
Identification of Cancer Cells in the Human Body by Anti-Telomerase Peptide Antibody: Towards the Isolation of Circulating Tumor Cells

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
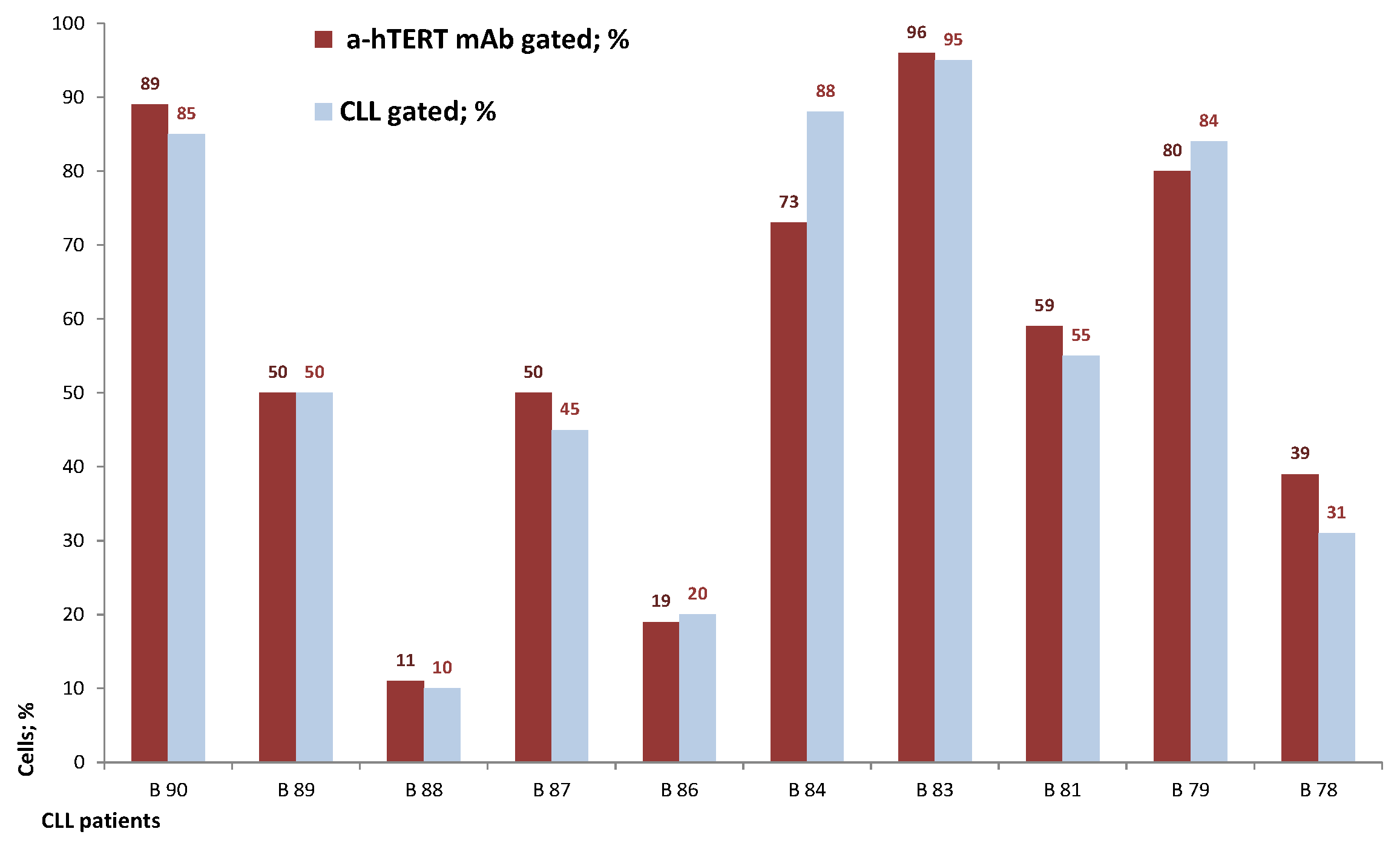{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
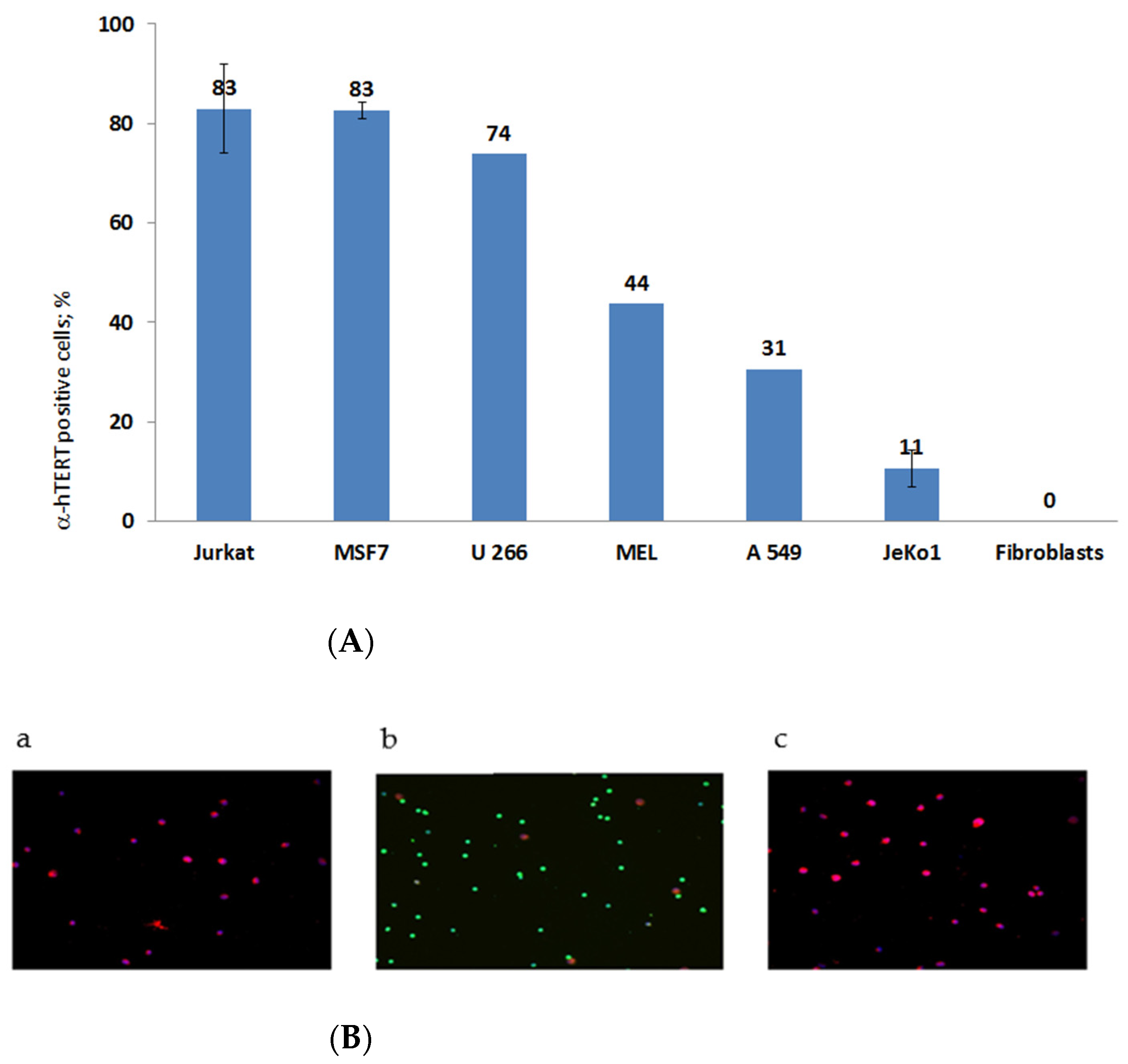
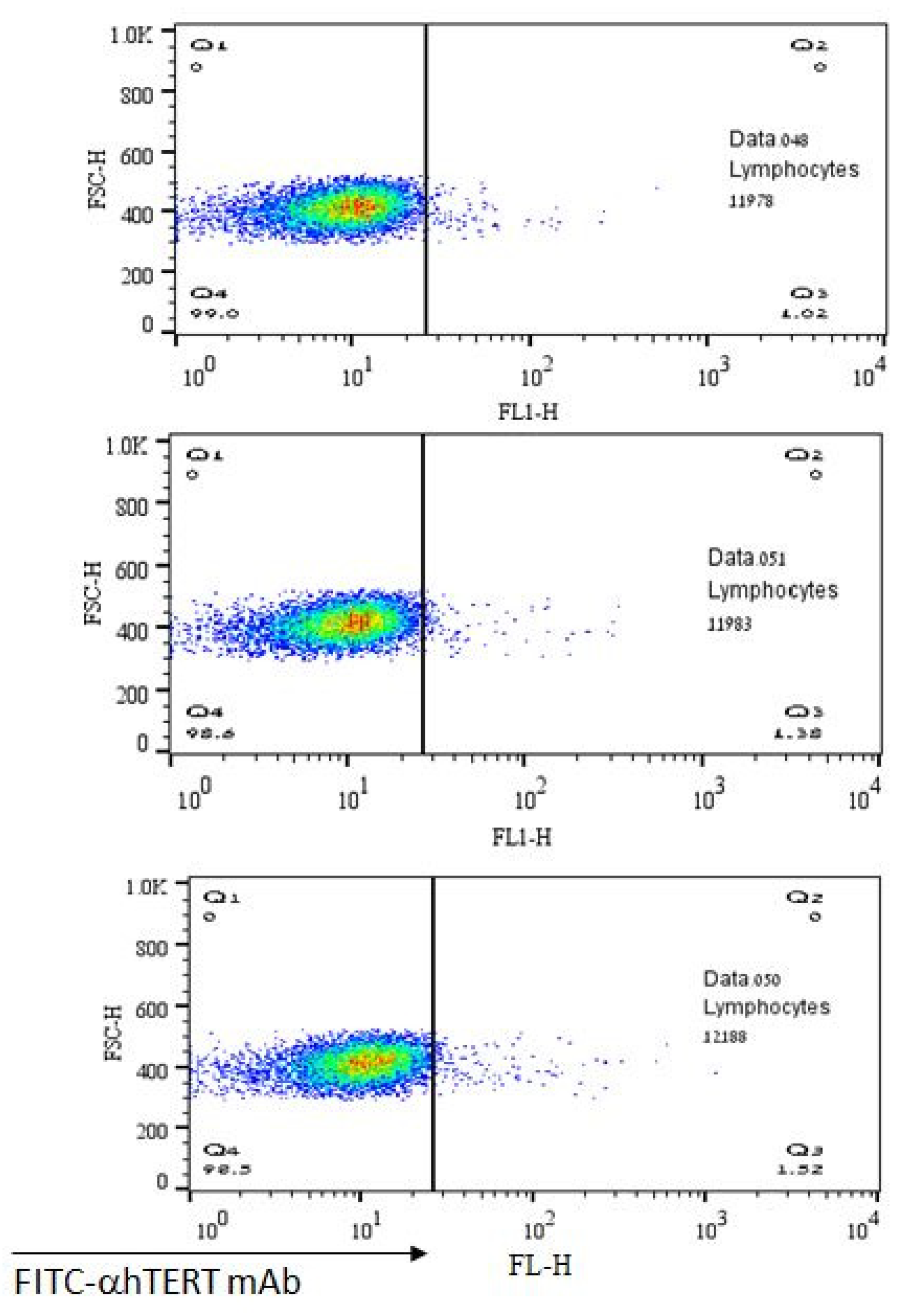
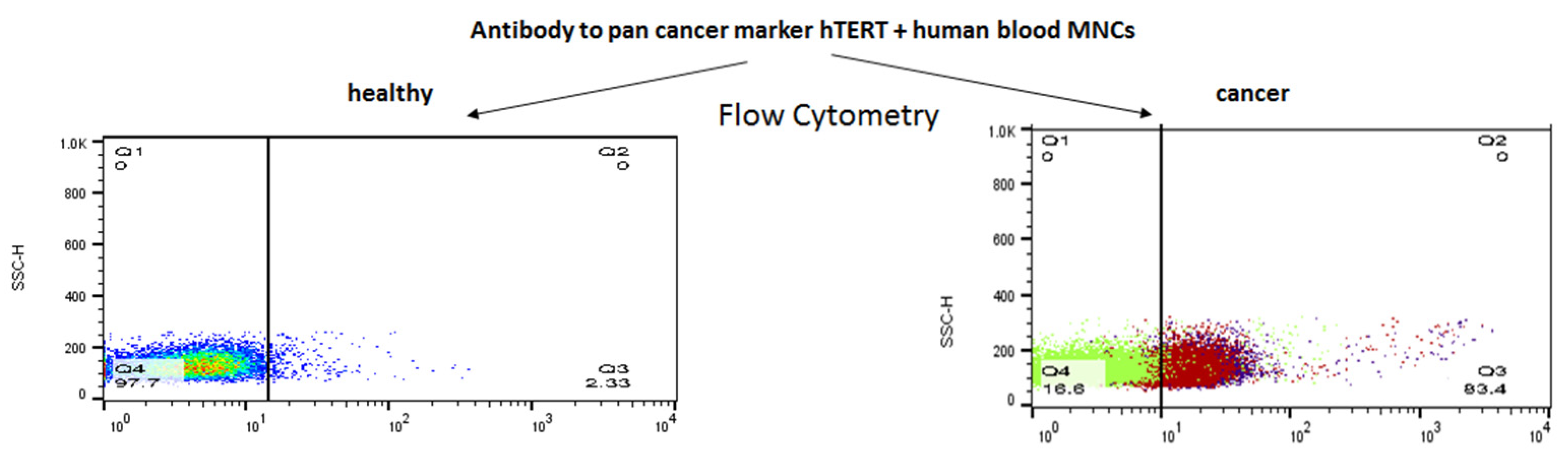
2.1. The α-hTERT Monoclonal Antibody Binds to Various Cancer Cells but Not to Fibroblasts
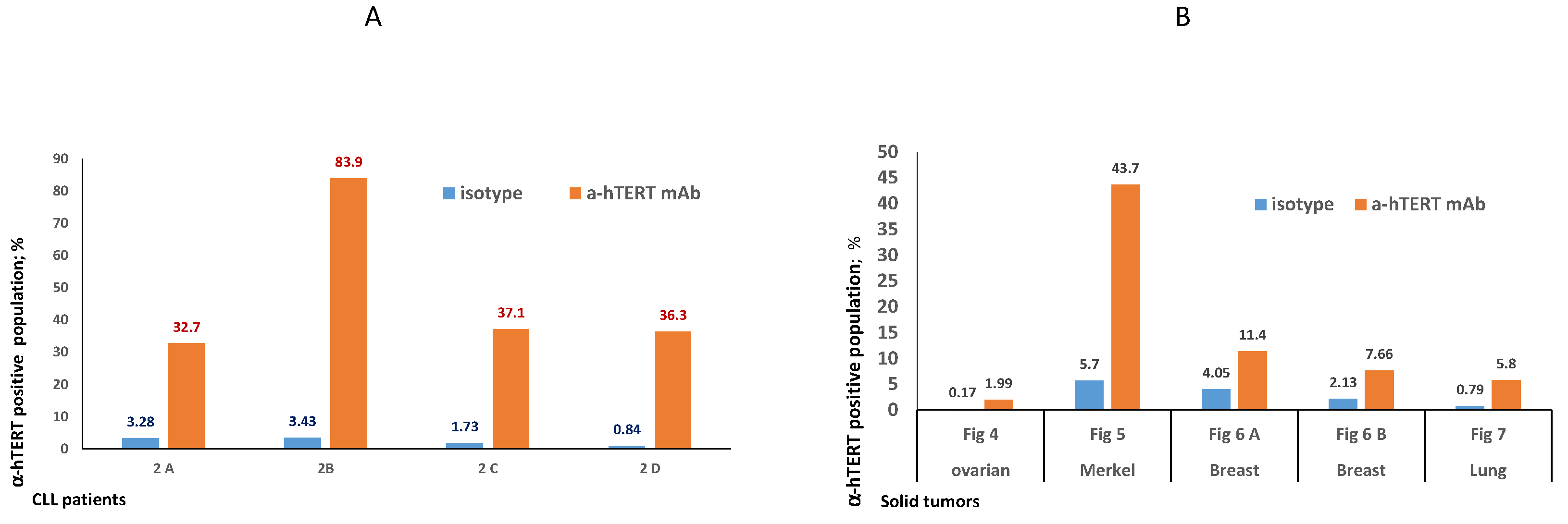
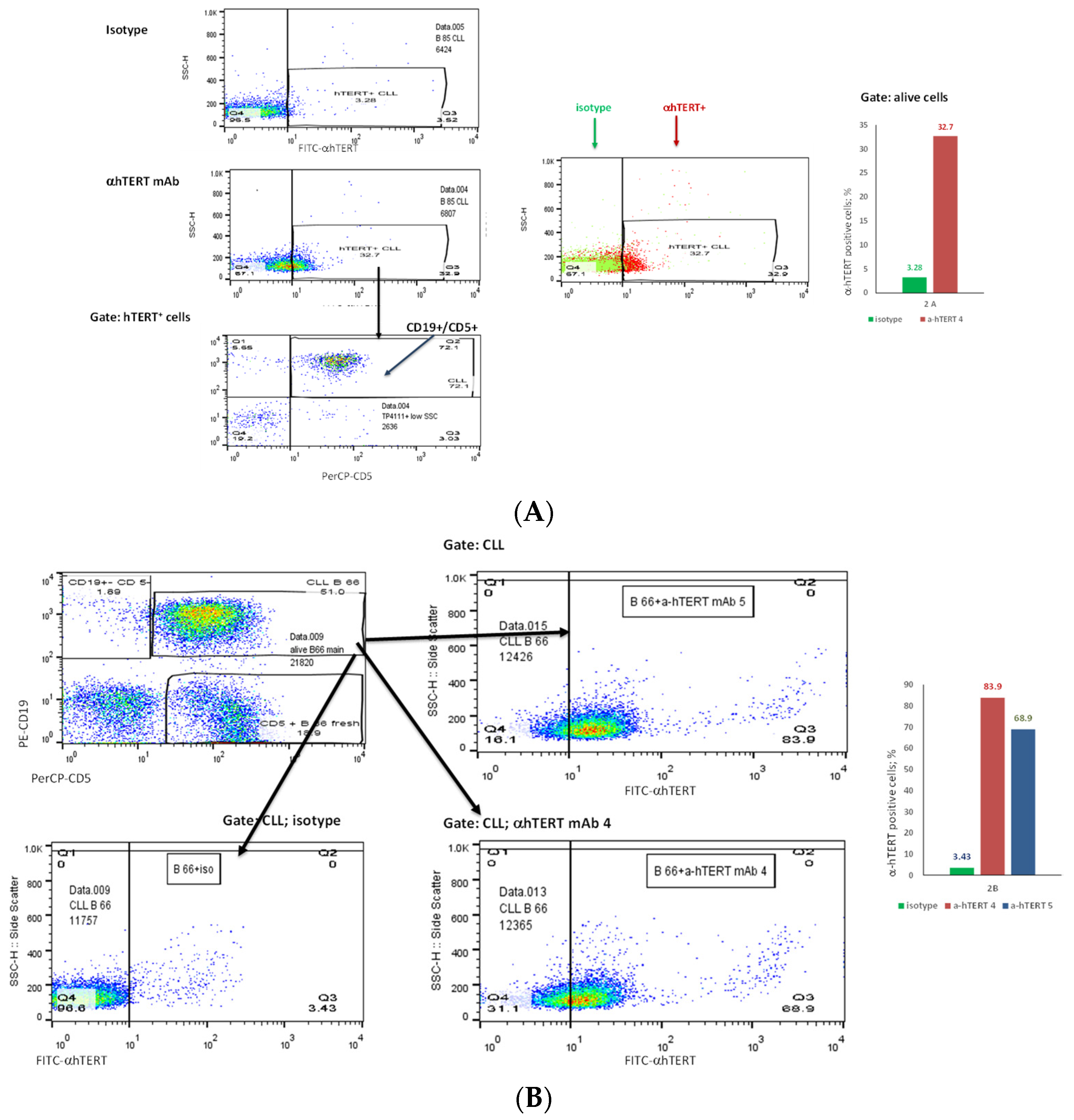
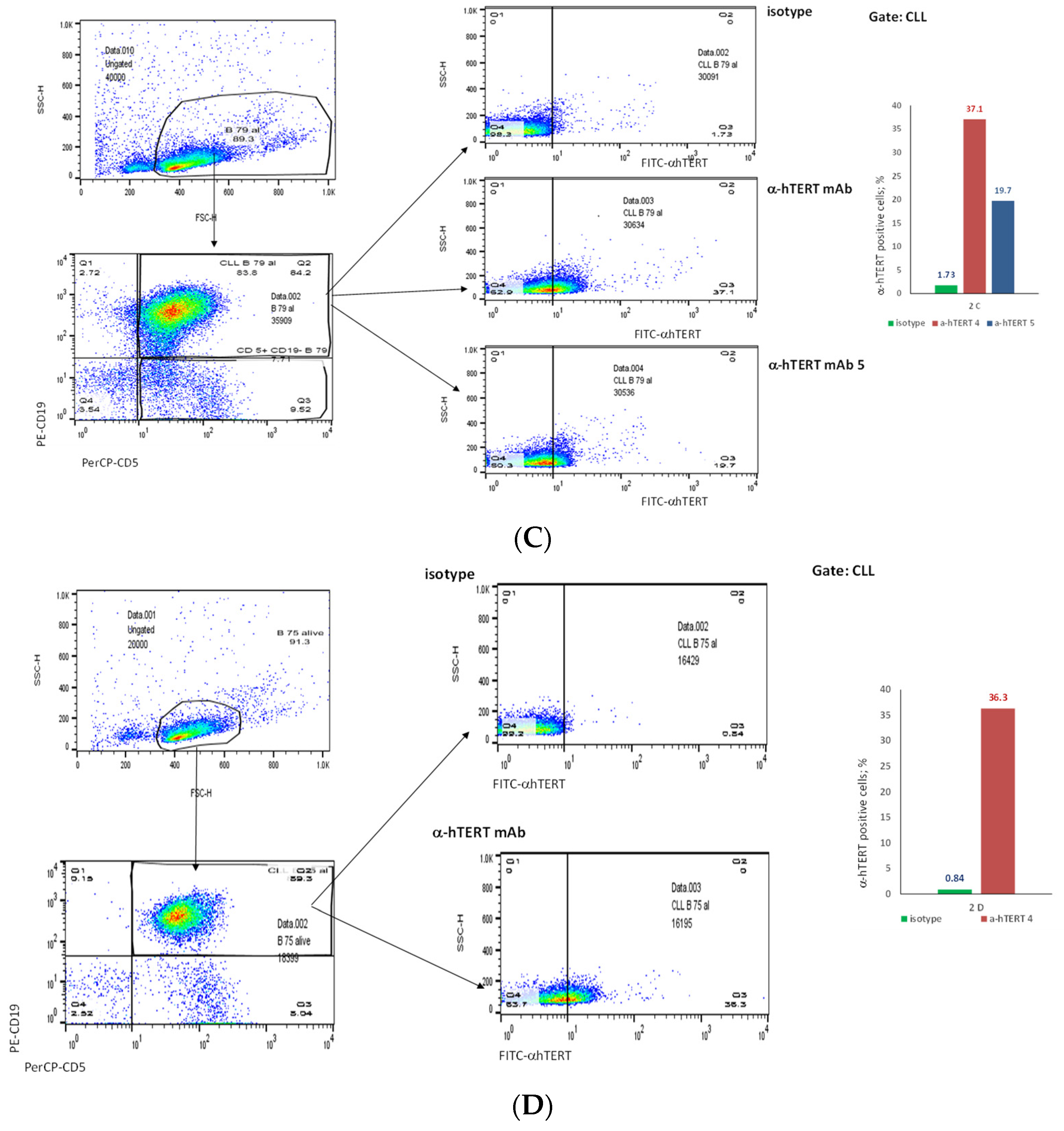
2.2. The α-hTERT mAb Binds to Cancer Cells Isolated from CLL Patients
2.3. Ex-Vivo Binding of α-hTERT mAb to Cancer Cells Isolated from Patients with Solid Tumors
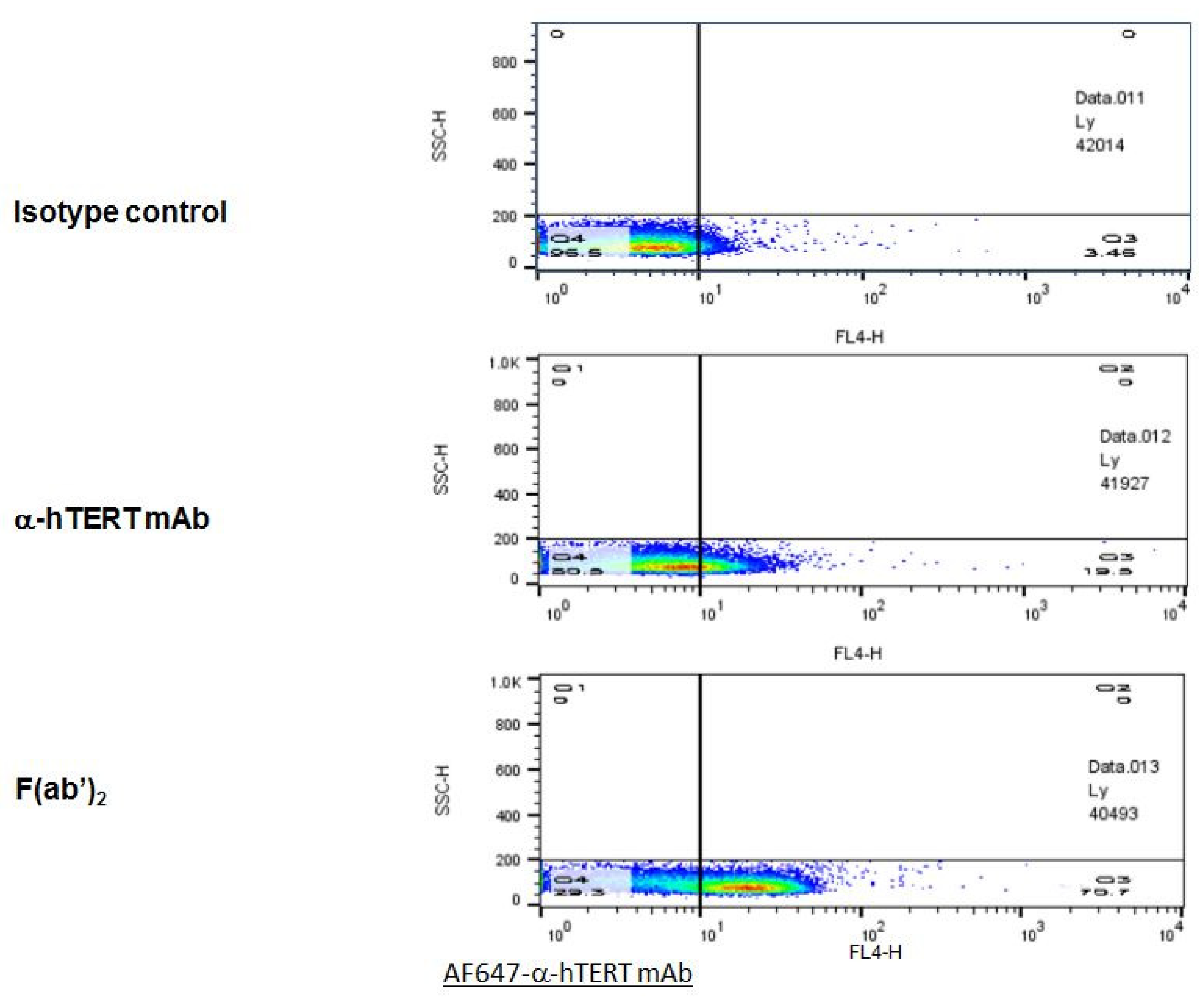
2.4. F(ab’)2 Fragment of the α-hTERT mAb: Purification and Ex-Vivo Binding to CLL Cells
2.5. Sequencing of the Anti-hTERT Monoclonal Antibody
- SEQ ID #1
- 5’-DVVMTQTPLTLSVTIGQPASISCKSSQNLLYSDGKTYLNWLLQRPGQSPKRLIYLVS
- KVDSGVPDRFTGSGSGTDFTLKISRVEAEDLGVYFCWQGTHLPYTFGGGTKLEIK-3’
- SEQ ID #2
- 5’VQLQQSGAELVRPGASVTLSCKASGYIFTDYEKHWVKQTPVHGLEWIGAIDPESG
- STVYNQRFKGKATLTADKSSGTAYMELRSLTSEDSAVYFCFLLRLFAYWGQGTLVTVSA -3’
- SEQ ID #3
- 5’-GATGTTGTGATGACCCAGACTCCACTCACTTTGTCGGTGACCATTGGACAACCA
- GCCTCCATCTCTTGCAAGTCAAGTCAGAACCTCTTATATAGTGATGGAAAGACATATTTGAATTGGTTGTTACAGAGGCCAGGCCAGTCTCCAAAGCGCCTAATCTATCTGGTGTCTAAAGTGGACTCTGGAGTCCCTGACAGGTTCACTGGCAGTGGATCAGGGACAGATTTCACACTGAAAATCAGCAGAGTGGAGGCTGAGGATTTGGGAGTTTATTTTTGCTGGCAAGGTACTCATCTTCCGTACACGTTCGGAGGGGGGACCAAGCTGGAAATAAAACGGGCTGATGCTGCACCAACTGTATCCATCTTCCCACCATCCAGTGAGCAGTTAACATCTGGAGGTGCCTCAGTCGTGTGCTTCTTGAACAACTTCTACCCCAGAGACATCAATGTC
- AAGTGGAAGATTGATGGCAGTGAACGACAAAATGGTGTCCTGAACAGTTGGACTGATCAGGACAGCAAAGACAGCACCTACAGCATGAGCAGCACCCTCACATTGACCAAGGACGAGTATGAACGACATAACAGCTATACCTGTGAGGCCACTCACAAGACATCAACTTCACCCATCGTCAAGAGCTTCAACAGGAATGAGTGTTAA-3’
- SEQ ID #4
- 5’-caggtgCAGCTGCAGCAGTCTGGGGCTGAACTGGTGAGGCCTGGGGCTTCAGTGACG
- CTGTCCTGCAAGGCTTCGGGCTACATATTTACTGATTATGAAAAGCACTGGGTGAAGCAGACACCTGTGCATGGCCTGGAGTGGATTGGAGCTATTGATCCTGAAAGTGGTAGTACTGTCTACAATCAGAGATTCAAGGGCAAGGCCACACTGACTGCAGACAAATCTTCCGGCACAGCCTACATGGAACTCCGCAGCCTGACATCTGAGGATTCTGCCGTCTATTTCTGCTTTTTACTACGGCTATTTGCTTACTGGGGCCAAGGGACTCTGGTCACTGTCTCTGCAGCC-3’
- SEQ ID #6
- 5’-GATGTTGTGATGACCCAGACTCCACTCACTTTGTCGGTGACCATTGGACAACCAGCCTCCATCTCTTGCAAGTCAAGTCAGAACCTCTTATATAGTGATGGAAAGACATATTTGAATTGGTTGTTACAGAGGCCAGGCCAGTCTCCAAAGCGCCTAATCTATCTGGTGTCTAAAGTGGACTCTGGAGTCCCTGACAGGTTCACTGGCAGTGGATCAGGGACAGATTTCACACTGAAAATCAGCAGAGTGGAGGCTGAGGATTTGGGAGTTTATTTTTGCTGGCAAGGTACTCATCTTCCGTACACGTTCGGAGGGGGGACCAAGCTGGAAATAAAACGGGCTGATGCTGCACCAACTGTATCCATCTTCCCACCATCCAGTGAGCAGTTAACATCTGGAGGTGCCTCAGTCGTGTGCTTCTTGAACAACTTC-3’
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Isolation of Mononuclear Cells
4.3. Antibody Production
4.4. F(ab’)2 Production
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Flow Cytometry and Sample Preparation
4.7. Protein Sequencing: (Edman Degradation)
4.8. Nucleic Acid Sequencing
4.9. Immunofluorescence
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tsimberidou, A.M.; Fountzilas, E.; Nikanjam, M.; Kurzrock, R. Review of precision cancer medicine: Evolution of the treatment paradigm. Cancer Treat. Rev. 2020, 86, 102019. [Google Scholar] [CrossRef]
- Haber, D.A.; Velculescu, V.E. Blood-based analyses of cancer: Circulating tumor cells and circulating tumor DNA. Cancer Discov. 2014, 4, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Galarza Fortuna, G.M.; Dvir, K. Circulating tumor DNA: Where are we now? A mini review of the literature. World J. Clin. Oncol. 2020, 11, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Riethdorf, S.; Fritsche, H.; Müller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Jänicke, F.; et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer:a validation study of the CellSearch system. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.J.; Uhr, J.W.; Terstappen, L.W.M.M. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Attard, G.; Swennenhuis, J.F.; Olmos, D.; Reid, A.H.; Vickers, E.; A’Hern, R.; Levink, R.; Coumans, F.; Moreira, J.; Riisnaes, R.; et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 2009, 69, 2912–2918. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef]
- Stott, S.L.; Hsu, C.H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Lin, H.K.; Zheng, S.; Williams, A.J.; Balic, M.; Groshen, S.; Scher, H.I.; Fleisher, M.; Stadler, W.; Datar, R.H.; Tai, Y.C.; et al. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin. Cancer Res. 2010, 16, 5011–5018. [Google Scholar] [CrossRef]
- Farace, F.; Massard, C.; Vimond, N.; Drusch, F.; Jacques, N.; Billiot, F.; Laplanche, A.; Chauchereau, A.; Lacroix, L.; Planchard, D.; et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br. J. Cancer 2011, 105, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, H.; Murray, M.; Turner, J.N.; Caggana, M. Isolation of tumor cells using size and deformation. J. Chromatogr. A 2009, 1216, 8289–8295. [Google Scholar] [CrossRef] [PubMed]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.P.; Schütze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M.; et al. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Tan, S.J.; Yobas, L.; Lee, G.Y.H.; Ong, C.N.; Lim, C.T. Microdevice for the isolation and enumeration of cancer cells from blood. Biomed. Microdevices 2009, 11, 883–892. [Google Scholar] [CrossRef]
- Gertler, R.; Rosenberg, R.; Fuehrer, K.; Dahm, M.; Nekarda, H.; Siewert, J.R. Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Recent Results Cancer Res. 2003, 162, 149–155. [Google Scholar]
- Gascoyne, P.R.C.; Noshari, J.; Anderson, T.J.; Becker, F.F. Isolation of rare cells from cell mixtures by dielectrophoresis. Electrophoresis 2009, 30, 1388–1398. [Google Scholar] [CrossRef]
- Zheng, S.; Lin, H.; Liu, J.-Q.; Balic, M.; Datar, R.; Cote, R.J.; Tai, Y.-C. Membrane micro-filter device for selective capture, electrolysis and genomic analysis of human circulating tumor cells. J. Chromatogr. A 2007, 1162, 154–161. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823. [Google Scholar] [CrossRef]
- Brunsvig, P.F.; Guren, T.K.; Nyakas, M.; Steinfeldt-Reisse, C.H.; Rasch, W.; Kyte, J.A.; Juul, H.V.; Aamdal, S.; Gaudernack, G.; Inderberg, E.M. Long-Term Outcomes of a Phase I Study With UV1, a Second Generation Telomerase Based Vaccine, in Patients With Advanced Non-Small Cell Lung Cancer. Front. Immunol. 2020, 11, 572172. [Google Scholar] [CrossRef]
- Suso, E.M.; Dueland, S.; Rasmussen, A.M.; Vetrhus, T.; Aamdal, S.; Kvalheim, G.; Gaudernack, G. hTERT mRNA dendritic cell vaccination: Complete response in a pancreatic cancer patient associated with response against several hTERT epitopes. Cancer Immunol. Immunother. 2011, 60, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Higashi, K.; Hazama, S.; Araki, A.; Yoshimura, K.; Iizuka, N.; Yoshino, S.; Noma, T.; Oka, M. A novel cancer vaccine strategy with combined IL-18 and HSV-TK gene therapy driven by the hTERT promoter in a murine colorectal cancer model. Int. J. Oncol. 2014, 45, 1412–1420. [Google Scholar] [CrossRef]
- Kim, H.; Seo, E.-H.; Lee, S.-H.; Kim, B.-J. The Telomerase-Derived Anticancer Peptide Vaccine GV1001 as an Extracellular Heat Shock Protein-Mediated Cell-Penetrating Peptide. Int. J. Mol. Sci. 2016, 17, 2054. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.J.; Schiemann, W.P. Telomerase in Cancer: Function, Regulation, and Clinical Translation. Cancers 2022, 14, 808. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Dudognon, C.; Nguyen, E.; Hillion, J.; Pendino, F.; Tarkanyi, I.; Aradi, J.; Lanotte, M.; Tong, J.-H.; Chen, G.-Q.; et al. Immunodetection of human telomerase reverse-transcriptase (hTERT) re-appraised: Nucleolin and telomerase cross paths. J. Cell Sci. 2006, 119, 2797–2806. [Google Scholar] [CrossRef]
- Gutkin, A.; Uziel, O.; Beery, E.; Nordenberg, J.; Pinchasi, M.; Goldvaser, H.; Henick, S.; Goldberg, M.; Lahav, M. Tumor cells derived exosomes contain hTERT mRNA and transform nonmalignant fibroblasts into telomerase positive cells. Oncotarget 2016, 7, 59173–59188. [Google Scholar] [CrossRef]
- Staff, C.; Mozaffari, F.; Frodin, J.-E.; Mellstedt, H.; Liljefors, M.G. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 2014, 45, 1293–1303. [Google Scholar] [CrossRef]
- Annels, N.E.; Shaw, V.E.; Gabitass, R.F.; Billingham, L.; Corrie, P.; Eatock, M.; Valle, J.; Smith, D.; Wadsley, J.; Cunningham, D.; et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol. Immunother. 2014, 63, 175–183. [Google Scholar] [CrossRef]
- Brunsvig, P.F.; Kyte, J.A.; Kersten, C.; Sundstrøm, S.; Møller, M.; Nyakas, M.; Hansen, G.L.; Gaudernack, G.; Aamdal, S. Telomerase peptide vaccination in NSCLC: A phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin. Cancer Res. 2011, 17, 6847–6857. [Google Scholar] [CrossRef]
- Hunger, R.E.; Kernland Lang, K.; Markowski, C.J.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Rasmussen, A.-M.; Braathen, L.R.; Gaudernack, G. Vaccination of patients with cutaneous melanoma with telomerase-specific peptides. Cancer Immunol. Immunother. 2011, 60, 1553–1564. [Google Scholar] [CrossRef]
- Choi, J.; Kim, H.; Kim, Y.; Jang, M.; Jeon, J.; Hwang, Y.-I.; Shon, W.J.; Song, Y.W.; Kang, J.S.; Lee, W.J. The anti-inflammatory effect of GV1001 mediated by the downregulation of ENO1-induced pro-inflammatory cytokine production. Immune Netw. 2015, 15, 291–303. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomerase activity, cell proliferation, and cancer. Proc. Natl. Acad. Sci. USA 1998, 95, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.J.; Baidoo, K.E.; Nayak, T.K.; Garmestani, K.; Brechbiel, M.W.; Milenic, D.E. In vitro and in vivo pre-clinical analysis of a F(ab’)2 fragment of panitumumab for molecular imaging and therapy of HER1-positive cancers. EJNMMI Res. 2011, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, D.E.; Sheoran, A.S.; Rich, C.M.; Richard, L.; Chapman-Bonofiglio, S.; Tzipori, S. Evaluation of Fab and F(ab’)2 fragments and isotype variants of a recombinant human monoclonal antibody against Shiga toxin 2. Infect. Immun. 2010, 78, 1376–1382. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpov, O.; Lahav, M.; Wolach, O.; Raanani, P.; Peer, D.; Kaplan, T.; Uziel, O. Identification of Cancer Cells in the Human Body by Anti-Telomerase Peptide Antibody: Towards the Isolation of Circulating Tumor Cells. Int. J. Mol. Sci. 2022, 23, 12872. https://doi.org/10.3390/ijms232112872
Karpov O, Lahav M, Wolach O, Raanani P, Peer D, Kaplan T, Uziel O. Identification of Cancer Cells in the Human Body by Anti-Telomerase Peptide Antibody: Towards the Isolation of Circulating Tumor Cells. International Journal of Molecular Sciences. 2022; 23(21):12872. https://doi.org/10.3390/ijms232112872
Chicago/Turabian StyleKarpov, Olga, Meir Lahav, Ofir Wolach, Pia Raanani, Dan Peer, Tal Kaplan, and Orit Uziel. 2022. "Identification of Cancer Cells in the Human Body by Anti-Telomerase Peptide Antibody: Towards the Isolation of Circulating Tumor Cells" International Journal of Molecular Sciences 23, no. 21: 12872. https://doi.org/10.3390/ijms232112872
APA StyleKarpov, O., Lahav, M., Wolach, O., Raanani, P., Peer, D., Kaplan, T., & Uziel, O. (2022). Identification of Cancer Cells in the Human Body by Anti-Telomerase Peptide Antibody: Towards the Isolation of Circulating Tumor Cells. International Journal of Molecular Sciences, 23(21), 12872. https://doi.org/10.3390/ijms232112872