, J.; Hochreiter, S. Deep Learning as an Opportunity in Virtual Screening. Proc. Deep Learn. Workshop NIPS 2014, 27, 1–9. [Google Scholar]
- Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol. Pharm. 2016, 13, 2524–2530. [Google Scholar] [CrossRef]
- Lusci, A.; Pollastri, G.; Baldi, P. Deep architectures and deep learning in chemoinformatics: The prediction of aqueous solubility for drug-like molecules. J. Chem. Inf. Model. 2013, 53, 1563–1575. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.S.; Maltarollo, V.G.; Oliveira, P.R.; Honorio, K.M. Transfer and Multi-task Learning in QSAR Modeling: Advances and Challenges. Front. Pharmacol. 2018, 9, 74. [Google Scholar] [CrossRef]
- Altae-Tran, H.; Ramsundar, B.; Pappu, A.S.; Pande, V. Low Data Drug Discovery with One-Shot Learning. ACS Cent. Sci. 2017, 3, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, K.; Deisenroth, M.P.; Brundage, M.; Bharath, A.A. Deep reinforcement learning: A brief survey. IEEE Signal. Process. Mag. 2017, 34, 26–38. [Google Scholar] [CrossRef]
- Arús-Pous, J.; Blaschke, T.; Ulander, S.; Reymond, J.L.; Chen, H.; Engkvist, O. Exploring the GDB-13 chemical space using deep generative models. J. Cheminform. 2019, 11, 20. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Chemical Data Formats, Fingerprints, and Other Molecular Descriptions for Database Analysis and Searching. In Comprehensive Medicinal Chemistry III; Elsevier Inc.: Amsterdam, The Netherlands, 2017; Volume 3–8, pp. 329–378. [Google Scholar] [CrossRef]
- Sunyaev, S.; Ramensky, V.; Bork, P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000, 16, 198–200. [Google Scholar] [CrossRef]
- Cargill, M.; Altshuler, D.; Ireland, J.; Sklar, P.; Ardlie, K.; Patil, N.; Lane, C.R.; Lim, E.P.; Kalyanaraman, N.; Nemesh, J.; et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 1999, 22, 231–238. [Google Scholar] [CrossRef]
- Collins, F.S.; Brooks, L.D.; Chakravarti, A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998, 8, 1229–1231. [Google Scholar] [CrossRef]
- Altshuler, D.L.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Hurles, M.E.; McVean, G.A. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of predicted and actual consequences of missense mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef]
- Saunders, C.T.; Baker, D. Evaluation of structural and evolutionary contributions to deleterious mutation prediction. J. Mol. Biol. 2002, 322, 891–901. [Google Scholar] [CrossRef]
- Stefl, S.; Nishi, H.; Petukh, M.; Panchenko, A.R.; Alexov, E. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013, 425, 3919–3936. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Chen, J.; Blundell, T.L.; Ascher, D.B. In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci. Rep. 2016, 6, 19848. [Google Scholar] [CrossRef] [PubMed]
- Castaldi, P.J.; Dahabreh, I.J.; Ioannidis, J.P.A. An empirical assessment of validation practices for molecular classifiers. Brief Bioinform. 2011, 12, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Baldi, P.; Brunak, S. Bioinformatics: The Machine Learning Approach, Bradford Books, 2nd ed.; The MIT Press: Cambridge, MA, USA, 2001. [Google Scholar]
- Thusberg, J.; Olatubosun, A.; Vihinen, M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011, 32, 358–368. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genom. Hum. Genet. 2006, 7, 61–80. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
deAndrés-Galiana, E.J.; Fernández-Martínez, J.L.; Fernández-Brillet, L.; Cernea, A.; Kloczkowski, A. Addressing Noise and Estimating Uncertainty in Biomedical Data through the Exploration of Chemical Space. Int. J. Mol. Sci. 2022, 23, 12975. https://doi.org/10.3390/ijms232112975
deAndrés-Galiana EJ, Fernández-Martínez JL, Fernández-Brillet L, Cernea A, Kloczkowski A. Addressing Noise and Estimating Uncertainty in Biomedical Data through the Exploration of Chemical Space. International Journal of Molecular Sciences. 2022; 23(21):12975. https://doi.org/10.3390/ijms232112975
Chicago/Turabian StyledeAndrés-Galiana, Enrique J., Juan Luis Fernández-Martínez, Lucas Fernández-Brillet, Ana Cernea, and Andrzej Kloczkowski. 2022. "Addressing Noise and Estimating Uncertainty in Biomedical Data through the Exploration of Chemical Space" International Journal of Molecular Sciences 23, no. 21: 12975. https://doi.org/10.3390/ijms232112975
APA StyledeAndrés-Galiana, E. J., Fernández-Martínez, J. L., Fernández-Brillet, L., Cernea, A., & Kloczkowski, A. (2022). Addressing Noise and Estimating Uncertainty in Biomedical Data through the Exploration of Chemical Space. International Journal of Molecular Sciences, 23(21), 12975. https://doi.org/10.3390/ijms232112975