
Glymphatic System Dysfunction and Sleep Disturbance May Contribute to the Pathogenesis and Progression of Parkinson’s Disease
 ,
, 
Abstract
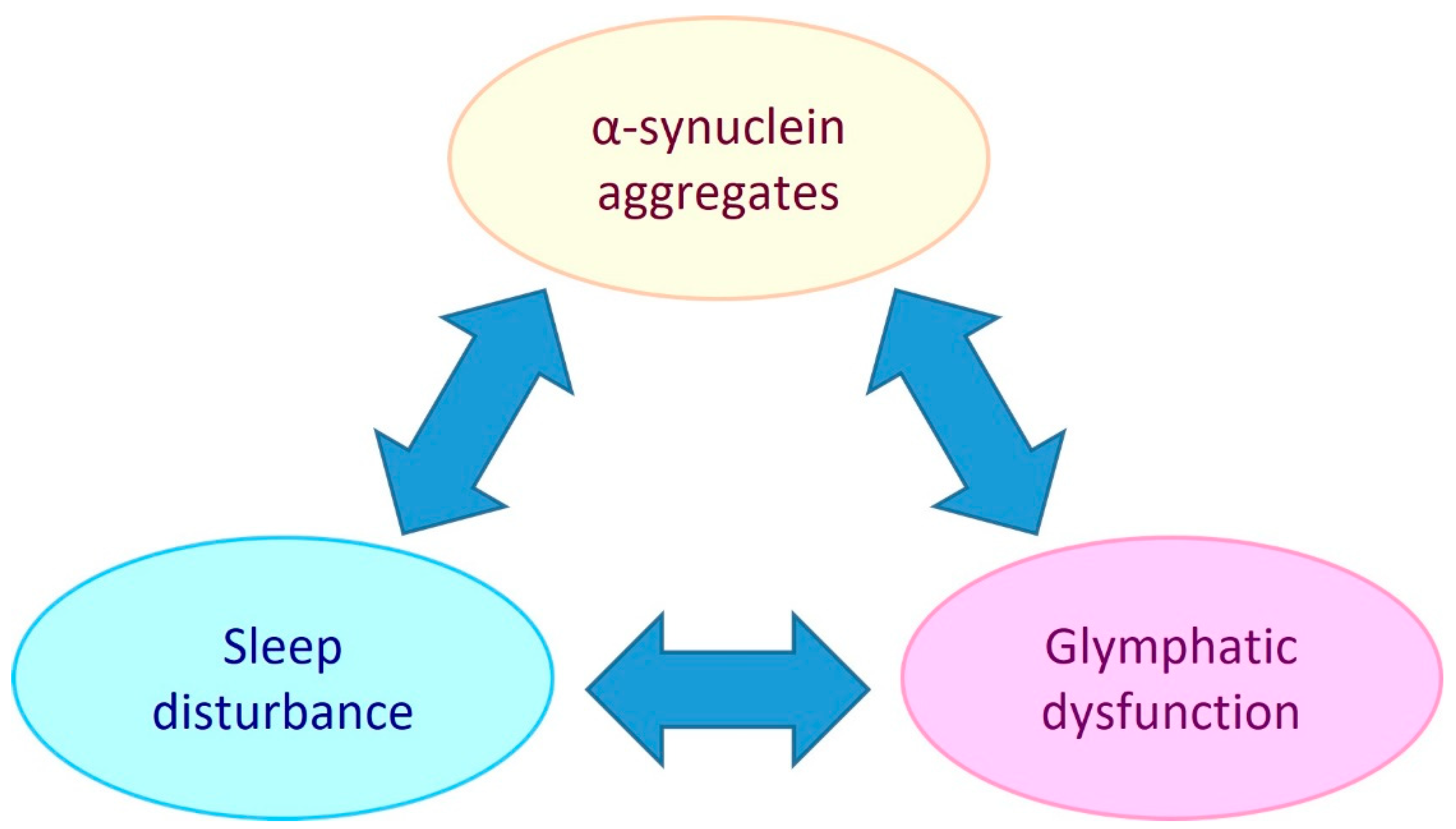
1. Introduction
2. Molecular Pathology
2.1. Role of Alpha-Synuclein
2.2. Neuroinflammation
2.2.1. Microglia
2.2.2. Astroglia
2.3. Aquaporin-4
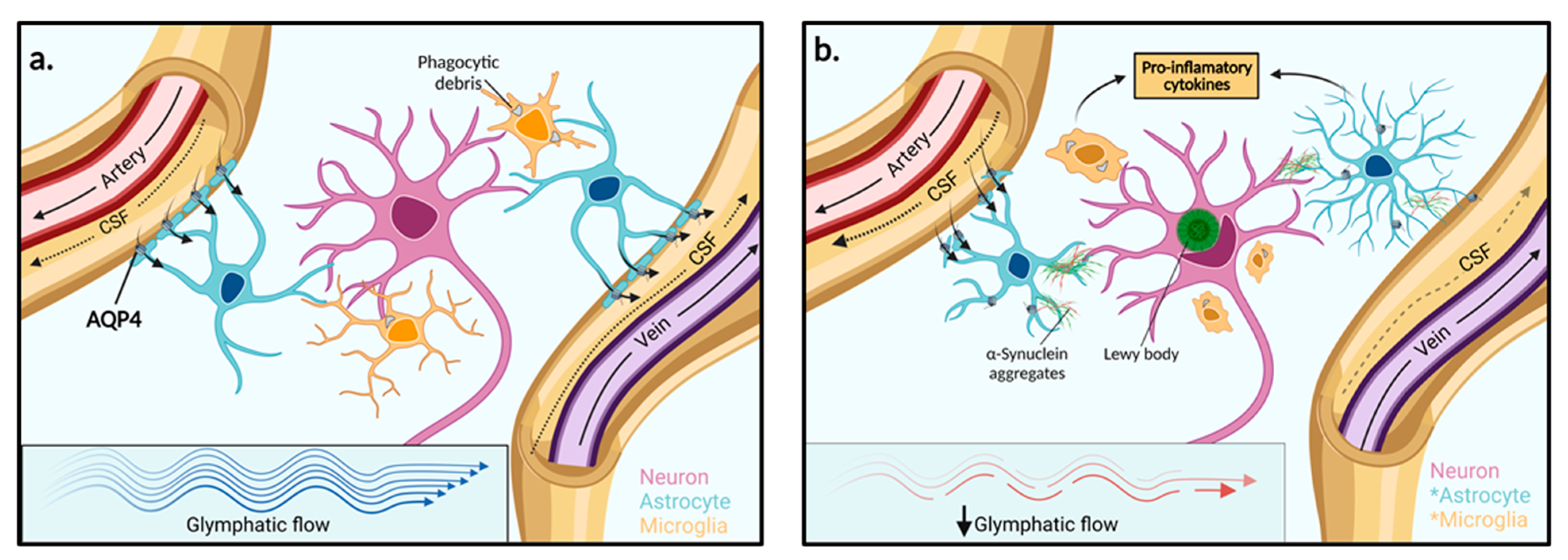
3. The Glymphatic System
3.1. Physiology and Aquaporin-4 Water Channels
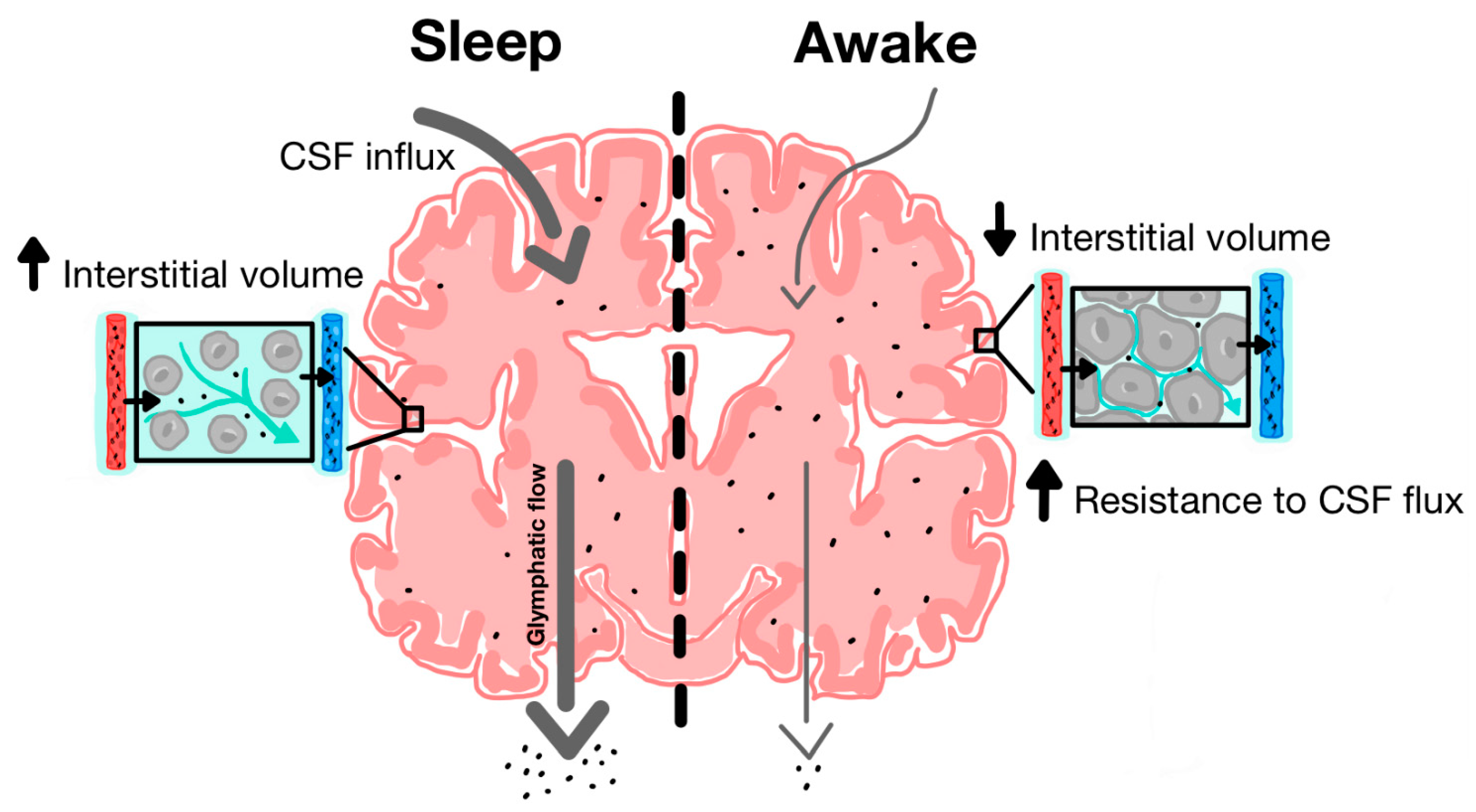
3.2. Physical Forces Driving Fluid Movement
4. Glymphatic System Dysfunction in Parkinson’s Disease Pathology
4.1. Compromised Aquaporin-4 Channels
4.2. Sleep Disruption
5. The Role of Sleep in Parkinson’s Disease
5.1. Lessons from Alzheimer’s Disease
5.2. Circadian Rhythm and Clock Gene Dysfunction
5.3. Sleep Disorders
6. Limitations of the Current Research
7. Potential Therapeutic Approaches
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The Emerging Evidence of the Parkinson Pandemic. J. Parkinson’s Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. Npj Parkinson’s Dis. 2020, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Noyce, A.J.; Bestwick, J.P.; Silveira-Moriyama, L.; Hawkes, C.H.; Giovannoni, G.; Lees, A.J.; Schrag, A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 2012, 72, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Hu, M.T.M. Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease. J. Parkinson’s Dis. 2019, 9, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Zahed, H.; Zuzuarregui, J.R.P.; Gilron, R.E.; Denison, T.; Starr, P.A.; Little, S. The Neurophysiology of Sleep in Parkinson’s Disease. Mov. Disord. 2021, 36, 1526–1542. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s Disease and Its Management: Part 1: Disease Entity, Risk Factors, Pathophysiology, Clinical Presentation, and Diagnosis. Pharm. Ther. 2015, 40, 504–532. [Google Scholar]
- Sonne, J.; Reddy, V.; Beato, M.R. Neuroanatomy, Substantia Nigra; StatPearls Publishing: Tampa, FL, USA, 2021. [Google Scholar]
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Barone, P.; Antonini, A.; Colosimo, C.; Marconi, R.; Morgante, L.; Avarello, T.P.; Bottacchi, E.; Cannas, A.; Ceravolo, G.; Ceravolo, R.; et al. The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov. Disord. 2009, 24, 1641–1649. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20 (Suppl. 1), S62–S67. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, S.; Hughes, R.L.; Peterson, E.; Müller-Oehring, E.M.; Brontë-Stewart, H.M.; Poston, K.L.; Faerman, A.; Bhowmick, C.; Schulte, T. Establishing a framework for neuropathological correlates and glymphatic system functioning in Parkinson’s disease. Neurosci. Biobehav. Rev. 2019, 103, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The Differential Diagnosis and Treatment of Atypical Parkinsonism. Dtsch. Ärzteblatt Int. 2016, 113, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Tsunematsu, T. Association between Sleep, Alzheimer’s, and Parkinson’s Disease. Biology 2021, 10, 1127. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-Synuclein as a Biomarker for Parkinson’s Disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Wegrzynowicz, M.; Bar-On, D.; Calo’, L.; Anichtchik, O.; Iovino, M.; Xia, J.; Ryazanov, S.; Leonov, A.; Giese, A.; Dalley, J.W.; et al. Depopulation of dense α-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol. 2019, 138, 575–595. [Google Scholar] [CrossRef]
- Power, J.H.T.; Barnes, O.L.; Chegini, F. Lewy Bodies and the Mechanisms of Neuronal Cell Death in Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2017, 27, 3–12. [Google Scholar] [CrossRef]
- Andersen, M.S.; Bandres-Ciga, S.; Reynolds, R.H.; Hardy, J.; Ryten, M.; Krohn, L.; Gan-Or, Z.; Holtman, I.R.; Pihlstrøm, L. Heritability Enrichment Implicates Microglia in Parkinson’s Disease Pathogenesis. Ann. Neurol. 2021, 89, 942–951. [Google Scholar] [CrossRef]
- Grozdanov, V.; Bousset, L.; Hoffmeister, M.; Bliederhaeuser, C.; Meier, C.; Madiona, K.; Pieri, L.; Kiechle, M.; McLean, P.J.; Kassubek, J.; et al. Increased Immune Activation by Pathologic α-Synuclein in Parkinson’s Disease. Ann. Neurol. 2019, 86, 593–606. [Google Scholar] [CrossRef]
- Harms, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standaert, D.G. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, C.; Gustin, A.; Birck, C.; Kirchmeyer, M.; Beaume, N.; Felten, P.; Grandbarbe, L.; Heuschling, P.; Heurtaux, T. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS ONE 2016, 11, e0162717. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Ettle, B.; Bruno, A.; Kulinich, A.; Hoffmann, A.-C.; Von Wittgenstein, J.; Winkler, J.; Xiang, W.; Schlachetzki, J.C.M. Alpha-synuclein activates BV2 microglia dependent on its aggregation state. Biochem. Biophys. Res. Commun. 2016, 479, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-T.; Lin, C.; Hsu, C.-T.; Wang, T.-F.; Ke, F.-Y.; Kuo, Y.-M. Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Struct. Funct. 2013, 218, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, B.; Sun, H.; Zhou, Y.; Liu, M.; Ding, J.; Fang, F.; Fan, Y.; Hu, G. Aquaporin-4 deficiency diminishes the differential degeneration of midbrain dopaminergic neurons in experimental Parkinson’s disease. Neurosci. Lett. 2016, 614, 7–15. [Google Scholar] [CrossRef]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M.; Hertz, L. Why are Astrocytes Important? Neurochem. Res. 2015, 40, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Sanz-Blasco, S.; Eisele, Y.S.; Ghatak, S.; Bodhinathan, K.; Akhtar, M.W.; Lynch, W.P.; Piña-Crespo, J.C.; Talantova, M.; Kelly, J.W.; et al. α-Synuclein Oligomers Induce Glutamate Release from Astrocytes and Excessive Extrasynaptic NMDAR Activity in Neurons, Thus Contributing to Synapse Loss. J. Neurosci. 2021, 41, 2264–2273. [Google Scholar] [CrossRef]
- Chou, T.-W.; Chang, N.P.; Krishnagiri, M.; Patel, A.P.; Lindman, M.; Angel, J.P.; Kung, P.-L.; Atkins, C.; Daniels, B.P. Fibrillar α-synuclein induces neurotoxic astrocyte activation via RIP kinase signaling and NF-κB. Cell Death Dis. 2021, 12, 756. [Google Scholar] [CrossRef]
- Plog, B.A.; Nedergaard, M. The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu. Rev. Pathol. 2018, 13, 379–394. [Google Scholar] [CrossRef]
- Zou, W.; Pu, T.; Feng, W.; Lu, M.; Zheng, Y.; Du, R.; Xiao, M.; Hu, G. Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated α-synuclein. Transl. Neurodegener. 2019, 8, 7. [Google Scholar] [CrossRef]
- Cui, H.; Wang, W.; Zheng, X.; Xia, D.; Liu, H.; Qin, C.; Tian, H.; Teng, J. Decreased AQP4 Expression Aggravates α-Synuclein Pathology in Parkinson’s Disease Mice, Possibly via Impaired Glymphatic Clearance. J. Mol. Neurosci. 2021, 71, 2500–2513. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Mestre, H.; Hablitz, L.M.; Xavier, A.L.; Feng, W.; Zou, W.; Pu, T.; Monai, H.; Murlidharan, G.; Castellanos Rivera, R.M.; Simon, M.J.; et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. eLife 2018, 7, e40070. [Google Scholar] [CrossRef]
- Bedussi, B.; Naessens, D.M.P.; De Vos, J.; Olde Engberink, R.; Wilhelmus, M.M.M.; Richard, E.; Ten Hove, M.; Vanbavel, E.; Bakker, E.N.T.P. Enhanced interstitial fluid drainage in the hippocampus of spontaneously hypertensive rats. Sci. Rep. 2017, 7, 744. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Lee, H.; Yu, M.; Feng, T.; Logan, J.; Nedergaard, M.; Benveniste, H. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J. Clin. Investig. 2013, 123, 1299–1309. [Google Scholar] [CrossRef]
- Faghih, M.M.; Sharp, M.K. Is bulk flow plausible in perivascular, paravascular and paravenous channels? Fluids Barriers CNS 2018, 15, 17. [Google Scholar] [CrossRef]
- Jin, B.-J.; Smith, A.J.; Verkman, A.S. Spatial model of convective solute transport in brain extracellular space does not support a “glymphatic” mechanism. J. Gen. Physiol. 2016, 148, 489–501. [Google Scholar] [CrossRef]
- Smith, A.J.; Yao, X.; Dix, J.A.; Jin, B.-J.; Verkman, A.S. Test of the ‘glymphatic’ hypothesis demonstrates diffusive and aquaporin-4-independent solute transport in rodent brain parenchyma. eLife 2017, 6, e27679. [Google Scholar] [CrossRef]
- Rey, J.; Sarntinoranont, M. Pulsatile flow drivers in brain parenchyma and perivascular spaces: A resistance network model study. Fluids Barriers CNS 2018, 15, 20. [Google Scholar] [CrossRef]
- Kiviniemi, V.; Wang, X.; Korhonen, V.; Keinänen, T.; Tuovinen, T.; Autio, J.; LeVan, P.; Keilholz, S.; Zang, Y.F.; Hennig, J.; et al. Ultra-fast magnetic resonance encephalography of physiological brain activity-Glymphatic pulsation mechanisms? J. Cereb. Blood Flow Metab. 2016, 36, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Miyazaki, M.; Yamashita, Y.; Ouyang, C.; Yui, M.; Nakahashi, M.; Shimizu, S.; Aoki, I.; Morohoshi, Y.; McComb, J.G. Influence of respiration on cerebrospinal fluid movement using magnetic resonance spin labeling. Fluids Barriers CNS 2013, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.; De Zélicourt, D.; Kurtcuoglu, V. Glymphatic solute transport does not require bulk flow. Sci. Rep. 2016, 6, 38635. [Google Scholar] [CrossRef] [PubMed]
- Holter, K.E.; Kehlet, B.; Devor, A.; Sejnowski, T.J.; Dale, A.M.; Omholt, S.W.; Ottersen, O.P.; Nagelhus, E.A.; Mardal, K.-A.; Pettersen, K.H. Interstitial solute transport in 3D reconstructed neuropil occurs by diffusion rather than bulk flow. Proc. Natl. Acad. Sci. USA 2017, 114, 9894–9899. [Google Scholar] [CrossRef]
- Koundal, S.; Elkin, R.; Nadeem, S.; Xue, Y.; Constantinou, S.; Sanggaard, S.; Liu, X.; Monte, B.; Xu, F.; Van Nostrand, W.; et al. Optimal Mass Transport with Lagrangian Workflow Reveals Advective and Diffusion Driven Solute Transport in the Glymphatic System. Sci. Rep. 2020, 10, 1990. [Google Scholar] [CrossRef]
- Ray, L.; Iliff, J.J.; Heys, J.J. Analysis of convective and diffusive transport in the brain interstitium. Fluids Barriers CNS 2019, 16, 6. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A.; et al. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef]
- Chen, H.; Wan, H.; Zhang, M.; Wardlaw, J.M.; Feng, T.; Wang, Y. Perivascular space in Parkinson’s disease: Association with CSF amyloid/tau and cognitive decline. Parkinsonism Relat. Disord. 2022, 95, 70–76. [Google Scholar] [CrossRef]
- Chung, S.J.; Yoo, H.S.; Shin, N.-Y.; Park, Y.W.; Lee, H.S.; Hong, J.-M.; Kim, Y.J.; Lee, S.-K.; Lee, P.H.; Sohn, Y.H. Perivascular Spaces in the Basal Ganglia and Long-term Motor Prognosis in Newly Diagnosed Parkinson Disease. Neurology 2021, 96, e2121–e2131. [Google Scholar] [CrossRef]
- Donahue, E.K.; Murdos, A.; Jakowec, M.W.; Sheikh-Bahaei, N.; Toga, A.W.; Petzinger, G.M.; Sepehrband, F. Global and Regional Changes in Perivascular Space in Idiopathic and Familial Parkinson’s Disease. Mov. Disord. 2021, 36, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, Z.; Chen, J.; Zhang, M.; Yang, Y.; Huang, P. Dilated Perivascular Space in the Midbrain May Reflect Dopamine Neuronal Degeneration in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Li, S.; Li, C.; Wang, R.; Chen, M.; Chen, H.; Su, W. Diffusion Tensor Imaging Along the Perivascular Space Index in Different Stages of Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 773951. [Google Scholar] [CrossRef]
- McKnight, C.D.; Trujillo, P.; Lopez, A.M.; Petersen, K.; Considine, C.; Lin, Y.-C.; Yan, Y.; Kang, H.; Donahue, M.J.; Claassen, D.O. Diffusion along perivascular spaces reveals evidence supportive of glymphatic function impairment in Parkinson disease. Parkinsonism Relat. Disord. 2021, 89, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Shin, N.Y.; Chung, S.J.; Kim, J.; Lim, S.M.; Lee, P.H.; Lee, S.K.; Ahn, K.J. Magnetic Resonance Imaging-Visible Perivascular Spaces in Basal Ganglia Predict Cognitive Decline in Parkinson’s Disease. Mov. Disord. 2019, 34, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Brown, G.L.; Zhu, Y.; Belkin-Rosen, A.E.; Lewis, M.M.; Du, G.; Gu, Y.; Eslinger, P.J.; Mailman, R.B.; Huang, X.; et al. Decoupling of Global Brain Activity and Cerebrospinal Fluid Flow in Parkinson’s Disease Cognitive Decline. Mov. Disord. 2021, 36, 2066–2076. [Google Scholar] [CrossRef]
- Fultz, N.E.; Bonmassar, G.; Setsompop, K.; Stickgold, R.A.; Rosen, B.R.; Polimeni, J.R.; Lewis, L.D. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science 2019, 366, 628–631. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Tuura, R.O.G.; Volk, C.; Callaghan, F.; Jaramillo, V.; Huber, R. Sleep-related and diurnal effects on brain diffusivity and cerebrospinal fluid flow. NeuroImage 2021, 241, 118420. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Vinitsky, H.S.; Sun, Q.; Stæger, F.F.; Sigurdsson, B.; Mortensen, K.N.; Lilius, T.O.; Nedergaard, M. Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci. Adv. 2019, 5, eaav5447. [Google Scholar] [CrossRef]
- Bishir, M.; Bhat, A.; Essa, M.M.; Ekpo, O.; Ihunwo, A.O.; Veeraraghavan, V.P.; Mohan, S.K.; Mahalakshmi, A.M.; Ray, B.; Tuladhar, S.; et al. Sleep Deprivation and Neurological Disorders. BioMed Res. Int. 2020, 2020, 5764017. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Holtzman, D.M. Bidirectional relationship between sleep and Alzheimer’s disease: Role of amyloid, tau, and other factors. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2020, 45, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.P.; Gamaldo, A.A.; An, Y.; Wu, M.N.; Simonsick, E.M.; Bilgel, M.; Zhou, Y.; Wong, D.F.; Ferrucci, L.; Resnick, S.M. Self-reported Sleep and β-Amyloid Deposition in Community-Dwelling Older Adults. JAMA Neurol. 2013, 70, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Bubu, O.M.; Brannick, M.; Mortimer, J.; Umasabor-Bubu, O.; Sebastião, Y.V.; Wen, Y.; Schwartz, S.; Borenstein, A.R.; Wu, Y.; Morgan, D.; et al. Sleep, Cognitive impairment, and Alzheimer’s disease: A Systematic Review and Meta-Analysis. Sleep 2017, 40, zsw032. [Google Scholar] [CrossRef]
- Shokri-Kojori, E.; Wang, G.-J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.; Reddy, A.; Barker, R. Sleep and Circadian Rhythm Regulation in Early Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 2015, 86, e4. [Google Scholar] [CrossRef]
- Ikeda, R.; Tsuchiya, Y.; Koike, N.; Umemura, Y.; Inokawa, H.; Ono, R.; Inoue, M.; Sasawaki, Y.; Grieten, T.; Okubo, N.; et al. REV-ERBα and REV-ERBβ function as key factors regulating Mammalian Circadian Output. Sci. Rep. 2019, 9, 10171. [Google Scholar] [CrossRef]
- Li, H.; Song, S.; Wang, Y.; Huang, C.; Zhang, F.; Liu, J.; Hong, J.-S. Low-Grade Inflammation Aggravates Rotenone Neurotoxicity and Disrupts Circadian Clock Gene Expression in Rats. Neurotox. Res. 2019, 35, 421–431. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, D.; Liu, W.; Li, S.; Chen, J.; Shen, Y.; Wang, F.; Hu, L.-F.; Liu, C.-F. Disruption of the Circadian Clock Alters Antioxidative Defense via the SIRT1-BMAL1 Pathway in 6-OHDA-Induced Models of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2018, 2018, 4854732. [Google Scholar] [CrossRef]
- Ono, K.; Mochizuki, H.; Ikeda, T.; Nihira, T.; Takasaki, J.-I.; Teplow, D.B.; Yamada, M. Effect of melatonin on α-synuclein self-assembly and cytotoxicity. Neurobiol. Aging 2012, 33, 2172–2185. [Google Scholar] [CrossRef]
- Hsiao, Y.H.; Chen, Y.T.; Tseng, C.M.; Wu, L.A.; Perng, D.W.; Chen, Y.M.; Chen, T.J.; Chang, S.C.; Chou, K.T. Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: A population-based cohort study. J. Sleep Res. 2017, 26, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Lysen, T.S.; Darweesh, S.K.L.; Ikram, M.K.; Luik, A.I.; Ikram, M.A. Sleep and risk of parkinsonism and Parkinson’s disease: A population-based study. Brain A J. Neurol. 2019, 142, 2013–2022. [Google Scholar] [CrossRef]
- Sohail, S.; Yu, L.; Schneider, J.A.; Bennett, D.A.; Buchman, A.S.; Lim, A.S.P. Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Durcan, R.; Wiblin, L.; Lawson, R.A.; Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Brooks, D.J.; Pavese, N.; Burn, D.J.; ICICLE-PD Study Group. Prevalence and duration of non-motor symptoms in prodromal Parkinson’s disease. Eur. J. Neurol. 2019, 26, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Belfer, S.J.; Bashaw, A.G.; Perlis, M.L.; Kayser, M.S. A Drosophila model of sleep restriction therapy for insomnia. Mol. Psychiatry 2021, 26, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Spurling, B.C.; Bollu, P.C. Chronic Insomnia; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Zou, G.; Li, Y.; Liu, J.; Zhou, S.; Xu, J.; Qin, L.; Shao, Y.; Yao, P.; Sun, H.; Zou, Q.; et al. Altered thalamic connectivity in insomnia disorder during wakefulness and sleep. Hum. Brain Mapp. 2021, 42, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Si, X.L.; Gu, L.Y.; Song, Z.; Zhou, C.; Fang, Y.; Jin, C.Y.; Wu, J.J.; Gao, T.; Guo, T.; Guan, X.J.; et al. Different Perivascular Space Burdens in Idiopathic Rapid Eye Movement Sleep Behavior Disorder and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 580853. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.H.; Duncan, G.W.; Firbank, M.J.; Yarnall, A.J.; Khoo, T.K.; Burn, D.J.; O’Brien, J.T. Rapid eye movement sleep behavior disorder in Parkinson’s disease: Magnetic resonance imaging study. Mov. Disord. 2013, 28, 832–836. [Google Scholar] [CrossRef]
- Shrestha, N.; Abe, R.A.M.; Masroor, A.; Khorochkov, A.; Prieto, J.; Singh, K.B.; Nnadozie, M.C.; Abdal, M.; Mohammed, L. The Correlation Between Parkinson’s Disease and Rapid Eye Movement Sleep Behavior Disorder: A Systematic Review. Cureus 2021, 13, e17026. [Google Scholar] [CrossRef]
- Iranzo, A.; Tolosa, E.; Gelpi, E.; Molinuevo, J.L.; Valldeoriola, F.; Serradell, M.; Sanchez-Valle, R.; Vilaseca, I.; Lomeña, F.; Vilas, D.; et al. Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol. 2013, 12, 443–453. [Google Scholar] [CrossRef]
- Lavault, S.; Leu-Semenescu, S.; du Montcel, S.T.; de Cock, V.C.; Vidailhet, M.; Arnulf, I. Does clinical rapid eye movement behavior disorder predict worse outcomes in Parkinson’s disease? J. Neurol. 2010, 257, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Boot, B.P.; Boeve, B.F.; Roberts, R.O.; Ferman, T.J.; Geda, Y.E.; Pankratz, V.S.; Ivnik, R.J.; Smith, G.; McDade, E.; Bsc, T.J.H.C.; et al. Probable rapid eye movement sleep behavior disorder increases risk for mild cognitive impairment and Parkinson disease: A population-based study. Ann. Neurol. 2012, 71, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Schenck, C.H.; Boeve, B.F.; Mahowald, M.W. Delayed emergence of a parkinsonian disorder or dementia in 81% of older men initially diagnosed with idiopathic rapid eye movement sleep behavior disorder: A 16-year update on a previously reported series. Sleep Med. 2013, 14, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain A J. Neurol. 2019, 142, 744–759. [Google Scholar] [CrossRef]
- Riemann, D.; Krone, L.B.; Wulff, K.; Nissen, C. Sleep, insomnia, and depression. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2020, 45, 74–89. [Google Scholar] [CrossRef]
- Wang, S.; Mao, S.; Xiang, D.; Fang, C. Association between depression and the subsequent risk of Parkinson’s disease: A meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 86, 186–192. [Google Scholar] [CrossRef]
- Ylikoski, A.; Martikainen, K.; Sieminski, M.; Partinen, M. Parkinson’s disease and insomnia. Neurol. Sci. 2015, 36, 2003–2010. [Google Scholar] [CrossRef]
- Tran, A.A.; De Smet, M.; Grant, G.D.; Khoo, T.K.; Pountney, D.L. Investigating the Convergent Mechanisms between Major Depressive Disorder and Parkinson’s Disease. Complex Psychiatry 2021, 6, 47–61. [Google Scholar] [CrossRef]
- Greely, H.T. Human Brain Surrogates Research: The Onrushing Ethical Dilemma. Am. J. Bioeth. 2021, 21, 34–45. [Google Scholar] [CrossRef]
- Farahany, N.A.; Greely, H.T.; Hyman, S.; Koch, C.; Grady, C.; Pașca, S.P.; Sestan, N.; Arlotta, P.; Bernat, J.L.; Ting, J.; et al. The ethics of experimenting with human brain tissue. Nature 2018, 556, 429–432. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed]
- Baumann-Vogel, H.; Imbach, L.L.; Sürücü, O.; Stieglitz, L.; Waldvogel, D.; Baumann, C.R.; Werth, E. The Impact of Subthalamic Deep Brain Stimulation on Sleep–Wake Behavior: A Prospective Electrophysiological Study in 50 Parkinson Patients. Sleep 2017, 40, zsx033. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-H.; Kim, H.-J.; Lee, J.-Y.; Yoo, D.; Im, J.H.; Paek, S.H.; Jeon, B. Long-term effects of bilateral subthalamic nucleus stimulation on sleep in patients with Parkinson’s disease. PLoS ONE 2019, 14, e0221219. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, L.; Chen, W.; Ling, Y.; Xu, M.; Li, Y.; Yang, C.; Liu, J.; Chen, L.; Jiang, N. Subthalamic nucleus deep brain stimulation improves sleep in Parkinson’s disease patients: A retrospective study and a meta-analysis. Sleep Med. 2020, 74, 301–306. [Google Scholar] [CrossRef]
- Pastukhov, Y.F.; Simonova, V.V.; Shemyakova, T.S.; Guzeev, M.A.; Polonik, S.G.; Ekimova, I.V. U-133, a heat shock proteins inducer, precludes sleep disturbances in a model of the preclinical stage of Parkinson’s disease in aged rats. Adv. Gerontol. 2019, 32, 935–940. [Google Scholar]
- Ahn, J.H.; Kim, M.; Park, S.; Jang, W.; Park, J.; Oh, E.; Cho, J.W.; Kim, J.S.; Youn, J. Prolonged-release melatonin in Parkinson’s disease patients with a poor sleep quality: A randomized trial. Parkinsonism Relat. Disord. 2020, 75, 50–54. [Google Scholar] [CrossRef]
- Delgado-Lara, D.L.; González-Enríquez, G.V.; Torres-Mendoza, B.M.; González-Usigli, H.; Cárdenas-Bedoya, J.; Macías-Islas, M.A.; de la Rosa, A.C.; Jiménez-Delgado, A.; Pacheco-Moisés, F.; Cruz-Serrano, J.A.; et al. Effect of melatonin administration on the PER1 and BMAL1 clock genes in patients with Parkinson’s disease. Biomed. Pharmacother. 2020, 129, 110485. [Google Scholar] [CrossRef]
- Videnovic, A. Management of sleep disorders in Parkinson’s disease and multiple system atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 659–668. [Google Scholar] [CrossRef]
- Lin, F.; Su, Y.; Weng, Y.; Lin, X.; Weng, H.; Cai, G.; Cai, G. The effects of bright light therapy on depression and sleep disturbances in patients with Parkinson’s disease: A systematic review and meta-analysis of randomized controlled trials. Sleep Med. 2021, 83, 280–289. [Google Scholar] [CrossRef]
- Martino, J.K.; Freelance, C.B.; Willis, G.L. The effect of light exposure on insomnia and nocturnal movement in Parkinson’s disease: An open label, retrospective, longitudinal study. Sleep Med. 2018, 44, 24–31. [Google Scholar] [CrossRef]
- Xue, X.; Zhang, W.; Zhu, J.; Chen, X.; Zhou, S.; Xu, Z.; Hu, G.; Su, C. Aquaporin-4 deficiency reduces TGF-β1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J. Cell. Mol. Med. 2019, 23, 2568–2582. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Dai, S.; Jin, C.; Si, X.; Gu, L.; Song, Z.; Gao, T.; Chen, Y.; Yan, Y.; Yin, X.; et al. Aquaporin-4 Polymorphisms Are Associated With Cognitive Performance in Parkinson’s Disease. Front. Aging Neurosci. 2022, 13, 740491. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Bidirectional Relationships | Article | Description |
|---|---|---|
| Glymphatic system dysfunction in PD | Chen and colleagues [50] | Basal ganglia perivascular spaces were associated with cognitive decline in PD patients at a 3-year follow up. BG-PVS and toxic protein levels in the CSF were significantly predictive of cognitive decline at 3-years. |
| Chung and colleagues [51] | PD patients with enlarged basal ganglia perivascular spaces were older, had greater small vessel disease burden, more severely decreased dopamine transporter availability and higher freezing of gait risk compared to PD patients without enlarged PVS. | |
| Cui and colleagues [33] | Decreased aquaporin-4 expression in mice leads to further alpha-synuclein accumulation, dopaminergic neurodegeneration, impaired motor ability and glymphatic transport. | |
| Ma and colleagues [54] | Late-stage PD patients had significantly lower ALPS scores than normal controls. | |
| McKnight and colleagues [55] | ALPS-index was more reduced in PD patients compared to essential tremor patients. ALPS-index decreased with age. | |
| Li and colleagues [53] | Compared to controls, PD patients had more dilated perivascular spaces, greater expression of tau and decreased dopamine transporter binding. | |
| Park and colleagues [56] | PD patients experienced higher severity of basal ganglia perivascular spaces, white matter hypersensitivity, higher levodopa-equivalent dose, hypertension, and low mini-mental state examination scores. These factors were independent positive predictors of cognitive decline. | |
| Zou and colleagues [32] | Glymphatic CSF influx was reduced in A53T mice, and this effect was also accompanied by alpha-synuclein aggregation and AQP4 depolarisation. Disruption of meningeal lymphatic flow exacerbated alpha-synuclein pathology as well as induced neuroinflammation and dopaminergic neurodegeneration. | |
| Iliff and colleagues [34] | There is perivascular CSF influx, ISF efflux and clearance throughout the brain. Aquaporin-4 channels facilitate fluid clearance in the parenchyma as well as interstitial amyloid-beta clearance. | |
| Role of sleep in Glymphatic clearance | Xie and colleagues [59] | Natural sleep or anaesthesia was associated with a 60% increase in interstitial space volume which contributed to a significant increase in convective CSF-ISF exchange and amyloid-beta clearance. |
| Sleep disturbance and PD pathology | Si and colleagues [79] | REM sleep behaviour disorder patients had a higher PVS burden than PD patients. |
| Lysen and colleagues [73] | Worsening sleep quality and reduction in sleep duration were found to be associated with increased PD risk in adults in the next 6 years. | |
| Hsiao and colleagues [72] | Non-apnoea sleep disorders, particularly chronic insomnia, are associated with a higher risk of developing PD. | |
| Sohail and colleagues [74] | Increased sleep fragmentation was associated with Lewy body pathology, substantia nigra neurodegeneration and PD diagnosis risk. | |
| Breen and colleagues [67] | PD patients experienced increased sleep latency, reduced sleep efficiency, REM sleep and melatonin levels, elevated cortisol and abnormal Bmal1 expression compared to controls. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massey, A.; Boag, M.K.; Magnier, A.; Bispo, D.P.C.F.; Khoo, T.K.; Pountney, D.L. Glymphatic System Dysfunction and Sleep Disturbance May Contribute to the Pathogenesis and Progression of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 12928. https://doi.org/10.3390/ijms232112928
Massey A, Boag MK, Magnier A, Bispo DPCF, Khoo TK, Pountney DL. Glymphatic System Dysfunction and Sleep Disturbance May Contribute to the Pathogenesis and Progression of Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(21):12928. https://doi.org/10.3390/ijms232112928
Chicago/Turabian StyleMassey, Andie, Matthew K. Boag, Annie Magnier, Dharah P. C. F. Bispo, Tien K. Khoo, and Dean L. Pountney. 2022. "Glymphatic System Dysfunction and Sleep Disturbance May Contribute to the Pathogenesis and Progression of Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 21: 12928. https://doi.org/10.3390/ijms232112928
APA StyleMassey, A., Boag, M. K., Magnier, A., Bispo, D. P. C. F., Khoo, T. K., & Pountney, D. L. (2022). Glymphatic System Dysfunction and Sleep Disturbance May Contribute to the Pathogenesis and Progression of Parkinson’s Disease. International Journal of Molecular Sciences, 23(21), 12928. https://doi.org/10.3390/ijms232112928