
Alterations in DNA Methylation in Orofacial Clefts

 ,
,
Abstract
1. Introduction
2. Alterations in DNA Methylation Found in Blood Samples of Patients with anOrofacial Cleft
3. Alterations in DNA Methylation Found in Lip, Palate, and Saliva of Patients with an Orofacial Cleft
4. Alterations in DNA Methylation in Animals with a Genetically Induced Orofacial Cleft
5. Alterations in DNA Methylation in Animals with a Genetically Induced Orofacial Cleft
6. Limitations and Strengths
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Stanier, P.; Moore, G.E. Genetics of cleft lip and palate: Syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 2004, 13, R73–R81. [Google Scholar] [CrossRef] [PubMed]
- Mossey, P.A.; Modell, B. Epidemiology of oral clefts 2012: An international perspective. Front. Oral. Biol. 2012, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lace, B.; Vasiljeva, I.; Dundure, I.; Barkane, B.; Akota, I.; Krumina, A. Mutation analysis of the MSX1 gene exons and intron in patients with nonsyndromic cleft lip and palate. Stomatologija 2006, 8, 21–24. [Google Scholar]
- Kohli, S.S.; Kohli, V.S. A comprehensive review of the genetic basis of cleft lip and palate. J. Oral Maxillofac. Pathol. 2012, 16, 64–72. [Google Scholar] [CrossRef]
- Spritz, R.A. The genetics and epigenetics of orofacial clefts. Curr. Opin. Pediatr. 2001, 13, 556–560. [Google Scholar] [CrossRef]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 1773–1785. [Google Scholar] [CrossRef]
- Garland, M.A.; Sun, B.; Zhang, S.; Reynolds, K.; Ji, Y.; Zhou, C.J. Role of epigenetics and miRNAs in orofacial clefts. Birth Defects Res. 2020, 112, 1635–1659. [Google Scholar] [CrossRef]
- Cantone, I.; Fisher, A.G. Epigenetic programming and reprogramming during development. Nat. Struct. Mol. Biol. 2013, 20, 282–289. [Google Scholar] [CrossRef]
- Sharp, G.C.; Stergiakouli, E.; Sandy, J.; Relton, C. Epigenetics and Orofacial Clefts: A Brief Introduction. Cleft Palate Craniofac. J. 2018, 55, 795–797. [Google Scholar] [CrossRef]
- Seelan, R.S.; Pisano, M.; Greene, R.M. Nucleic acid methylation and orofacial morphogenesis. Birth Defects Res. 2019, 111, 1593–1610. [Google Scholar] [CrossRef]
- Lei, H.; Oh, S.P.; Okano, M.; Jüttermann, R.; Goss, K.A.; Jaenisch, R.; Li, E. De novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development 1996, 122, 3195–3205. [Google Scholar] [CrossRef]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Greene, R.M.; Pisano, M.M. Palate morphogenesis: Current understanding and future directions. Birth Defects Res. Part C Embryo Today Rev. 2010, 90, 133–154. [Google Scholar] [CrossRef]
- Rogers, J.M.; Francis, B.M.; Sulik, K.K.; Alles, A.J.; Massaro, E.J.; Zucker, R.M.; Elstein, K.H.; Rosen, M.B.; Chernoff, N. Cell death and cell cycle perturbation in the developmental toxicity of the demethylating agent, 5-aza-2’-deoxycytidine. Teratology 1994, 50, 332–339. [Google Scholar] [CrossRef]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef]
- Sharp, G.C.; Lawlor, D.A.; Richmond, R.C.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.A.; Lyttleton, O.; McArdle, W.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 2015, 44, 1288–1304. [Google Scholar] [CrossRef]
- Alvizi, L.; Ke, X.; Brito, L.A.; Seselgyte, R.; Moore, G.E.; Stanier, P.; Passos-Bueno, M.R. Differential methylation is associated with non-syndromic cleft lip and palate and contributes to penetrance effects. Sci. Rep. 2017, 7, 2441. [Google Scholar] [CrossRef]
- Howe, L.J.; Richardson, T.G.; Arathimos, R.; Alvizi, L.; Passos-Bueno, M.R.; Stanier, P.; Nohr, E.; Ludwig, K.U.; Mangold, E.; Knapp, M.; et al. Evidence for DNA methylation mediating genetic liability to non-syndromic cleft lip/palate. Epigenomics 2019, 11, 133–145. [Google Scholar] [CrossRef]
- Xu, Z.; Lie, R.T.; Wilcox, A.J.; Saugstad, O.D.; Taylor, J.A. A comparison of DNA methylation in newborn blood samples from infants with and without orofacial clefts. Clin. Epigenetics 2019, 11, 40. [Google Scholar] [CrossRef]
- Li, Y.; Deng, Y.; Deng, C.; Xie, L.; Yu, L.; Liu, L.; Yuan, Y.; Liu, H.; Dai, L. Association of long interspersed nucleotide element-1 and interferon regulatory factor 6 methylation changes with nonsyndromic cleft lip with or without cleft palate. Oral Dis. 2019, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wu, X.; Li, Y.; Yang, X.; Hu, J.; Zheng, M.; Tian, J. CITED2 mutation and methylation in children with congenital heart disease. J. Biomed. Sci. 2014, 21, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, J.; Lei, Y.; Zou, J.; Lu, X.; Bao, Y.; Wu, L.; Wu, J.; Zheng, X.; Shen, Y.; et al. Global DNA hypomethylation is associated with NTD-affected pregnancy: A case-control study. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, F.; Guan, J.; Le, J.; Wu, L.; Zou, J.; Zhao, H.; Pei, L.; Zheng, X.; Zhang, T. Relation between hypomethylation of long interspersed nucleotide elements and risk of neural tube defects. Am. J. Clin. Nutr. 2010, 91, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Sharp, G.C.; Ho, K.; Davies, A.; Stergiakouli, E.; Humphries, K.; McArdle, W.; Sandy, J.; Davey Smith, G.; Lewis, S.J.; Relton, C.L. Distinct DNA methylation profiles in subtypes of orofacial cleft. Clin. Epigenetics 2017, 9, 63. [Google Scholar] [CrossRef]
- Khan, M.F.J.; Little, J.; Mossey, P.A.; Steegers-Theunissen, R.P.; Autelitano, L.; Lombardo, I.; Andreasi, R.B.; Rubini, M. Evaluating LINE-1 methylation in cleft lip tissues and its association with early pregnancy exposures. Epigenomics 2018, 10, 105–113. [Google Scholar] [CrossRef]
- Khan, M.F.J.; Little, J.; Aleotti, V.; Mossey, P.A.; Steegers-Theunissen, R.P.M.; Autelitano, L.; Meazzini, M.C.; Ravaei, A.; Rubini, M. LINE-1 methylation in cleft lip tissues: Influence of infant MTHFR c.677C>T genotype. Oral Dis. 2019, 25, 1668–1671. [Google Scholar] [CrossRef]
- Mills, J.L.; Kirke, P.N.; Molloy, A.M.; Burke, H.; Conley, M.R.; Lee, Y.J.; Mayne, P.D.; Weir, D.G.; Scott, J.M. Methylenetetrahydrofolate reductase thermolabile variant and oral clefts. Am. J. Med. Genet. 1999, 86, 71–74. [Google Scholar] [CrossRef]
- Prescott, N.J.; Winter, R.M.; Malcolm, S. Maternal MTHFR genotype contributes to the risk of non-syndromic cleft lip and palate. J. Med. Genet. 2002, 39, 368–369. [Google Scholar] [CrossRef]
- Behunova, J.; Klimcakova, L.; Podracka, L. Urinary tract anomalies associated with MTHFR gene polymorphism C677T in girls. Kidney Blood Press. Res. 2011, 34, 465–471. [Google Scholar] [CrossRef]
- Blanton, S.H.; Patel, S.; Hecht, J.T.; Mulliken, J.B. MTHFR is not a risk factor in the development of isolated nonsyndromic cleft lip and palate. Am. J. Med. Genet. 2002, 110, 404–405. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Tilling, K.; Davey Smith, G.; Relton, C.L. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum. Mol. Genet. 2015, 24, 3752–3763. [Google Scholar] [CrossRef]
- Acevedo, N.; Reinius, L.E.; Vitezic, M.; Fortino, V.; Söderhäll, C.; Honkanen, H.; Veijola, R.; Simell, O.; Toppari, J.; Ilonen, J.; et al. Age-associated DNA methylation changes in immune genes, histone modifiers and chromatin remodeling factors within 5 years after birth in human blood leukocytes. Clin. Epigenetics 2015, 7, 34. [Google Scholar] [CrossRef]
- Young, J.I.; Slifer, S.; Hecht, J.T.; Blanton, S.H. DNA Methylation Variation Is Identified in Monozygotic Twins Discordant for Non-syndromic Cleft Lip and Palate. Front. Cell. Dev. Biol. 2021, 9, 656865. [Google Scholar] [CrossRef]
- Smith, A.K.; Kilaru, V.; Klengel, T.; Mercer, K.B.; Bradley, B.; Conneely, K.N.; Ressler, K.J.; Binder, E.B. DNA extracted from saliva for methylation studies of psychiatric traits: Evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2015, 168, 36–44. [Google Scholar] [CrossRef]
- Patel, V.N.; Hoffman, M.P. Salivary gland development: A template for regeneration. Semin. Cell Dev. Biol. 2014, 25-26, 52–60. [Google Scholar] [CrossRef]
- Green, R.M.; Leach, C.L.; Diewert, V.M.; Aponte, J.D.; Schmidt, E.J.; Cheverud, J.M.; Roseman, C.C.; Young, N.M.; Marcucio, R.S.; Hallgrimsson, B. Nonlinear gene expression-phenotype relationships contribute to variation and clefting in the A/WySn mouse. Dev. Dyn. 2019, 248, 1232–1242. [Google Scholar] [CrossRef]
- Shu, X.; Dong, Z.; Cheng, L.; Shu, S. DNA hypermethylation of Fgf16 and Tbx22 associated with cleft palate during palatal fusion. J. Appl. Oral Sci. 2019, 27, e20180649. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J. Mouse genetic models of cleft lip with or without cleft palate. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 63–77. [Google Scholar] [CrossRef]
- Juriloff, D.M.; Harris, M.J.; Dewell, S.L.; Brown, C.J.; Mager, D.L.; Gagnier, L.; Mah, D.G. Investigations of the genomic region that contains the clf1 mutation, a causal gene in multifactorial cleft lip and palate in mice. Birth Defects Res. Part A Clin. Mol. Teratol. 2005, 73, 103–113. [Google Scholar] [CrossRef]
- Plamondon, J.A.; Harris, M.J.; Mager, D.L.; Gagnier, L.; Juriloff, D.M. The clf2 gene has an epigenetic role in the multifactorial etiology of cleft lip and palate in the A/WySn mouse strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2011, 91, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Mager, D.L.; Gagnier, L. Epigenetic mechanism causes Wnt9b deficiency and nonsyndromic cleft lip and palate in the A/WySn mouse strain. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 772–788. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Greene, R.M.; Pisano, M.M. Cigarette smoke induces proteasomal-mediated degradation of DNA methyltransferases and methyl CpG-/CpG domain-binding proteins in embryonic orofacial cells. Reprod. Toxicol. 2015, 58, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Higashihori, N.; Yahiro, K.; Moriyama, K. Retinoic acid inhibits histone methyltransferase Whsc1 during palatogenesis. Biochem. Biophys. Res. Commun. 2015, 458, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, M.; Udagawa, A.; Yoshimoto, S.; Ichinose, M.; Sato, K.; Yamazaki, K.; Matsuno, Y.; Shiota, K.; Mori, C. DNA methylation changes during cleft palate formation induced by retinoic acid in mice. Cleft Palate Craniofac. J. 2008, 45, 545–551. [Google Scholar] [CrossRef]
- Shu, X.; Shu, S.; Zhai, Y.; Zhu, L.; Ouyang, Z. Genome-Wide DNA Methylation Profile of Gene cis-Acting Element Methylations in All-trans Retinoic Acid-Induced Mouse Cleft Palate. DNA Cell Biol. 2018, 37, 993–1002. [Google Scholar] [CrossRef]
- Erickson, R.P. Genes, environment, and orofacial clefting: N-acetyltransferase and folic acid. J. Craniofac. Surg. 2010, 21, 1384–1387. [Google Scholar] [CrossRef]
- Liu, H.Y.; Liu, S.M.; Zhang, Y.Z. Maternal Folic Acid Supplementation Mediates Offspring Health via DNA Methylation. Reprod. Sci. 2020, 27, 963–976. [Google Scholar] [CrossRef]
- Richmond, R.C.; Sharp, G.C.; Herbert, G.; Atkinson, C.; Taylor, C.; Bhattacharya, S.; Campbell, D.; Hall, M.; Kazmi, N.; Gaunt, T.; et al. The long-term impact of folic acid in pregnancy on offspring DNA methylation: Follow-up of the Aberdeen Folic Acid Supplementation Trial (AFAST). Int. J. Epidemiol. 2018, 47, 928–937. [Google Scholar] [CrossRef]
- López-Gordillo, Y.; Maldonado, E.; Nogales, L.; Del Río, A.; Barrio, M.C.; Murillo, J.; Martínez-Sanz, E.; Paradas-Lara, I.; Alonso, M.I.; Partearroyo, T.; et al. Maternal folic acid supplementation reduces the severity of cleft palate in Tgf-β(3) null mutant mice. Pediatr. Res. 2019, 85, 566–573. [Google Scholar] [CrossRef]
- Gonseth, S.; Shaw, G.M.; Roy, R.; Segal, M.R.; Asrani, K.; Rine, J.; Wiemels, J.; Marini, N.J. Epigenomic profiling of newborns with isolated orofacial clefts reveals widespread DNA methylation changes and implicates metastable epiallele regions in disease risk. Epigenetics 2019, 14, 198–213. [Google Scholar] [CrossRef]
- Bliek, B.J.; Steegers-Theunissen, R.P.; Blok, L.J.; Santegoets, L.A.; Lindemans, J.; Oostra, B.A.; Steegers, E.A.; de Klein, A. Genome-wide pathway analysis of folate-responsive genes to unravel the pathogenesis of orofacial clefting in man. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 627–635. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Samples (Population) | Number of Cases: Gender (Controls: Gender) | Major Findings in Methylation | Interpretation | Ref | ||||
|---|---|---|---|---|---|---|---|---|
| Gene (Chromosome) | CpG | DMR | Methylation Level (%) | Risk of Orofacial Cleft | ||||
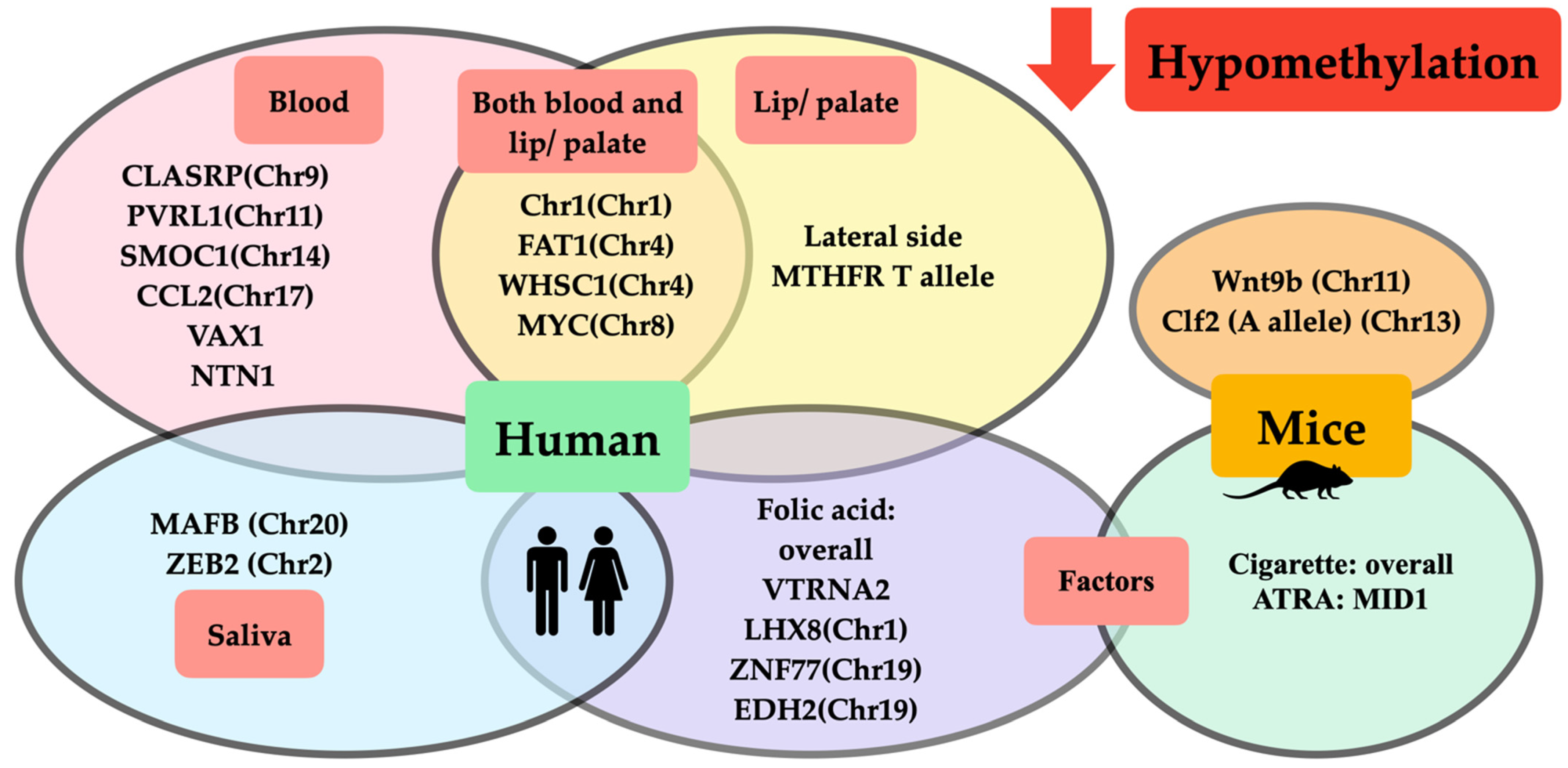
| Whole blood (Brazil) Whole blood (UK) | 67 CL/P: M = 37, F = 30 (59: M = 28, F = 31) 171 CL/P: M = 107, F = 64 (177: M = 100, F = 77) | (Chr8) (Chr4) (Chr1) (Chr4) | cg00611675 cg00405769 cg15897635 cg03150409 | MYC FAT1 Chr1 WHSC1 | ↓ ↓ ↓ ↓ | NA | Hypomethylation at MYC, FAT1, Chr1, WHSC1 contributed to CLP. | Alvizi et al., 2017 [18] |
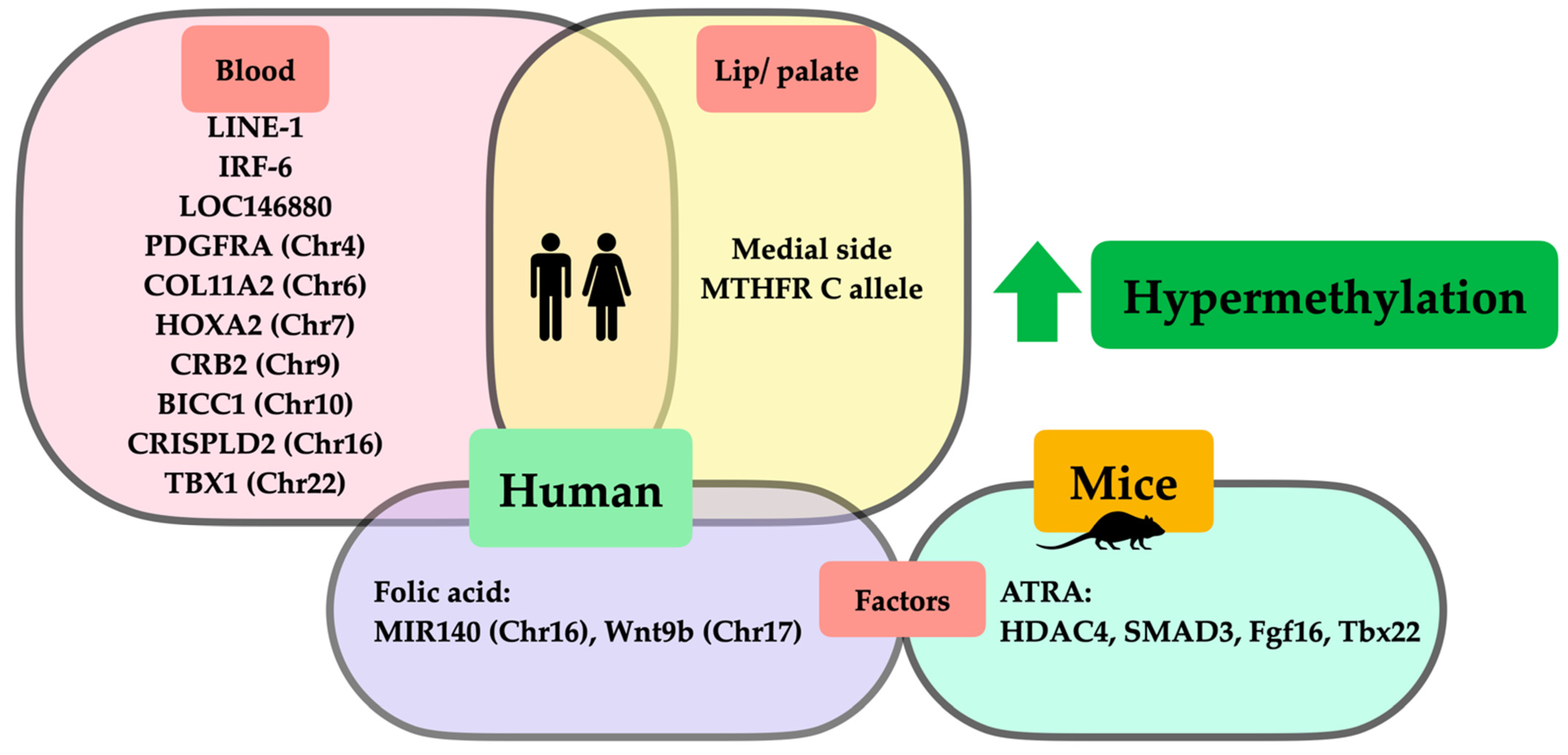
| Leukocytes (UK) | Total 150: 50 CLO: M = 31, F = 19 50 CPO: M = 27, F = 23 50 CL/P: M = 42, F = 8 (-) | TBX1(Chr22) COL11A2 (Chr6) HOXA2 (Chr7) CRB2 (Chr9) PDGFRA (Chr4) CRISPLD2 (Chr16) SMOC1 (Chr14) PVRL1 (Chr11) CCL2 (Chr17) | 6 15 7 2 2 2 5 2 6 | 19750918–19752870 33132086–33132728 27143046–27143807 126130901–126131310 55090812–55091179 84870066–84870204 70316898–70317240 119630144–119630]3 32582128–32582829 | ↑ CPO vs. CLO ↑ CPO vs. CLO ↑ CPO vs. CLO, CL/P ↑ CPO vs. CLO ↑ CPO vs. CLO ↑ CPO vs. CLO ↓ CPO vs. CL/P ↓ CPO vs. CLO ↓ CPO vs. CLO | NA | Methylation level associated with orofacial cleft subtype, mostly CPO, was hypermethylated to a greater extent than CLO (dependent on regions of methylation). | Sharp et al., 2017 [25] |
| Leukocytes (China) | Total 37: M = 28, F = 9 20 CL/P: - 17 CLO: - (60: M = 40, F = 20) | LINE-1 (-) IRF-6 (-) | L1_11.12 I6_34.35 | NA NA | Total (↑ 1.78%) >CL/P (↑ 1.77%) >CLO (↑ 1.75%) >control CL/P (↑ 3.42%) >Total (↑ 2.99%) >CLO (↑ 2.48%) >control | OR: ↑ CLO 12.07 OR: ↑ Total 6.89 OR: ↑ CL/P 4.83 OR: ↑ CL/P 6.00 OR: ↑ Total 4.67 OR: ↑ CLO 3.60 | Hypermethylation of LINE-1 and IRF-6 increased CL/P risk. | Li et al., 2019 [21] |
| Whole blood (UK) | Central European: 399 CL/P: - (1318: -) European ICC duos/trios: 816 CL/P: - (1454: -) | VAX1 (-) NTN1 (-) LOC146880 (-) | cg11398452 cg16197528 cg02598441 | NA | ↓ ↓ ↑ | Mediation causal effect: SNP → DNA methylation → CL/P | Genetic variants caused hypomethylation of VAX1 and NTN1 and hypermethylation of LOC146880; both induced CL/P. | Howe et al., 2019 [19] |
| Whole blood (Norway) | 92 CLO: M = 51, F = 41; 84 CPO: M = 37, F = 47; 132 CL/P: M = 95, F = 37 (436: M = 238, F = 198) | BICC1 (Chr10) (CLO + CL/P) CLASRP (Chr9) (CPO + CL/P) | cg09696939 cg26985354 | 26 31 | ↓ (average 0.02) ↑ (average 0.89) | NA NA | BICC1 hypermethylation was associated with cleft lip subtype; CLASRP hypomethylation was associated with cleft palate subtype. | Xu et al., 2019 [20] |
| Samples (Population) | Number of Cases (Controls) | Major Findings in Methylation | Interpretation | Ref | |||
|---|---|---|---|---|---|---|---|
| Gene (Chromosome) | CpG (DMR) | Methylation Level (%) | Correlation with Blood Methylation | ||||
| Lip (UK) | 18 CL/P lip (18 CL/P blood from same individuals) | NA | NA | NA | Highly positive | A correlation was shown between methylation and lip and blood samples | Alvizi et al., 2017 [18] |
| Lip (UK) Palate (UK) | 48 CLO (43 CL/P) 7 CPO (43 CL/P) | LOC154882 (Chr7) PARK2 (Chr6) OR2L13 (Chr1) KIAA0415 (Chr7) VAMP3 (Chr1) NA | - (158789723–158790116) - (161796785–161796855) - (248100183–248100615) - (4832112–4832536) - (7842159–7842407) NA | NA NA NA CLO 4% > CL/P NA NA | 84% positive 73% positive | Blood methylation levels showed a strong correlation with tissue-specific cleft subtype: - Between blood and lip in CLO - Between blood and palate in CPO | Sharp et al., 2017 [25] |
| Lip (China) | Total 23 CL 13 CL/P 10 (-) | LINE-1 (-) | NA | Medial > lateral (↑1.87%) | NA | Two separated embryologic origins and timing of development of CL caused LINE-1 methylation profile differences in which the medial side was hypermethylated more than the lateral side. | Khan et al., 2018 [26] |
Lip (China) | 45 CL/P: 23 left, 6 right, 7 bilateral (-) | LINE-1 (-) MTHFR c.677C > T genotype -CC -CT -TT | NA | Medial > lateral (↑4.3%) Medial > lateral (↑3.14%) ↔ | NA | LINE-1 methylation in the medial side of CL was hypermethylated to a greater extent than the lateral side, which was possibly influenced by MTHFR c.677C > T (C allele). | Khan et al., 2019 [27] |
| Saliva (US) | 6 affected CLP twin (6 unaffected normal twin) | MAFB (Chr20) ZEB2 (Chr2) | NA | ↓ | NA | Comparison of differential methylation between monozygotic twin pairs discordant for NSCLP found hypomethylation in MAFB and ZEB2. | Young et al., 2021 [34] |
| Animal Model Cases (Control) | Number of Cases (Control) | Age of Embryo (Organ Specificity) | Major Findings in Methylation | Interpretation | Ref | ||
|---|---|---|---|---|---|---|---|
| Gene (Chromosome) | Transcription Level | Methylation Level (Mean %) | |||||
| A/WySn mice, C57BL/6J mice (-) | 1146 (-) | E14 (lip formation) | Clf2 (Chr13) Genotype BB, AB, AA | NA | BB > AB > AA A/WySn lip phenotype - ↑ Normal lip (30–60%) -↓ CL/P (0–20%) | - Clf2 gene polymorphism, especially in A allele, induced hypomethylation and an increased risk of CL/P in A/WySn mice strain. - Severity of lip phenotype varied throughout methylation levels of Clf2 (lower methylation directly correlated with increased severity of lip phenotypes). | Plamondon et al., 2011 [41] |
A/WySn mice (C57BL/6J mice as normal strain) | Transcription level - 13/ (6) Methylation level - 12/ (10) | E10 (nasal pit invagination) E11 (medial and lateral prominence expansion) E12 (primary palate formation) | Wnt9b (Chr11) | A/WySn < C57BL/6J 1.6 fold A/WySn < C57BL/6J 2.2 fold A/WySn < C57BL/6J 1.5 fold | ↓ Overall A/WySn lip phenotype (compared with C57BL/6J) - ↑ Normal lip (50%) - ↓ Unilateral CL (10–40%) - ↓↓ Bilateral CL (0%) | - A/WySn mice had low Wnt9b transcription level and increased risk of CL. - Hypomethylation of Wnt9b was correlated with the severity of CL phenotypes (lower methylation was associated with more severe phenotypes). | Juriloff et al., 2014 [42] |
A/WySn mice (-) | 50 (-) | E11 (medial nasal process) | Wnt9b (Chr11) | NA | ↓ (0–20%) | Hypomethylation at Wnt9b in A/WySn mice nonlinearly affected facial shape variation in CLP (lower methylation was associated with increased facial shape variation). | Green et al., 2019 [37] |
| Species /Organ Specificity (Age) /Population | Risk Factors /Exposure duration /Vehicle | Number of Cases (Control) | Major Findings in Methylation | Interpretation | Ref | |||
|---|---|---|---|---|---|---|---|---|
| Gene (Chromosome) | CpG (DMR) | Gene Expression Level (Fold Change) | Methylation Level (% Change) | |||||
| ICR mice (normal strain) /1st branchial arch cultured cell (E10.5) /- | Cigarette smoke extract (CSE) 20 mcg/mL 40 mcg/mL 80 mcg/mL /24 h /Phosphate-buffered saline Proteasome inhibitor (MG-132) 1.5 mcM /3 h prior to 24 h of CSE /Dimethyl sulfoxide | NA NA | Overall Overall Overall DNMT-1 (-) DNMT-3A (-) DNMR-3B (-) MeCP-2 (-) MBD-2 (-) MBD-3 (-) DNMT-1 (-) DNMT-3A (-) MeCP-2 (-) MBD-3 (-) | NA NA | NA NA NA ↓ 2.21 ↓ 1.85 ↓ 1.80 ↓ 2.43 ↓ 1.50 ↓ 1.52 ↑ ↑ ↑ ↑ | ↔ ↓ 1.8 % ↓ 13% NA NA NA NA NA NA NA NA NA NA | Cigarette smoke extract decreased overall gene expression level of DNA methyltransferase and methyl-CPG-binding domain protein, leading to global DNA hypomethylation in a dose-dependent manner, which induced occurrence of CLP. Pretreatment with proteasomal inhibitor can reverse global DNA methyltransferase and methyl-CPG-binding domain protein degradation from cigarette smoke extract via inhibition of the proteomic degradation pathway. | Mukhopadhyay et al., 2015 [43] |
| C57BL/6J mice (normal strain) /palatal shelves (E14.5) /- | All-trans retinoic acid 70 mg/kg /4 days oral gavage /corn oil | 3 (3) | HDAC4 (-) SMAD3 (-) MID1 (-) | 92051600–92053400 63658601–63660200 169978000–169980801 | NA | ↑ ↑ ↓ | All-trans retinoic acid caused hypermethylation of both HDAC4 and SMAD3 and hypomethylation of MID1, which disrupted palatogenesis, resulting in an increased risk of CP. | Shu et al., 2018 [46] |
| C57BL/6J mice (normal strain) /palatal shelves (E14.5) /- | All-trans retinoic acid 70 mg/kg /oral gavage /corn oil | 3 (3) | Fgf16 (ChrX) Tbx22 (-) | 105725515–105764278 CCGG exon sequences | ↓ ↓ | ↑ ↑ | All-trans retinoic acid caused reciprocal relation (hypermethylation but decreased gene expression) of Fgf16 and Tbx22, which caused developmental failure of the palate, leading to CP. | Shu et al., 2019 [38] |
| Human /newborn archived bloodspot /USA | Without folic acid (prior to mandatory folate dietary) /- /- | 94 CL/P: M = 57, F = 37 (88: M = 51, F = 37) | NA ZNF77 (Chr19) EDH2 (Chr19) VTRNA2-1 (-) LHX8 (Chr1) MIR140 (Chr16) WNT9B (Chr17) | 63.5% of overall CpG cg19689947 cg02718229 NA NA NA NA | NA | ↓ ↓↓ ↓↓ ↓ ↓ ↑ ↑ | Periconceptional folic acid deficiency induced widespread hypomethylation, except hypermethylation of MIR140 and WNT9B, which increased the risk of CL/P. | Gonseth et al., 2019 [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charoenvicha, C.; Sirimaharaj, W.; Khwanngern, K.; Chattipakorn, N.; Chattipakorn, S.C. Alterations in DNA Methylation in Orofacial Clefts. Int. J. Mol. Sci. 2022, 23, 12727. https://doi.org/10.3390/ijms232112727
Charoenvicha C, Sirimaharaj W, Khwanngern K, Chattipakorn N, Chattipakorn SC. Alterations in DNA Methylation in Orofacial Clefts. International Journal of Molecular Sciences. 2022; 23(21):12727. https://doi.org/10.3390/ijms232112727
Chicago/Turabian StyleCharoenvicha, Chirakan, Wimon Sirimaharaj, Krit Khwanngern, Nipon Chattipakorn, and Siriporn C. Chattipakorn. 2022. "Alterations in DNA Methylation in Orofacial Clefts" International Journal of Molecular Sciences 23, no. 21: 12727. https://doi.org/10.3390/ijms232112727
APA StyleCharoenvicha, C., Sirimaharaj, W., Khwanngern, K., Chattipakorn, N., & Chattipakorn, S. C. (2022). Alterations in DNA Methylation in Orofacial Clefts. International Journal of Molecular Sciences, 23(21), 12727. https://doi.org/10.3390/ijms232112727