
PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination
 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
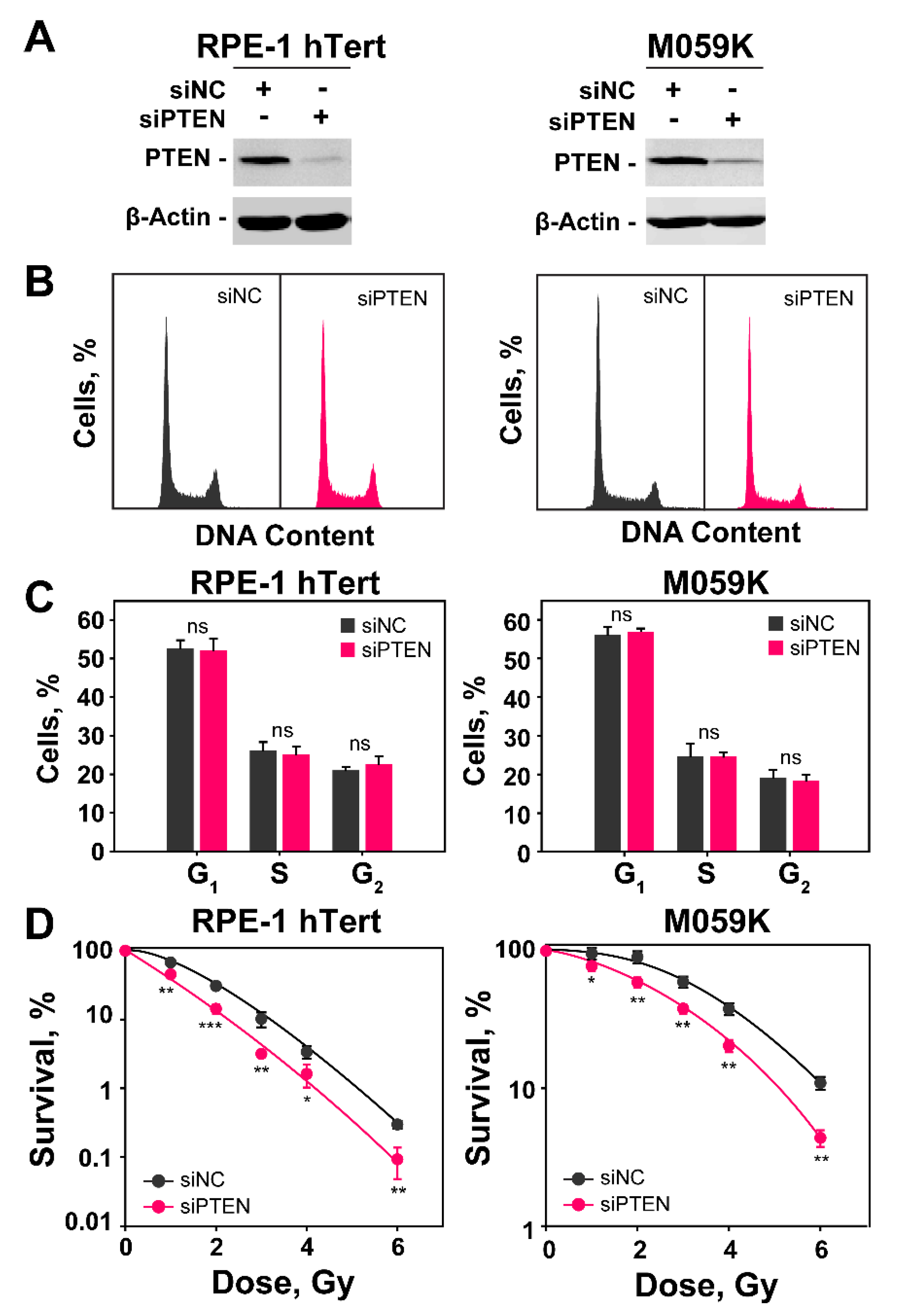
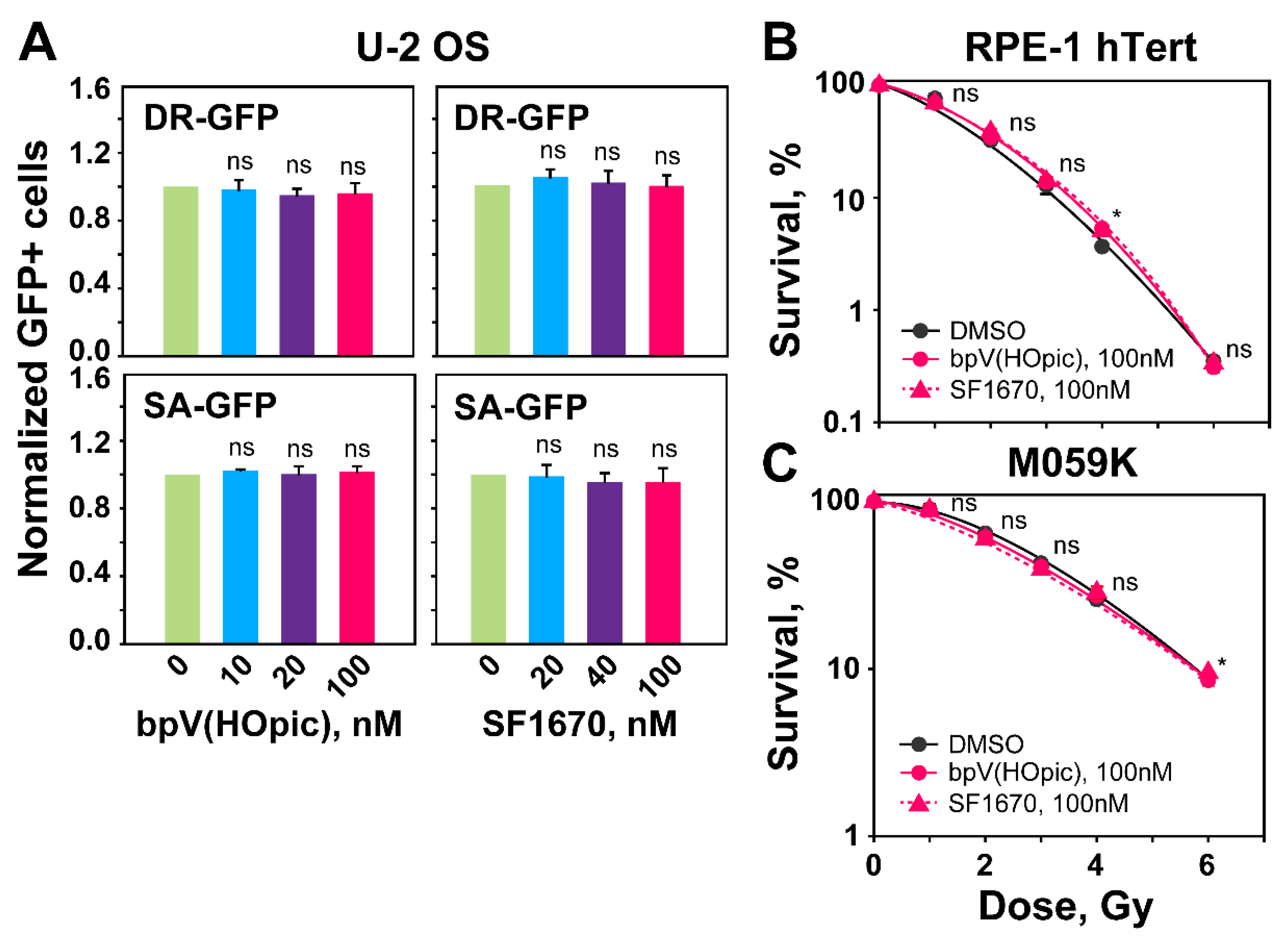
2.1. PTEN Knockdown Radiosensitizes Normal Epithelial RPE-1 and Glioblastoma M059K Cells
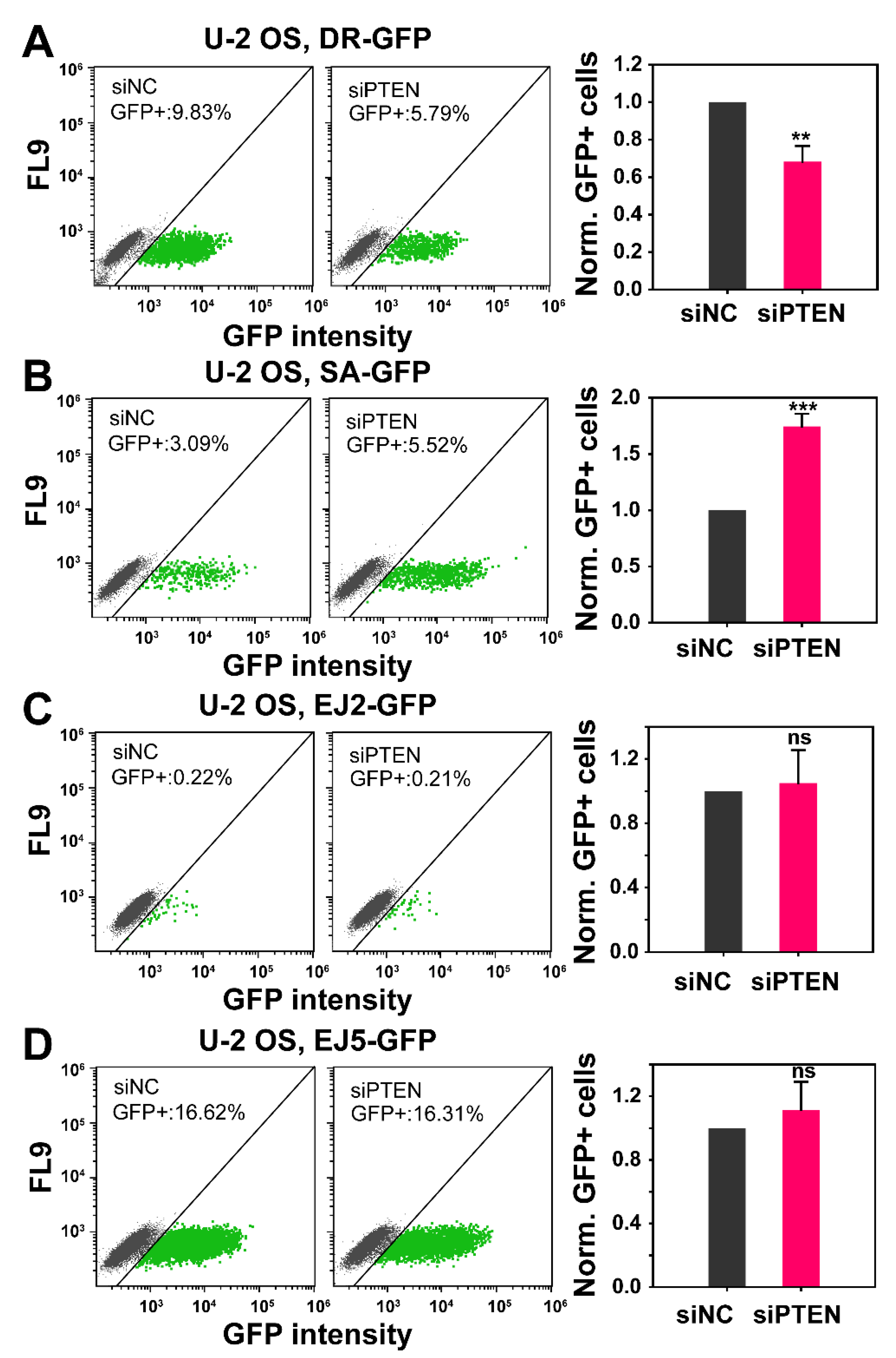
2.2. The Role of PTEN in HR, SSA, c-NHEJ and alt-EJ
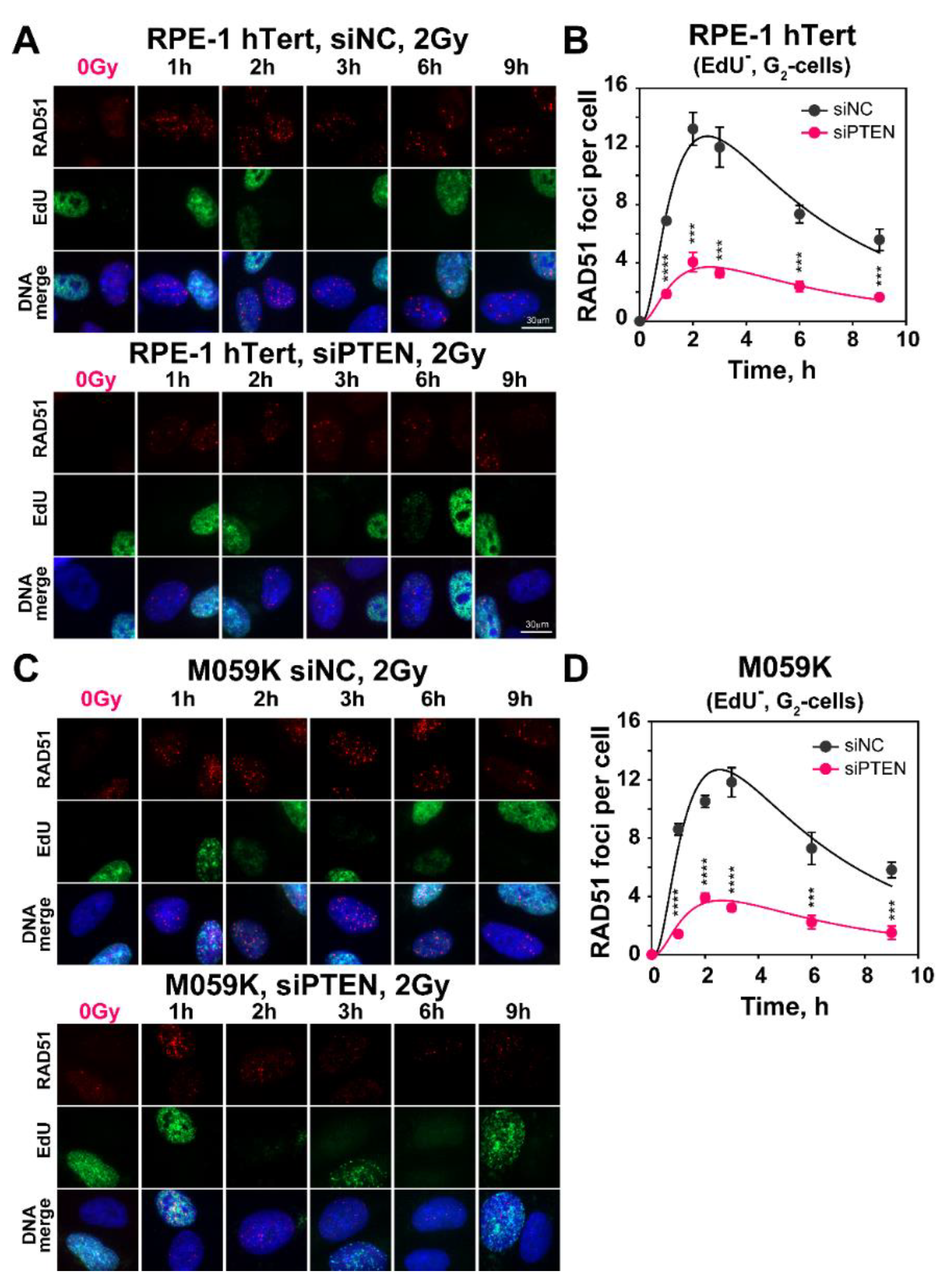
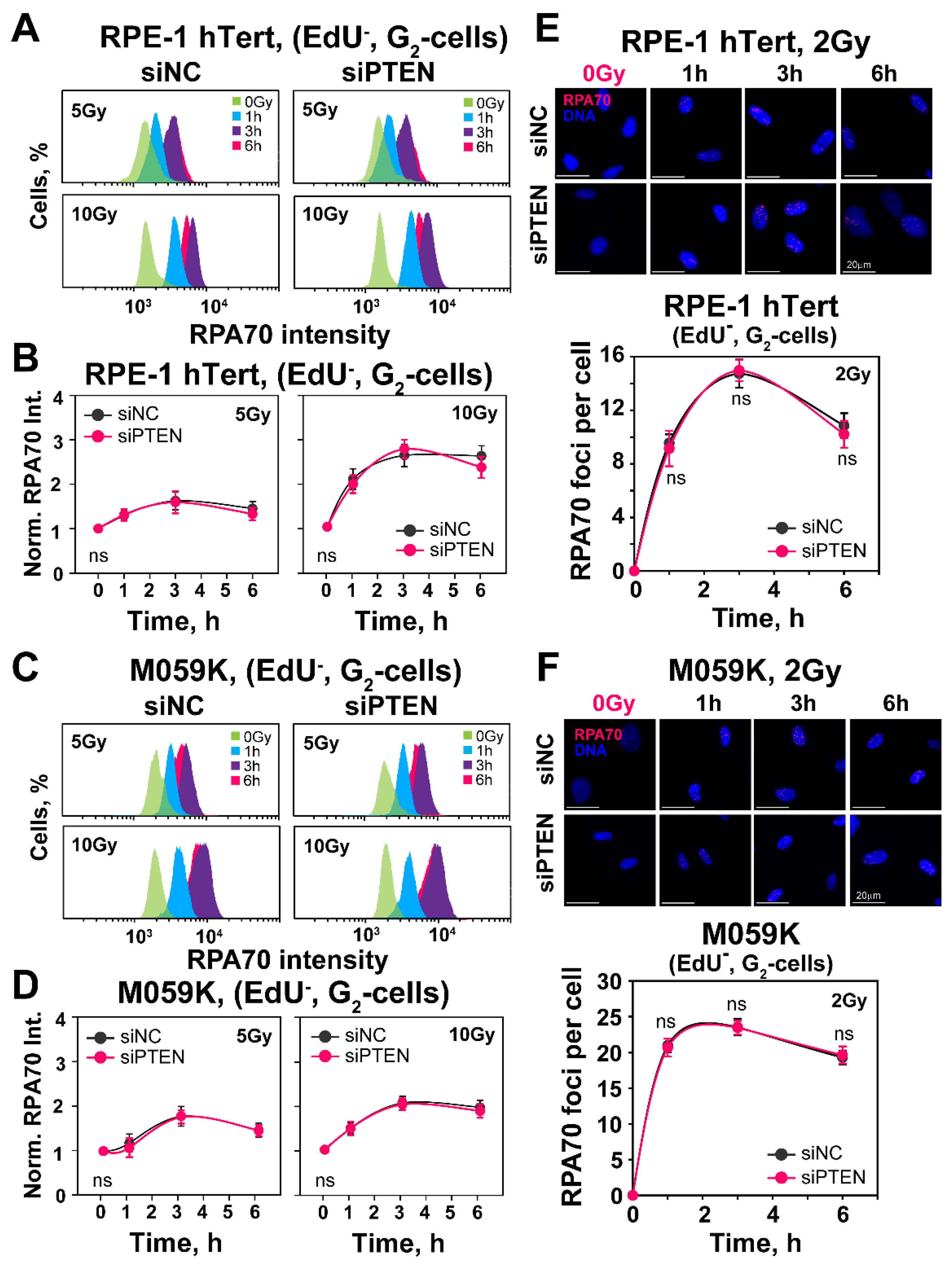
2.3. Effect of PTEN on RAD51 Foci Formation
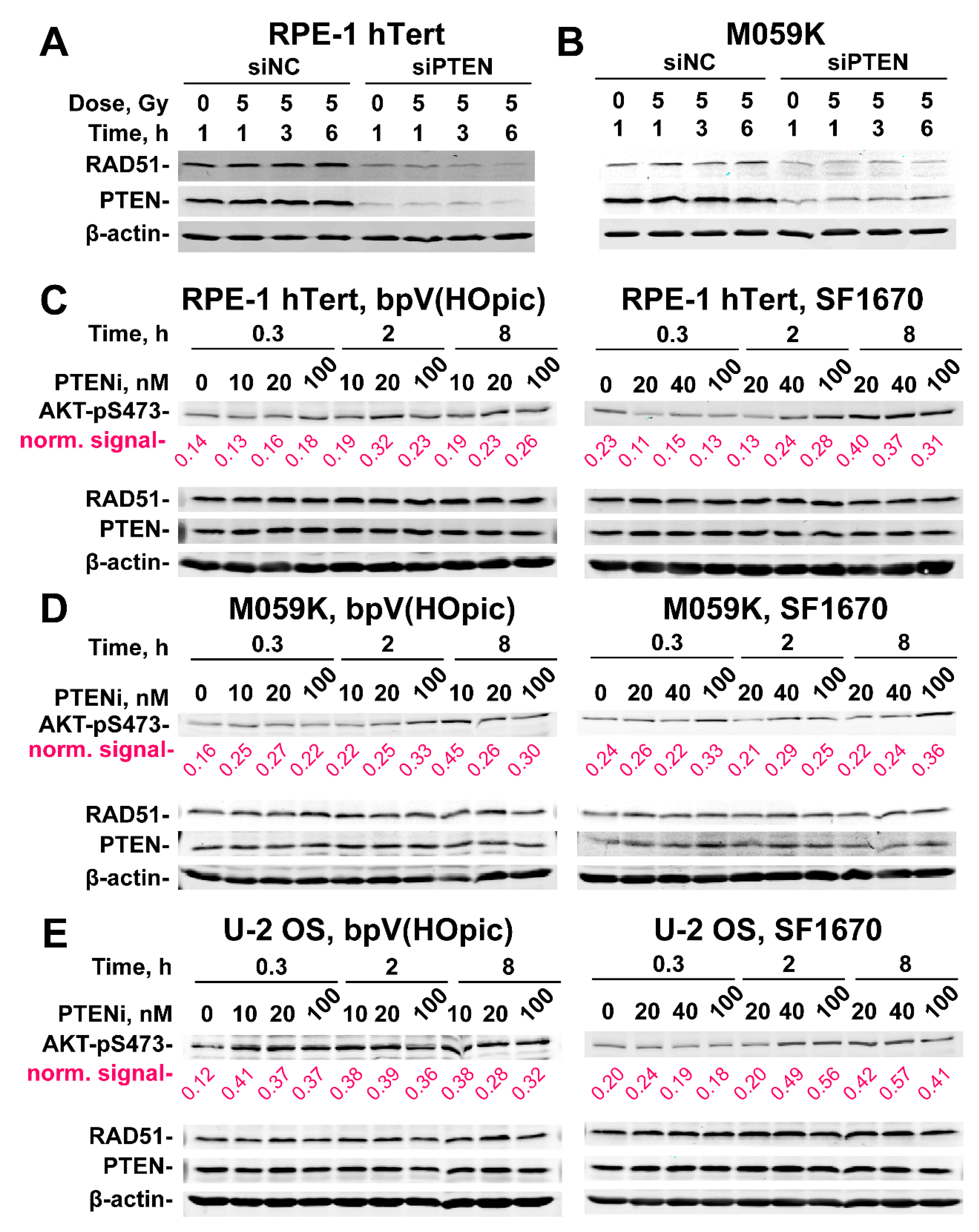
2.4. PTEN Knockdown Suppresses RAD51 Expression
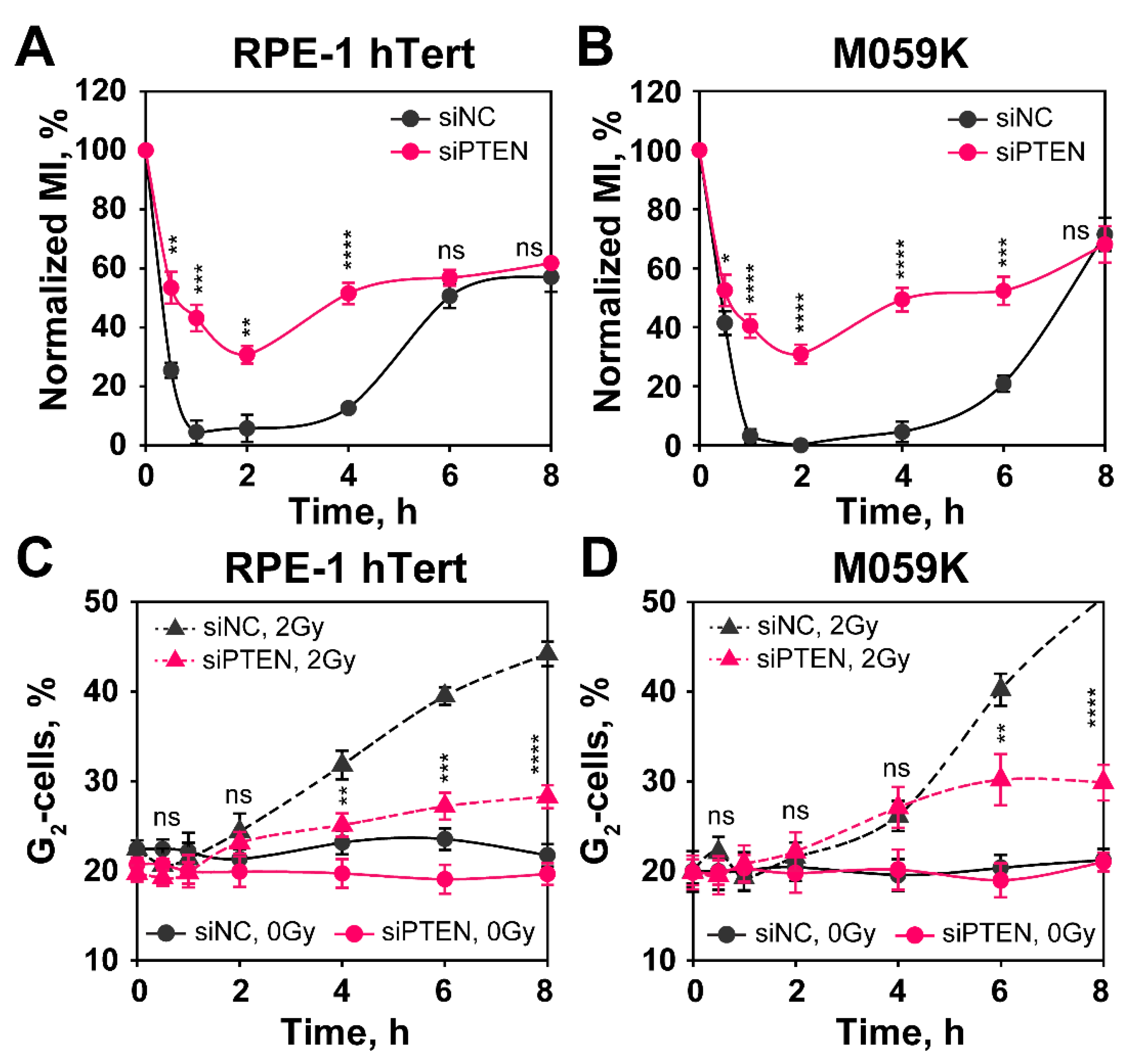
2.5. Effect of PTEN on the Regulation of G2-Checkpoint
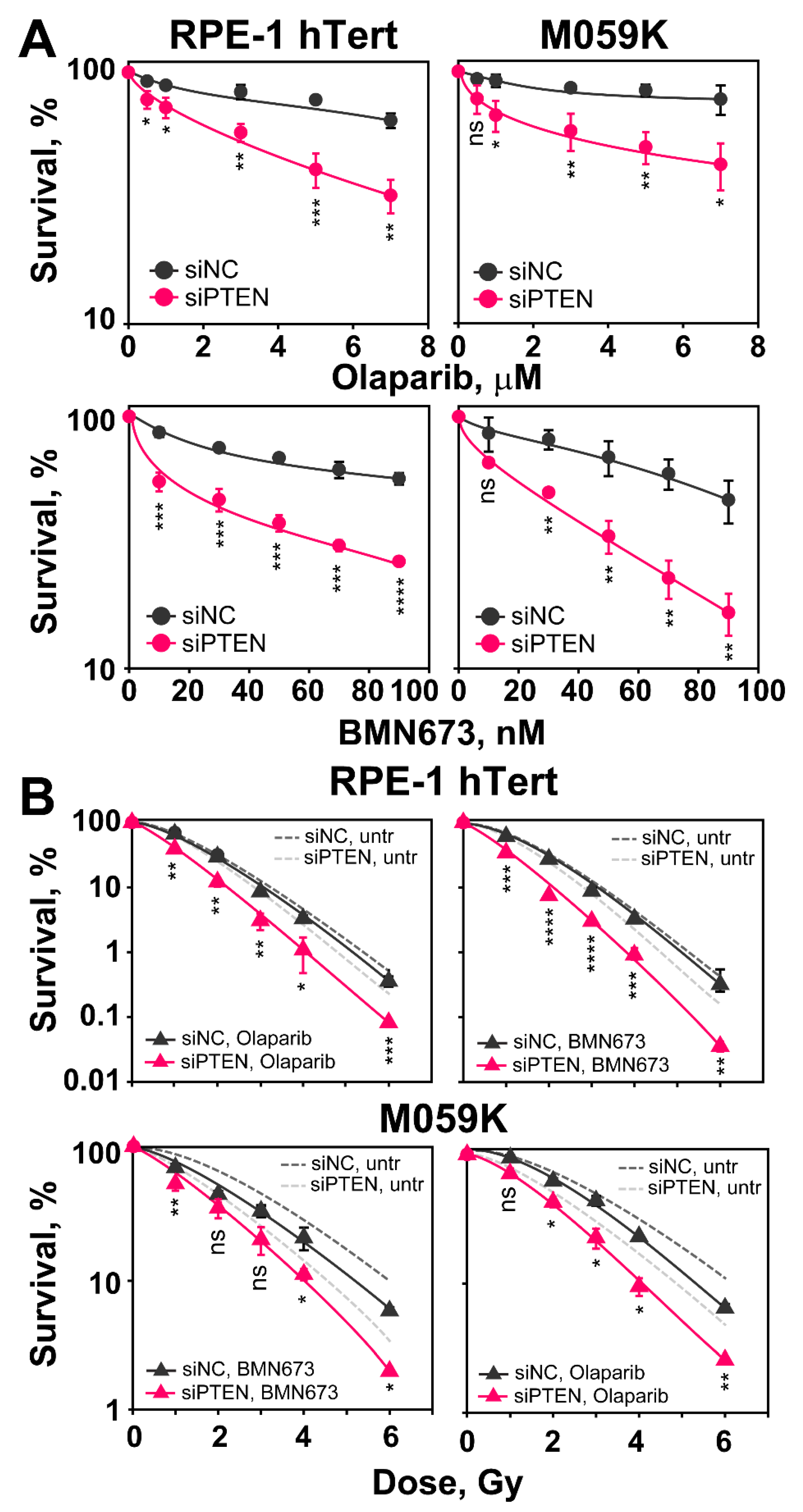
2.6. PTEN Depletion Sensitizes Cells to Olaparib and BMN673
3. Discussion
3.1. PTEN Is a Tumor Suppressor with Functions in DSB Repair
3.2. The Role of PTEN in DSB Repair Pathway Balance
3.3. The Role of PTEN on the G2/M Checkpoint
3.4. PTEN Deficient Cells Are Sensitized to Olaparib or BMN673 with or without IR
4. Materials and Methods
4.1. Cell Lines, X-ray Irradiation and Drug Treatments
4.2. RNA Interference
4.3. Clonogenic Survival Assays
4.4. Flow Cytometry (FC) Analyses of Cell Cycle Distribution, Mitotic Index (H3-pS10) and DNA End-Resection
4.5. Indirect Immunofluorescence Staining and Quantitative Image-Based Cytometry (QIBC) Foci Analysis
4.6. SDS-PAGE and Immunoblotting
4.7. GFP Reporter Cell Lines to Measure HR, SSA, alt-EJ, and NHEJ Activity at I-SceI Induced DSBs
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Steck, P.A.; Pershouse, M.A.; Jasser, S.A.; Yung, W.K.; Lin, H.; Ligon, A.H.; Langford, L.A.; Baumgard, M.L.; Hattier, T.; Davis, T.; et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 1997, 15, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Dragoo, D.D.; Taher, A.; Wong, V.K.; Elsaiey, A.; Consul, N.; Mahmoud, H.S.; Mujtaba, B.; Stanietzky, N.; Elsayes, K.M. PTEN Hamartoma Tumor Syndrome/Cowden Syndrome: Genomics, Oncogenesis, and Imaging Review for Associated Lesions and Malignancy. Cancers 2021, 13, 3120. [Google Scholar] [CrossRef]
- Magana, M.; Landeta-Sa, A.P.; Lopez-Flores, Y. Cowden Disease: A Review. Am. J. Dermatopathol. 2022, 44, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, L.A.J.; Schuurs-Hoeijmakers, J.; Spier, I.; Haadsma, M.L.; Eijkelenboom, A.; Cremer, K.; Mensenkamp, A.R.; Aretz, S.; Vos, J.R.; Hoogerbrugge, N. Catch them if you are aware: PTEN postzygotic mosaicism in clinically suspicious patients with PTEN Hamartoma Tumour Syndrome and literature review. Eur. J. Med. Genet. 2022, 65, 104533. [Google Scholar] [CrossRef]
- Yehia, L.; Eng, C. PTEN Hamartoma Tumor Syndrome. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews: Seattle, WA, USA, 1993. [Google Scholar]
- Worby, C.A.; Dixon, J.E. Pten. Annu. Rev. Biochem. 2014, 83, 641–669. [Google Scholar] [CrossRef]
- Chow, J.T.; Salmena, L. Recent advances in PTEN signalling axes in cancer. Fac. Rev. 2020, 9, 31. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, J.; Xiang, H.; Ding, P.; Wu, T.; Ji, G. The biochemical and clinical implications of phosphatase and tensin homolog deleted on chromosome ten in different cancers. Am. J. Cancer Res. 2021, 11, 5833–5855. [Google Scholar]
- Kim, D.; Dan, H.C.; Park, S.; Yang, L.; Liu, Q.; Kaneko, S.; Ning, J.; He, L.; Yang, H.; Sun, M.; et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front. Biosci. 2005, 10, 975–987. [Google Scholar] [CrossRef]
- Sansal, I.; Sellers, W.R. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef]
- Choudhury, A.D. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate 2022, 82 (Suppl. S1), S60–S72. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Lester, A.; Rapkins, R.; Nixdorf, S.; Khasraw, M.; McDonald, K. Combining PARP inhibitors with radiation therapy for the treatment of glioblastoma: Is PTEN predictive of response? Clin. Transl. Oncol. 2017, 19, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wei, L.; Huang, Y.; Wen, S.; Liu, T.; Duan, X.; Wang, Y.; Zhang, H.; Fan, B.; Hu, B. Identification of distinct genomic features reveals frequent somatic AHNAK and PTEN mutations predominantly in primary malignant melanoma presenting in the ureter. Jpn. J. Clin. Oncol. 2022, 52, 922–935. [Google Scholar] [CrossRef]
- Gupta, S.; Kumar, M.; Chaudhuri, S.; Kumar, A. The non-canonical nuclear functions of key players of the PI3K-AKT-MTOR pathway. J. Cell. Physiol. 2022, 237, 3181–3204. [Google Scholar] [CrossRef]
- Ma, J.; Benitez, J.A.; Li, J.; Miki, S.; Ponte de Albuquerque, C.; Galatro, T.; Orellana, L.; Zanca, C.; Reed, R.; Boyer, A.; et al. Inhibition of Nuclear PTEN Tyrosine Phosphorylation Enhances Glioma Radiation Sensitivity through Attenuated DNA Repair. Cancer Cell 2019, 35, 504–518.e7. [Google Scholar] [CrossRef]
- Hunt, C.R.; Gupta, A.; Horikoshi, N.; Pandita, T.K. Does PTEN Loss Impair DNA Double-Strand Break Repair by Homologous Recombination? Clin. Cancer Res. 2012, 18, 920–922. [Google Scholar] [CrossRef]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. PTEN Deletion in Prostate Cancer Cells Does Not Associate with Loss of RAD51 Function: Implications for Radiotherapy and Chemotherapy. Clin. Cancer Res. 2012, 18, 1015–1027. [Google Scholar] [CrossRef]
- Gupta, A.; Yang, Q.; Pandita, R.K.; Hunt, C.R.; Xiang, T.; Misri, S.; Zeng, S.; Pagan, J.; Jeffery, J.; Puc, J.; et al. Cell cycle checkpoint defects contribute to genomic instability in PTEN deficient cells independent of DNA DSB repair. Cell Cycle 2009, 8, 2198–2210. [Google Scholar] [CrossRef]
- Sinha, A.; Saleh, A.; Endersby, R.; Yuan, S.H.; Chokshi, C.R.; Brown, K.R.; Kuzio, B.; Kauppinen, T.; Singh, S.K.; Baker, S.J.; et al. RAD51-Mediated DNA Homologous Recombination Is Independent of PTEN Mutational Status. Cancers 2020, 12, 3178. [Google Scholar] [CrossRef]
- Noll, A.; Illuzzi, G.; Ame, J.C.; Dantzer, F.; Schreiber, V. PARG deficiency is neither synthetic lethal with BRCA1 nor PTEN deficiency. Cancer Cell Int. 2016, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Wetterskog, D.; Mendes-Pereira, A.M.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Vatcheva, R.; Savage, K.; Mackay, A.; Lord, C.J.; et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci. Transl. Med. 2010, 2, 53ra75. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Choudhary, G.S.; Sharma, A.; Singh, K.K.; Heston, W.D.; Ciezki, J.; Klein, E.A.; Almasan, A. PARP Inhibition Sensitizes to Low Dose-Rate Radiation TMPRSS2-ERG Fusion Gene-Expressing and PTEN-Deficient Prostate Cancer Cells. PLoS ONE 2013, 8, e60408. [Google Scholar] [CrossRef] [PubMed]
- Srividya, M.R.; Thota, B.; Shailaja, B.C.; Arivazhagan, A.; Thennarasu, K.; Chandramouli, B.A.; Hegde, A.S.; Santosh, V. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: A prospective translational study on a uniformly treated cohort of adult patients. Neuropathology 2011, 31, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.-S.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Misra, S.; Ghosh, G.; Chowdhury, S.G.; Karmakar, P. Non-canonical function of nuclear PTEN and its implication on tumorigenesis. DNA Repair 2021, 107, 103197. [Google Scholar] [CrossRef]
- Schmid, A.C.; Byrne, R.D.; Vilar, R.; Woscholski, R. Bisperoxovanadium compounds are potent PTEN inhibitors. FEBS Lett. 2004, 566, 35–38. [Google Scholar] [CrossRef]
- Li, Y.; Prasad, A.; Jia, Y.; Roy, S.G.; Loison, F.; Mondal, S.; Kocjan, P.; Silberstein, L.E.; Ding, S.; Luo, H.R. Pretreatment with phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibitor SF1670 augments the efficacy of granulocyte transfusion in a clinically relevant mouse model. Blood J. Am. Soc. Hematol. 2011, 117, 6702–6713. [Google Scholar] [CrossRef]
- Soni, A.; Mladenov, E.; Iliakis, G. Proficiency in homologous recombination repair is prerequisite for activation of G2-checkpoint at low radiation doses. DNA Repair 2021, 101, 103076. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Stucki, M. How Cells Respond to DNA Breaks in Mitosis. Trends Biochem. Sci. 2020, 45, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-Driven Poly-ADP-Ribosylation Regulates BRCA1 Function in Homologous Recombination–Mediated DNA Repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Li, F.; Wang, Y.; Grabos, M.; Krieger, L.M.; Chaudhary, S.; Hasan, M.S.M.; Ahmed, M.; Coleman, C.N.; Teicher, B.A.; et al. Inhibition of Parp1 by BMN673 Effectively Sensitizes Cells to Radiotherapy by Upsetting the Balance of Repair Pathways Processing DNA Double-Strand Breaks. Mol. Cancer Ther. 2018, 17, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, P.L.; Scanlon, S.E.; Oeck, S.; Glazer, P.M. PTEN Regulates Nonhomologous End Joining By Epigenetic Induction of NHEJ1/XLF. Mol. Cancer Res. MCR 2018, 16, 1241–1254. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Krejci, L.; Van Komen, S.; Sehorn, M.G. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003, 278, 42729–42732. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Ronato, D.A.; Mersaoui, S.Y.; Busatto, F.F.; Affar, E.B.; Richard, S.; Masson, J.-Y. Limiting the DNA Double-Strand Break Resectosome for Genome Protection. Trends Biochem. Sci. 2020, 45, 779–793. [Google Scholar] [CrossRef]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20 (Suppl. S4), iv1–iv86. [Google Scholar] [CrossRef]
- Wu, Z.; Cho, H.; Hampton, G.M.; Theodorescu, D. Cdc6 and cyclin E2 are PTEN-regulated genes associated with human prostate cancer metastasis. Neoplasia 2009, 11, 66–76. [Google Scholar] [CrossRef]
- Mise-Omata, S.; Obata, Y.; Iwase, S.; Mise, N.; Doi, T.S. Transient strong reduction of PTEN expression by specific RNAi induces loss of adhesion of the cells. Biochem. Biophys. Res. Commun. 2005, 328, 1034–1042. [Google Scholar] [CrossRef]
- Coant, N.; Ben Mkaddem, S.; Pedruzzi, E.; Guichard, C.; Treton, X.; Ducroc, R.; Freund, J.N.; Cazals-Hatem, D.; Bouhnik, Y.; Woerther, P.L.; et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol. Cell. Biol. 2010, 30, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- Gunn, A.; Stark, J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. 2012, 920, 379–391. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, X.; Mladenov, E.; Soni, A.; Li, F.; Stuschke, M.; Iliakis, G. PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination. Int. J. Mol. Sci. 2022, 23, 12876. https://doi.org/10.3390/ijms232112876
Pei X, Mladenov E, Soni A, Li F, Stuschke M, Iliakis G. PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination. International Journal of Molecular Sciences. 2022; 23(21):12876. https://doi.org/10.3390/ijms232112876
Chicago/Turabian StylePei, Xile, Emil Mladenov, Aashish Soni, Fanghua Li, Martin Stuschke, and George Iliakis. 2022. "PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination" International Journal of Molecular Sciences 23, no. 21: 12876. https://doi.org/10.3390/ijms232112876
APA StylePei, X., Mladenov, E., Soni, A., Li, F., Stuschke, M., & Iliakis, G. (2022). PTEN Loss Enhances Error-Prone DSB Processing and Tumor Cell Radiosensitivity by Suppressing RAD51 Expression and Homologous Recombination. International Journal of Molecular Sciences, 23(21), 12876. https://doi.org/10.3390/ijms232112876