
Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives
 , ,
, , 
Abstract
1. Introduction
2. Epigenetic Changes in Prion-Like Diseases
2.1. DNA Methylation
2.1.1. DNA Methylation Profiles
2.1.2. Biomarkers Based on DNA Methylation
2.1.3. In Vitro Studies
2.1.4. Mitochondrial DNA Methylation

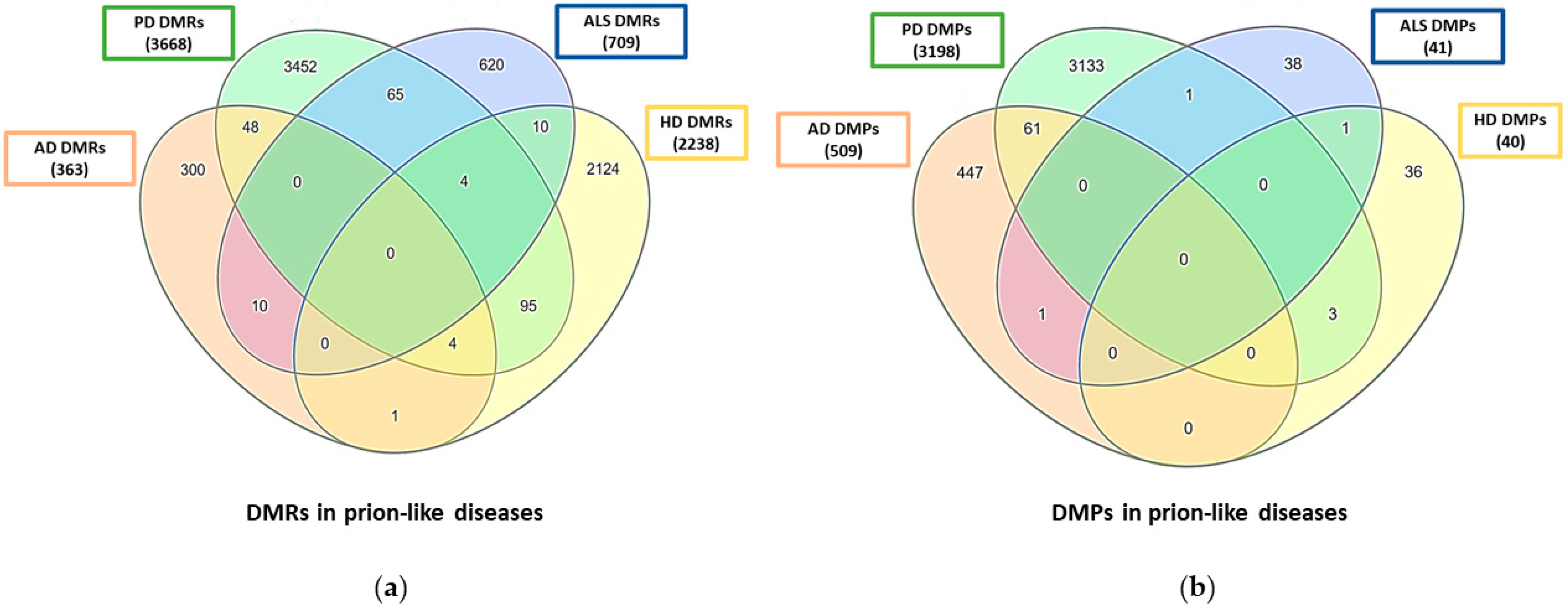{kind=link}
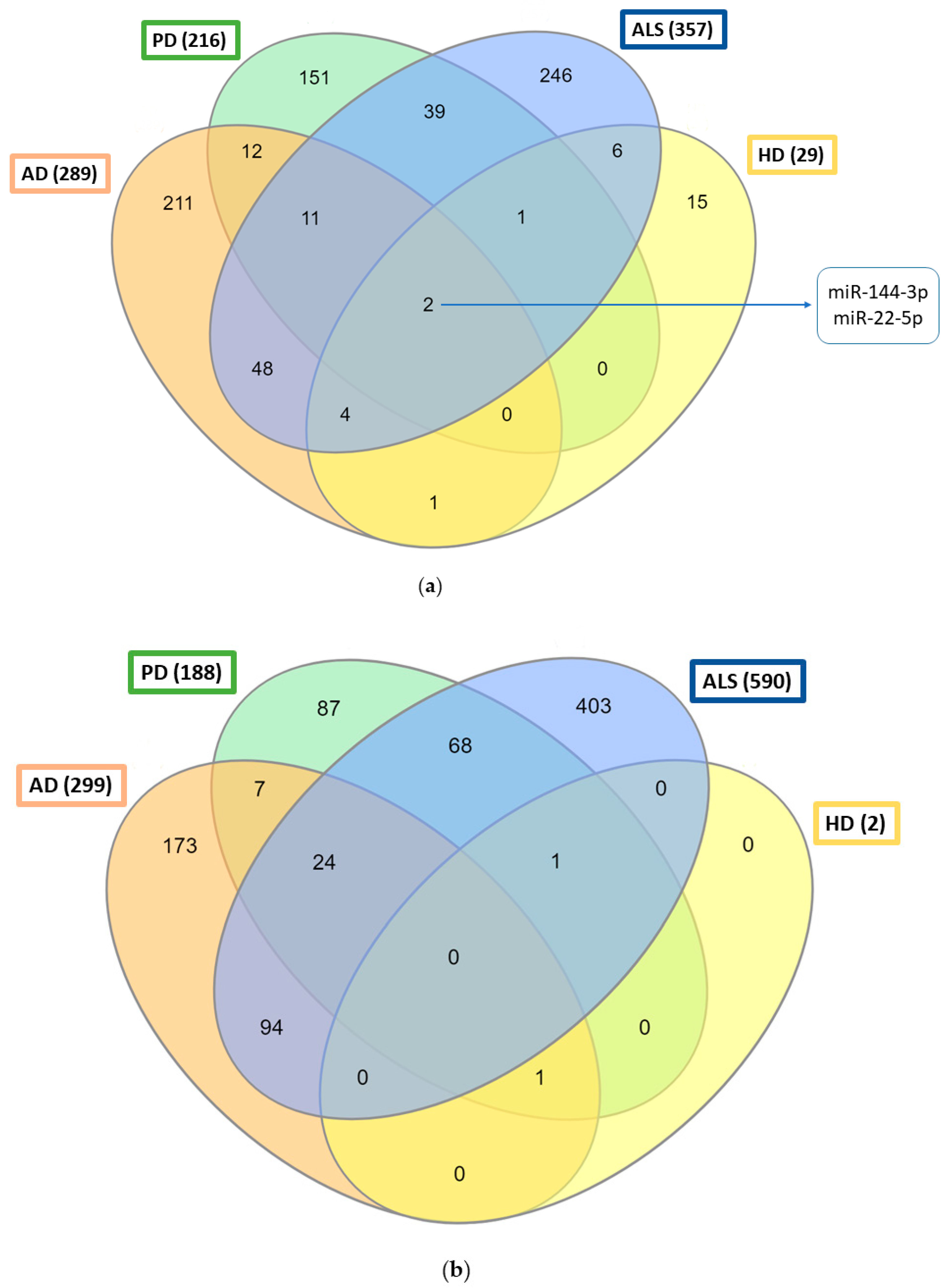{kind=link}
| Disease | Species/Model | Tissue Type | Methylation Finding | References |
|---|---|---|---|---|
| Alzheimer’s disease | Human | Brain, peripheral blood | Methylation profiles of AD patients. Identification of differentially methylated positions (DMPs) | [19,20,30,31] |
| Human | Brain | Methylation profile of AD patients. Identification of differentially methylated regions (DMRs) | [27] | |
| Human | Superior temporal gyrus | Hypermethylated DMRs | [28,29] | |
| APP/PS1 mice | Hippocampus | Changes in mitochondrial DNA methylation | [68] | |
| Human | Neurons | Hypomethylated enhancers in DSCAML1 gene | [41] | |
| Human | Hippocampus, cerebellum, peripheral blood | Hypomethylation in TOMM40 and APOE gene promoters | [61] | |
| Human | Frontal cortex | Methylation of PM20D1 gene | [42] | |
| Human | Peripheral blood | Hypomethylation of B3GALT4 and ZADH2 genes | [57] | |
| Human | Peripheral blood | Hypomethylation of BIN1 gene | [58] | |
| Human | Peripheral blood | Hypermethylation of ERα gene promoter | [59] | |
| Parkinson’s disease | Human | Peripheral blood | Identification of DMRs between PD patients and healthy controls | [22] |
| Human | Brain | Identification of DMRs in PD-affected brain areas | [32] | |
| Human | iPSC-derived dopaminergic neurons | Global DNA hypermethylation changes | [36,37] | |
| Human | Peripheral blood, saliva | Global DNA hypomethylation changes | [21,23] | |
| Human | Peripheral blood, iPSC-derived dopaminergic neurons | Hypomethylation of SNCA gene promoter. Reversion of disease symptoms via CRISPR/Cas9-mediated SNCA hypermethylation | [43,65] | |
| Human | Leukocytes | Hypomethylation of NPAS2 and DRD2 genes | [60,63] | |
| Amyotrophic lateral sclerosis | Human | Peripheral blood | Methylation profile of ALS patients. Identification of DMPs | [24] |
| Human | Brain | Methylation profile of ALS patients. Identification of DMRs | [33] | |
| Human | Peripheral blood | Increased global 5-methylcytosines levels in sALS and FALS | [53] | |
| Human | Peripheral blood | Hypermethylation of the C9orf72 promoter and association of DNA methylation age-acceleration with disease duration and age of onset in C9orf72 expansion carriers | [54,55] | |
| Human | Peripheral blood | Increase in global DNA methylation and demethylation of the mitochondrial D-loop region in SOD1 mutation carriers | [56,67] | |
| Human | hESCs, iPSCs | hESCs unmethylated and iPSCs hypermethylated at the C9orf72 repeats | [64] | |
| Human | Brain, motor neurons | Differential methylation of KIAA1147, IGHMBP2, COL15A1, TARDBP, RANGAP1, IGHMBP2, OGG1, APEX1, PNKP, and APTX | [44,45,46] | |
| Huntington’s disease | YAC128 mice | Brain | Methylation profile. Identification of DMRs | [34] |
| Human | Peripheral blood | Methylation profile of HD patients. Identification of DMPs | [26] | |
| Human | Brain | Minimal association of DNA methylation with HD status | [39] | |
| Human | Peripheral blood | No significant changes between patients and controls | [25] | |
| Human | Brain | 11 co-methylation modules associated with HD status | [35] | |
| Human | Brain | HES4 promoter hypermethylation | [47] | |
| Human | Plasma, saliva | BDNF promoter hypermethylation | [48] |
2.2. Histone Modifications
| Disease | Species/Model | Tissue Type | Main Finding | References |
|---|---|---|---|---|
| Alzheimer’s disease | Human | Brain | Widespread acetylomic variation associated with AD, possibly induced by pathological tau | [69,70] |
| Human | Frontal cortex, hippocampus | Decreased levels of HDAC1 | [73] | |
| Human | Frontal cortex | Decreased levels of HDAC2 contributing to neuronal dysfunction, neurofibrillary tangles pathology, and cognitive decline | [73,74] | |
| 3xTg-AD mice, Human | Human iPSC-derived neurons | HDAC3 inhibition decreases pathological tau phosphorylation and acetylation | [75] | |
| ADLPAPT mice, Human | Human-derived brain organoids | HDAC6 inhibition stimulates pathological tau degradation | [77] | |
| Parkinson’s disease | Human | Dopaminergic neurons | PD neurotoxins increase histone acetylation through an autophagy-mediated HDACs reduction mechanism | [71] |
| Human | Fibroblasts | Imbalance between total HATs and HDACs activities | [72] | |
| LRRK2 R1441G mice | Brain | HDAC inhibition has a neuroprotective effect through modulation of neuroinflammation and improvement of PD-like behaviors | [79] | |
| Sirt2 −/− C57-BL6 mice, Human | Brain | Sirtuin-2 deacetylase inhibition protects degenerating dopaminergic neurons, reduces microglial activation, and facilitates the trafficking and clearance of misfolded proteins | [80,81] | |
| Human | Microglia from the substantia nigra | Upregulation of HDAC2 | [83] | |
| E13-14 mice | Dopaminergic neurons | HDAC4 accumulation promotes neuronal apoptosis | [84] | |
| C57-BL6 mice | Brain | HDAC6 could contribute to oxidative injury | [86] | |
| Amyotrophic lateral sclerosis | SOD1-ALS mice | Skeletal muscle | Increase in class II HDACs (4, 5, and 6) involved in modulating the expression and function of glutamate transporter | [85,86] |
| Human | iPSC-derived motor neurons | HDAC inhibition rescues the DNA repair response | [90] | |
| Tg FUS +/+ mice | Spinal cord | Global histone hypoacetylation. Restoration of histone acetylation ameliorates the disease phenotype | [91] | |
| Human | iPSC-derived motor neurons | HDAC6 inhibition restores axonal transport defects and mitochondrial and endoplasmic reticulum vesicle transport defects | [92,93] | |
| Human | Brain | No significant differences in HDAC expression levels between patients and controls | [97] | |
| SOD1-ALS mice, Human | Skeletal muscle | Decreased expression of HDAC4 associated with an earlier onset of the disease | [87,88] | |
| Huntington’s disease | tgHD rats, BACHD, R6/2, YAC128 mice | Brain | HDAC inhibition produces neuroprotective beneficial effects | [94,95,96,97] |
| HttQ20, HttQ140 mice | Brain | HDAC4 regulates synaptic vesicle trafficking and interacts with htt | [98] | |
| YAC128 mice | Brain | Reduction of HDAC2 could contribute to improving the disease phenotype | [99] | |
| N171-82Q, HdhQ111 knock-in mice | Brain | HDAC3 inhibition improves motor deficits, suppresses striatal CAG repeat expansions, and reduces accumulation of mutant huntingtin oligomeric forms | [100,101] | |
| R6/1 mice | In vivo assessment | HDAC6 deletion exacerbates social impairments and hypolocomotion | [102] | |
| Human | Brain | Differentially enrichment of H3K4me3 mark | [103] | |
| Drosophila melanogaster | Brain, eye | Activity reduction of Utx ameliorates neurodegeneration and diminishes htt aggregation. dSETDB1/ESET might be a mediator of mutant htt-induced degeneration | [104,105] |
2.3. MicroRNAs
2.3.1. MicroRNA Profiles
2.3.2. The Role of microRNAs in Prion-Like Diseases
| Disease | Species/Model | Tissue Type | miRNA | Main Finding | References |
|---|---|---|---|---|---|
| Alzheimer’s disease | Human | Brain, peripheral blood, serum, CSF, serum and CSF exosomes, and plasma extracellular vesicles | miRNA profiles | Differential miRNA expression profiles in AD patients | [111,112,113,114,115,116,117,118,119,120,121,122] |
| APP/PS1 and 5XFAD mice | Brain and urinary exosomes | miRNA profiles | Differential miRNA expression profiles in AD mouse models | [123,124,125] | |
| Human | Serum | miR-193a-3p, miR-133b | Neuroprotective roles attenuating Aβ accumulation and its associated neurotoxicity | [170,171] | |
| Human | Brain | miR-335-5p, miR-361-3p | [172,173] | ||
| SAMP8 mice | Hippocampus | miR-340 | [175] | ||
| Human | SH-SY5Y, SK-N-SH cells | miR-107 | [176] | ||
| Human | Blood plasma | miR-200a-3p | [174] | ||
| Rat | Hippocampus | miR-134-5p | Involved in rescuing AD synaptic plasticity | [177] | |
| C57BL/6J, Tg2576 mice | Hippocampus | miR-124 | Alleviates tau pathology and mediates synaptic and memory deficits | [178,180] | |
| SAMP8 mice, Human | Hippocampus, serum | miR-34c | Mediates synaptic and memory deficits | [179] | |
| Human | Blood, Brain | miR-146a, miR-181a, miR-142-3p | Associated with AD risk and progression | [181,182] | |
| Parkinson’s disease | Human | Brain, gut, plasma, serum, CSF, saliva, plasma exosomes, serum extracellular vesicles, and iPSC-derived dopaminergic neurons | miRNA profiles | Differential miRNA expression profiles in PD patients | [126,127,128,129,130,131,132,133,134,135,136,137] |
| [139] | |||||
| Rat | Peripheral blood | miRNA profile | Differential miRNA expression profile in a PD rat model | [138] | |
| Human | Serum | miR-150 | Amelioration of PD-associated neuroinflammation | [183] | |
| C57BL/6 mice | Brain | miR-let-7a, miR-190 | [184,185] | ||
| Human, rat | SH-SY5Y, PC-12 cells | miR-135b | [186] | ||
| Wistar rats, C57BL/6 mice, Human | Brain, SH-SY5Y cells | miR-375, miR-29c | Decrease in dopaminergic neurons’ damage and neuroinflammatory response | [187,188] | |
| Sprague–Dawley rats | Brain | miR-3557, miR-324 | Involved in delaying PD neurodegeneration | [189] | |
| Human, rat | SH-SY5Y, PC-12 cells | miR-410 | Overexpression exerts neuroprotective effects against apoptosis and reactive oxygen species production | [190] | |
| C57BL/6 mice | Brain | miR-326 | Overexpression promotes autophagy of dopaminergic neurons | [191] | |
| Mouse | BV2 cells | miR-195 | Downregulation might induce microglia-mediated neuroinflammation activation | [192] | |
| Sprague–Dawley rats | Peripheral blood, brain | miR-7 | Involved in regulating brain-derived neurotrophic factor expression in early stages of PD | [193] | |
| Human | PBMCs, SH-SY5Y cells | miR-376a | Implicated in PD pathogenesis regulating the expression of mitochondrial-related genes | [194] | |
| Amyotrophic lateral sclerosis | Human | Brain, spinal cord, skeletal muscle, neuromuscular junctions, plasma, serum, leukocytes, CSF, plasma, serum, brain and spinal cord extracellular vesicles, motor neuron-derived exosomes, iPSC-derived motor neurons, and motor neuron progenitors | miRNA profiles | Differential miRNA expression profiles in ALS patients | [140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155] |
| [158] | |||||
| SOD1G86R and SOD1G93A mice | Serum, skeletal muscle | miRNA profiles | Differential miRNA expression profiles in ALS mouse models | [156,157] | |
| Human | Serum | miR-335-5p | Downregulation may enhance mitophagy, autophagy, and apoptosis pathways | [195] | |
| Wobbler mice | Spinal cord | miR-375-3p | Regulates target structures that intervene at the apoptosis pathway | [196] | |
| Wobbler mice | Cerebellum | miR-29b-3p | Downregulates proapoptotic factors, leading to neuroprotection | [197] | |
| SOD1G93A mice | Muscle | miR-126-5p | Involved in axon degeneration | [198] | |
| Human | Astrocytes | miR-494-3p | [199] | ||
| Human | CNS | miR-1825 | [200] | ||
| SOD1 mice | Spinal cord | miR-338-3p | Upregulation decreases glycogenolysis causing glycogen accumulation within the spinal cord | [201] | |
| Human | Spinal cord | miR-105, miR-9 | Contribute to intermediate filament aggregation in ALS | [202] | |
| Mouse | Astrocytes | miR-218 | Affects astrocyte function negatively | [203] | |
| Human, SOD1G93A mice | Skeletal muscle, serum, plasma | miR-206 | Upregulated. Involved in neuromuscular junction, regeneration, and muscle atrophy | [79,156] | |
| Human | Skeletal muscle | miR-27a, miR-221, miR-155 | Increased in FALS patients | [79] | |
| Human, SOD1G93A mice | Motor neurons | miR-17~92 cluster | Associated with vulnerability of motor neurons to ALS-related degeneration | [205] | |
| Huntington’s disease | C57BL/6, R6/1 and BACHD mice | Brain | miRNA profiles | Differential miRNA expression profiles in HD mouse models | [164,165,166,167] |
| Human | Brain, plasma, CSF | miRNA profiles | Differential miRNA expression profiles in HD patients. Association between CSF-miRNAs expression profiles and the earliest prodromal stages of HD | [172,173,174,175,176] | |
| Hdh mice | Striatum, cortex | miRNA profiles | miRNA regulation may have a limited global role in responding to HD | [206] | |
| R6/2 mice | Brain | miR-132 | Decreased levels, whose restoration confers amelioration in motor function and lifespan | [207] | |
| FVB mouse embryos, HD1/HD7/WT monkey | Neural progenitors, neural cells | miR-196a | Neuroprotective effects | [208,209] | |
| R6/2 mice | Neuronal stem cells | miR-27a | Reduces mutant htt aggregation | [210] | |
| Hu128/21 mice, sheep | Striatum | Artificial miRNAs | Reduce mutant htt levels | [211,212] |
3. Epigenetic Changes in Prion Diseases
3.1. DNA Methylation Profiles
3.2. Histone Modifications
3.3. MicroRNA Profiles
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jing, L.; Li, M.; He, L.; Guo, Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol. Med. Rep. 2019, 19, 3963–3971. [Google Scholar] [CrossRef] [PubMed]
- Huang, W. MicroRNAs: Biomarkers, diagnostics, and therapeutics. Methods Mol. Biol. 2017, 1617, 57–67. [Google Scholar]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef]
- Bottani, M.; Banfi, G.; Lombardi, G. Perspectives on miRNAs as Epigenetic Markers in Osteoporosis and Bone Fracture Risk: A Step Forward in Personalized Diagnosis. Front. Genet. 2019, 10, 1044. [Google Scholar] [CrossRef]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L.A.; et al. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Heyward, F.D.; Sweatt, J.D. DNA Methylation in Memory Formation: Emerging Insights. Neuroscientist 2015, 21, 475–489. [Google Scholar] [CrossRef]
- Miller, C.A.; Sweatt, J.D. Covalent Modification of DNA Regulates Memory Formation. Neuron 2007, 53, 857–869. [Google Scholar] [CrossRef]
- Hu, Z.; Li, Z. miRNAs in synapse development and synaptic plasticity. Curr. Opin. Neurobiol. 2017, 45, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Woldemichael, B.T.; Mansuy, I.M. Micro-RNAs in cognition and cognitive disorders: Potential for novel biomarkers and therapeutics. Biochem. Pharmacol. 2016, 104, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Aksoy-Aksel, A.; Zampa, F.; Schratt, G. MicroRNAs and synaptic plasticity-a mutual relationship. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130515. [Google Scholar] [CrossRef]
- Jaunmuktane, Z.; Brandner, S. Invited Review: The role of prion-like mechanisms in neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2019, 46, 522–545. [Google Scholar] [CrossRef]
- Masnata, M.; Sciacca, G.; Maxan, A.; Bousset, L.; Denis, H.L.; Lauruol, F.; David, L.; Saint-Pierre, M.; Kordower, J.H.; Melki, R.; et al. Demonstration of prion-like properties of mutant huntingtin fibrils in both in vitro and in vivo paradigms. Acta Neuropathol. 2019, 137, 981–1001. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, F. Prion disease and the ‘protein-only hypothesis’. Essays Biochem. 2014, 56, 181–191. [Google Scholar] [CrossRef]
- Prusiner, S. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Chakravarty, A.K.; Jarosz, D.F. More than Just a Phase: Prions at the Crossroads of Epigenetic Inheritance and Evolutionary Change. J. Mol. Biol. 2018, 430, 4607–4618. [Google Scholar] [CrossRef]
- Li, H.; Guo, Z.; Guo, Y.; Li, M.; Yan, H.; Cheng, J.; Wang, C.; Hong, G. Common DNA methylation alterations of Alzheimer’s disease and aging in peripheral whole blood. Oncotarget 2016, 7, 19089–19098. [Google Scholar] [CrossRef]
- Li, Q.S.; Vasanthakumar, A.; Davis, J.W.; Idler, K.B.; Nho, K.; Waring, J.F.; Saykin, A.J.; for the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Association of peripheral blood DNA methylation level with Alzheimer’s disease progression. Clin. Epigenetics 2021, 13, 191. [Google Scholar] [CrossRef]
- Wang, C.; Chen, L.; Yang, Y.; Zhang, M.; Wong, G. Identification of potential blood biomarkers for Parkinson’s disease by gene expression and DNA methylation data integration analysis. Clin. Epigenet. 2019, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R.; Wang, Q.; Meechoovet, B.; Siniard, A.L.; Naymik, M.; de Both, M.; Huentelman, M.J.; Caselli, R.J.; Driver-Dunckley, E.; Dunckley, T. DNA Methylation and Expression Profiles of Whole Blood in Parkinson’s Disease. Front. Genet. 2021, 12, 640266. [Google Scholar] [CrossRef] [PubMed]
- Chuang, Y.-H.; Paul, K.C.; Bronstein, J.M.; Bordelon, Y.; Horvath, S.; Ritz, B. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Hop, P.J.; Zwamborn, R.A.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.; Dekker, A.M.; Westeneng, H.-J.; et al. Genome-wide study of DNA methylation shows alterations in metabolic, inflammatory, and cholesterol pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef]
- Zadel, M.; Maver, A.; Kovanda, A.; Peterlin, B. DNA methylation profiles in whole blood of Huntington’s disease patients. Front. Neurol. 2018, 9, 655. [Google Scholar] [CrossRef]
- Lu, A.T.; Narayan, P.; Grant, M.J.; Langfelder, P.; Wang, N.; Kwak, S.; Wilkinson, H.; Chen, R.Z.; Chen, J.; Bawden, C.S.; et al. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun. 2020, 11, 4529. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, Y.; Yang, J.; Li, L.; Wu, H.; de Jager, P.L.; Jin, P.; Bennett, D.A. A genome-wide profiling of brain DNA hydroxymethylation in Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 674–688. [Google Scholar] [CrossRef]
- Watson, C.T.; Roussos, P.; Garg, P.; Ho, D.J.; Azam, N.; Katsel, P.L.; Haroutunian, V.; Sharp, A.J. Genome-wide12 DNA methylation profiling in the superior temporal gyrus reveals epigenetic signatures associated with Alzheimer’s disease. Genome Med. 2016, 8, 5. [Google Scholar] [CrossRef]
- Gao, Z.; Fu, H.; Zhao, L.; Sun, Z.; Yang, Y.; Zhu, H. Aberrant DNA methylation associated with Alzheimer’s disease in the superior temporal gyrus. Exp. Ther. Med. 2017, 15, 103–108. [Google Scholar] [CrossRef]
- Altuna, M.; Casado, A.U.; de Gordoa, J.S.-R.; Zelaya, M.V.; Labarga, A.; Lepesant, J.M.J.; Roldán, M.; Blanco-Luquin, I.; Perdones, Á.; Larumbe, R.; et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin. Epigenetics 2019, 11, 91. [Google Scholar] [CrossRef]
- Smith, A.; Smith, R.G.; Pishva, E.; Hannon, E.; Roubroeks, J.A.Y.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Sloan, C.; Mill, J.; et al. Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer’s disease. Clin. Epigenetics 2019, 11, 52. [Google Scholar] [CrossRef] [PubMed]
- Young, J.I.; Sivasankaran, S.K.; Wang, L.; Ali, A.; Mehta, A.; Davis, D.A.; Dykxhoorn, D.M.; Petito, C.K.; Beecham, G.W.; Martin, E.R.; et al. Genome-wide brain DNA methylation analysis suggests epigenetic reprogramming in Parkinson disease. Neurol. Genet. 2019, 5, e342. [Google Scholar] [CrossRef] [PubMed]
- Appleby-Mallinder, C.; Schaber, E.; Kirby, J.; Shaw, P.; Cooper-Knock, J.; Heath, P.R.; Highley, J.R. TDP43 proteinopathy is associated with aberrant DNA methylation in human amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2020, 47, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Lin, X.; Wang, J.-Q.; Wu, Y.-S.; Xie, W.; Wang, D.; Zhu, S.; Liao, Y.-Q.; Sun, Q.; et al. Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3641–3653. [Google Scholar] [CrossRef]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.M.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging 2016, 8, 1485–1512. [Google Scholar] [CrossRef]
- Fernández-Santiago, R.; Carballo-Carbajal, I.; Castellano, G.; Torrent, R.; Richaud, Y.; Sánchez-Danés, A.; Vilarrasa-Blasi, R.; Sánchez-Pla, A.; Mosquera, J.L.; Soriano, J.; et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 2015, 7, 1529–1546. [Google Scholar] [CrossRef]
- Fernández-Santiago, R.; Merkel, A.; Castellano, G.; Heath, S.; Raya, A.; Tolosa, E.; Martí, M.J.; Consiglio, A.; Ezquerra, M. Whole-genome DNA hyper-methylation in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. Clin. Epigenet. 2019, 11, 108. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; Da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A Web-Based Tool for the Analysis of Sets through Venn Diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- De Souza, R.A.; Islam, S.A.; McEwen, L.M.; Mathelier, A.; Hill, A.; Mah, S.M.; Wasserman, W.W.; Kobor, M.S.; Leavitt, B.R. DNA methylation profiling in human Huntington’s disease brain. Hum. Mol. Genet. 2016, 25, 2013–2030. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Glauser, L.; Monk, D.; Gräff, J. Comprehensive analysis of PM20D1 QTL in Alzheimer’s disease. Clin. Epigenet. 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Eryilmaz, I.E.; Cecener, G.; Erer, S.; Egeli, U.; Tunca, B.; Zarifoglu, M.; Elibol, B.; Tokcaer, A.B.; Saka, E.; Demirkiran, M.; et al. Epigenetic approach to early-onset Parkinson’s disease: Low methylation status of SNCA and PARK2 promoter regions. Neurol. Res. 2017, 39, 965–972. [Google Scholar] [CrossRef]
- Taskesen, E.; Mishra, A.; van der Sluis, S.; Ferrari, R.; International FTD-Genomics Consortium; Veldink, J.H.; van Es, M.A.; Smit, A.B.; Posthuma, D.; Pijnenburg, Y. Susceptible genes and disease mechanisms identified in frontotemporal dementia and frontotemporal dementia with Amyotrophic Lateral Sclerosis by DNA-methylation and GWAS. Sci. Rep. 2017, 7, 8899. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, M.; Ross, C.A.; Pregent, L.J.; Lank, R.J.; Zhang, C.; Katzman, R.B.; Jansen-West, K.; Song, Y.; da Rocha, E.L.; Palmucci, C.; et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72-associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol. 2017, 134, 715–728. [Google Scholar] [CrossRef]
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol. Commun. 2020, 8, 7. [Google Scholar] [CrossRef]
- Bai, G.; Cheung, I.; Shulha, H.P.; Coelho, J.E.; Li, P.; Dong, X.; Jakovcevski, M.; Wang, Y.; Grigorenko, A.; Jiang, Y.; et al. Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum. Mol. Genet. 2014, 24, 1441–1456. [Google Scholar] [CrossRef]
- Gutierrez, A.; Corey-Bloom, J.; Thomas, E.A.; Desplats, P. Evaluation of Biochemical and Epigenetic Measures of Peripheral Brain-Derived Neurotrophic Factor (BDNF) as a Biomarker in Huntington’s Disease Patients. Front. Mol. Neurosci. 2020, 12, 335. [Google Scholar] [CrossRef]
- Bachman, M.; Uribe-Lewis, S.; Yang, X.; Williams, M.; Murrell, A.; Balasubramanian, S. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat. Chem. 2014, 6, 1049–1055. [Google Scholar] [CrossRef]
- Ellison, E.M.; Abner, E.L.; Lovell, M.A. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J. Neurochem. 2016, 140, 383–394. [Google Scholar] [CrossRef]
- Kaut, O.; Kuchelmeister, K.; Moehl, C.; Wüllner, U. 5-methylcytosine and 5-hydroxymethylcytosine in brains of patients with multiple system atrophy and patients with Parkinson’s disease. J. Chem. Neuroanat. 2019, 96, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.H.; Derrington, I.M.; Brinkerhoff, H.; Langford, K.W.; Nova, I.C.; Samson, J.M.; Bartlett, J.J.; Pavlenok, M.; Gundlach, J.H. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl. Acad. Sci. USA 2013, 110, 18904–18909. [Google Scholar] [CrossRef] [PubMed]
- Hamzeiy, H.; Savaş, D.; Tunca, C.; Şen, N.E.; Gündoǧdu Eken, A.; Şahbaz, I.; Calini, D.; Tiloca, C.; Ticozzi, N.; Ratti, A.; et al. Elevated Global DNA Methylation Is Not Exclusive to Amyotrophic Lateral Sclerosis and Is Also Ob-served in Spinocerebellar Ataxia Types 1 and 2. Neurodegener. Dis. 2018, 18, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.L.; Finch, N.A.; Baker, M.C.; Kachergus, J.M.; DeJesus-Hernandez, M.; Pereira, K.; Christopher, E.; Prudencio, M.; Heckman, M.G.; Thompson, E.A.; et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol. Neurodegener. 2020, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Tartaglia, M.C.; Moreno, D.; Sato, C.; McKeever, P.; Weichert, A.; Keith, J.; Robertson, J.; Zinman, L.; Rogaeva, E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta. Neuropathol. 2017, 134, 271–279. [Google Scholar] [CrossRef]
- Coppedè, F.; Stoccoro, A.; Mosca, L.; Gallo, R.; Tarlarini, C.; Lunetta, C.; Marocchi, A.; Migliore, L.; Penco, S. Increase in DNA methylation in patients with amyotrophic lateral sclerosis carriers of not fully penetrant SOD1 mutations. Amyotroph. Lateral Scler. Front. Degener. 2018, 19, 93–101. [Google Scholar] [CrossRef]
- Madrid, A.; Hogan, K.J.; Papale, L.A.; Clark, L.R.; Asthana, S.; Johnson, S.C.; Alisch, R.S. DNA Hypomethylation in Blood Links B3GALT4 and ZADH2 to Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 66, 927–934. [Google Scholar] [CrossRef]
- Salcedo-Tacuma, D.; Melgarejo, J.D.; Mahecha, M.F.; Ortega-Rojas, J.; Arboleda-Bustos, C.E.; Pardo-Turriago, R.; Arboleda, H. Differential Methylation Levels in CpGs of the BIN1 Gene in Individuals with Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2019, 33, 321–326. [Google Scholar] [CrossRef]
- Li, K.-X.; Sun, Q.; Wei, L.-L.; Du, G.-H.; Huang, X.; Wang, J.-K. ERα Gene Promoter Methylation in Cognitive Function and Quality of Life of Patients with Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2019, 32, 221–228. [Google Scholar] [CrossRef]
- Mao, W.; Zhao, C.; Ding, H.; Liang, K.; Xue, J.; Chan, P.; Cai, Y. Pyrosequencing analysis of methylation levels of clock genes in leukocytes from Parkinson’s disease patients. Neurosci. Lett. 2018, 668, 115–119. [Google Scholar] [CrossRef]
- Shao, Y.; Shaw, M.; Todd, K.; Khrestian, M.; D’Aleo, G.; Barnard, P.J.; Zahratka, J.; Pillai, J.; Yu, C.-E.; Keene, C.D.; et al. DNA methylation of TOMM40-APOE-APOC2 in Alzheimer’s disease. J. Hum. Genet. 2018, 63, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Usefulness of DNA Methylation Levels in COASY and SPINT1 Gene Promoter Regions as Biomarkers in Diagnosis of Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. PLoS ONE 2016, 11, e0168816. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Yoshino, Y.; Yamazaki, K.; Ochi, S.; Iga, J.; Nagai, M.; Nomoto, M.; Ueno, S. DRD2 methylation to differentiate dementia with Lewy bodies from Parkinson’s disease. Acta Neurol. Scand. 2019, 141, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Hadad, Y.; Altarescu, G.; Eldar-Geva, T.; Levi-Lahad, E.; Zhang, M.; Rogaeva, E.; Gotkine, M.; Bartok, O.; Ashwal-Fluss, R.; Kadener, S.; et al. Marked Differences in C9orf72 Methylation Status and Isoform Expression between C9/ALS Human Embryonic and Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 7, 927–940. [Google Scholar] [CrossRef][Green Version]
- Kantor, B.; Tagliafierro, L.; Gu, J.; Zamora, M.E.; Ilich, E.; Grenier, C.; Huang, Z.Y.; Murphy, S.; Chiba-Falek, O. Downregulation of SNCA Expression by Targeted Editing of DNA Methylation: A Potential Strategy for Precision Therapy in PD. Mol. Ther. 2018, 26, 2638–2649. [Google Scholar] [CrossRef]
- Park, H.; Shin, J.; Kim, Y.; Saito, T.; Saido, T.C.; Kim, J. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 41. [Google Scholar] [CrossRef]
- Stoccoro, A.; Mosca, L.; Carnicelli, V.; Cavallari, U.; Lunetta, C.; Marocchi, A.; Migliore, L.; Coppedè, F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics 2018, 10, 1431–1443. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, L.; Han, M.; Liu, X.; Li, F.; Zhou, X.; Wang, Y.; Bi, J. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an APP/PS1 transgenic mouse model of Alzheimer disease. Biochem. Biophys. Res. Commun. 2019, 520, 41–46. [Google Scholar] [CrossRef]
- Marzi, S.; Leung, S.K.; Ribarska, T.; Hannon, E.; Smith, A.R.; Pishva, E.; Poschmann, J.; Moore, K.; Troakes, C.; Al-Sarraj, S.; et al. A histone acetylome-wide association study of Alzheimer’s disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat. Neurosci. 2018, 21, 1618–1627. [Google Scholar] [CrossRef]
- Klein, H.-U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 2018, 22, 37–46. [Google Scholar] [CrossRef]
- Park, G.; Tan, J.; Garcia, G.; Kang, Y.; Salvesen, G.; Zhang, Z. Regulation of Histone Acetylation by Autophagy in Parkinson Disease. J. Biol. Chem. 2016, 291, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Yakhine-Diop, S.M.S.; Niso-Santano, M.; Rodríguez-Arribas, M.; Gómez-Sánchez, R.; Martínez-Chacón, G.; Uribe-Carretero, E.; Navarro-García, J.A.; Ruiz-Hurtado, G.; Aiastui, A.; Cooper, J.M.; et al. Impaired Mitophagy and Protein Acetylation Levels in Fibroblasts from Parkinson’s Disease Patients. Mol. Neurobiol. 2019, 56, 2466–2481. [Google Scholar] [CrossRef] [PubMed]
- Schueller, E.; Paiva, I.; Blanc, F.; Wang, X.L.; Cassel, J.C.; Boutillier, A.L.; Bousiges, O. Dysregulation of histone acetylation pathways in hippocampus and frontal cortex of Alzheimer’s disease patients. Eur. Neuropsychopharmacol. 2020, 33, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Mahady, L.; Nadeem, M.; Malek-Ahmadi, M.; Chen, K.; Perez, S.E.; Mufson, E.J. HDAC 2 dysregulation in the nucleus basalis of Meynert during the progression of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2019, 45, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Delvaux, E.; Nolz, J.; Coleman, P.D.; Chen, S.; Mastroeni, D. Upregulation of histone deacetylase 2 in laser capture nigral microglia in Parkinson’s disease. Neurobiol. Aging 2018, 68, 134–141. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, X.; Zhang, L.; Zhang, Y.; Feng, L. Nuclear Accumulation of Histone Deacetylase 4 (HDAC4) Exerts Neurotoxicity in Models of Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6970–6983. [Google Scholar] [CrossRef]
- Buonvicino, D.; Felici, R.; Ranieri, G.; Caramelli, R.; Lapucci, A.; Cavone, L.; Muzzi, M.; Di Pietro, L.; Bernardini, C.; Zwergel, C.; et al. Effects of Class II-Selective Histone Deacetylase Inhibitor on Neuromuscular Function and Disease Progression in SOD1-ALS Mice. Neuroscience 2018, 379, 228–238. [Google Scholar] [CrossRef]
- Lapucci, A.; Cavone, L.; Buonvicino, D.; Felici, R.; Gerace, E.; Zwergel, C.; Valente, S.; Mai, A.; Chiarugi, A. Effect of Class II HDAC inhibition on glutamate transporter expression and survival in SOD1-ALS mice. Neurosci. Lett. 2017, 656, 120–125. [Google Scholar] [CrossRef]
- Pegoraro, V.; Marozzo, R.; Angelini, C. MicroRNAs and HDAC4 protein expression in the skeletal muscle of ALS patients. Clin. Neuropathol. 2020, 39, 105–114. [Google Scholar] [CrossRef]
- Pigna, E.; Simonazzi, E.; Sanna, K.; Bernadzki, K.M.; Proszynski, T.; Heil, C.; Palacios, D.; Adamo, S.; Moresi, V. Histone deacetylase 4 protects from denervation and skeletal muscle atrophy in a murine model of amyo-trophic lateral sclerosis. eBioMedicine 2019, 40, 717–732. [Google Scholar] [CrossRef]
- Dios, A.M.; Babu, S.; Granucci, E.J.; Mueller, K.; Mills, A.; Alshikho, M.J.; Zürcher, N.R.; Cernasov, P.; Gilbert, T.; Glass, J.D.; et al. Class I and II histone deacetylase expression is not altered in human amyotrophic lateral sclerosis: Neuropathological and positron emission tomography molecular neuroimaging evidence. Muscle Nerve 2019, 60, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Boutillier, A.-L.; Tzeplaeff, L.; Dupuis, L. The dark side of HDAC inhibition in ALS. eBioMedicine 2019, 41, 38–39. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.; Palma, A.; Gomes, R.; Santos, D.; Silva, D.; Cardoso, S. Acetylation as a major determinant to microtubule-dependent autophagy: Relevance to Alzheimer’s and Parkinson disease pathology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1865, 2008–2023. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Kim, H.J.; Yang, J.; Chae, S.; Lee, W.; Chung, S.; Kim, J.; Choi, H.; Song, H.; Lee, C.K.; et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell. 2020, 19, e13081. [Google Scholar] [CrossRef]
- Jian, W.; Wei, X.; Chen, L.; Wang, Z.; Sun, Y.; Zhu, S.; Lou, H.; Yan, S.; Li, X.; Zhou, J.; et al. Inhibition of HDAC6 increases acetylation of peroxiredoxin1/2 and ameliorates 6-OHDA induced dopaminergic injury. Neurosci. Lett. 2017, 658, 114–120. [Google Scholar] [CrossRef]
- Kim, T.; Song, S.; Park, Y.; Kang, S.; Seo, H. HDAC inhibition by valproic acid induces neuroprotection and improvement of PD-like behaviors in LRRK2 R1441G transgenic mice. Exp. Neurobiol. 2019, 28, 504–515. [Google Scholar] [CrossRef]
- Harrison, I.F.; Smith, A.D.; Dexter, D.T. Pathological histone acetylation in Parkinson’s disease: Neuroprotection and inhibition of microglial activation through SIRT 2 inhibition. Neurosci. Lett. 2017, 666, 48–57. [Google Scholar] [CrossRef]
- Esteves, A.R.; Arduíno, D.M.; Silva, D.F.; Viana, S.D.; Pereira, F.C.; Cardoso, S.M. Mitochondrial Metabolism Regulates Microtubule Acetylome and Autophagy Trough Sirtuin-2: Impact for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 1440–1462. [Google Scholar] [CrossRef]
- Kuta, R.; Larochelle, N.; Fernandez, M.; Pal, A.; Minotti, S.; Tibshirani, M.; St.Louis, K.; Gentil, B.J.; Nalbantoglu, J.N.; Hermann, A.; et al. Depending on the stress, histone deacetylase inhibitors act as heat shock protein co-inducers in motor neurons and potentiate arimoclomol, exerting neuroprotection through multiple mechanisms in ALS models. Cell Stress Chaperones 2020, 25, 173–191. [Google Scholar] [CrossRef]
- Rossaert, E.; Pollari, E.; Jaspers, T.; van Helleputte, L.; Jarpe, M.; van Damme, P.; de Bock, K.; Moisse, M.; van den Bosch, L. Restoration of histone acetylation ameliorates disease and metabolic abnormalities in a FUS mouse model. Acta Neuropathol. Commun. 2019, 7, 107. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Bosch, L.V.D. Therapeutic potential of HDAC6 in amyotrophic lateral sclerosis. Cell Stress 2018, 2, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrübl, F.A.; Raber, K.A.; Urbach, Y.K.; Schulze-Krebs, A.; Canneva, F.; Moceri, S.; Habermeyer, J.; Achoui, D.; Gupta, B.; Steindler, D.A.; et al. Early postnatal behavioral, cellular, and molecular changes in models of Huntington disease are reversible by HDAC inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E8765–E8774. [Google Scholar] [CrossRef] [PubMed]
- Chopra, V.; Quinti, L.; Khanna, P.; Paganetti, P.; Kuhn, R.; Young, A.B.; Kazantsev, A.G.; Hersch, S. LBH589, A Hydroxamic Acid-Derived HDAC Inhibitor, is Neuroprotective in Mouse Models of Huntington’s Disease. J. Huntingtons. Dis. 2016, 5, 347–355. [Google Scholar] [CrossRef]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. USA 2014, 112, E56–E64. [Google Scholar] [CrossRef]
- Naia, L.; Cunha-Oliveira, T.; Rodrigues, J.; Rosenstock, T.R.; Oliveira, A.; Ribeiro, M.; Carmo, C.; Oliveira-Sousa, S.I.; Duarte, A.I.; Hayden, M.R.; et al. Histone Deacetylase Inhibitors Protect Against Pyruvate Dehydrogenase Dysfunction in Huntington’s Disease. J. Neurosci. 2017, 37, 2776–2794. [Google Scholar] [CrossRef]
- Federspiel, J.D.; Greco, T.M.; Lum, K.K.; Cristea, I.M. Hdac4 Interactions in Huntington’s Disease Viewed Through the Prism of Multiomics. Mol. Cell. Proteom. 2019, 18, S92–S113. [Google Scholar] [CrossRef]
- Moreno, C.L.; Ehrlich, M.E.; Mobbs, C.V. Protection by dietary restriction in the YAC128 mouse model of Huntington’s disease: Relation to genes regulating histone acetylation and HTT. Neurobiol. Dis. 2016, 85, 25–34. [Google Scholar] [CrossRef][Green Version]
- Jia, H.; Wang, Y.; Morris, C.D.; Jacques, V.; Gottesfeld, J.; Rusche, J.R.; Thomas, E.A. The Effects of Pharmacological Inhibition of Histone Deacetylase 3 (HDAC3) in Huntington’s Disease Mice. PLoS ONE 2016, 11, e0152498. [Google Scholar] [CrossRef]
- Suelves, N.; Kirkham-McCarthy, L.; Lahue, R.S.; Ginés, S. A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington’s disease mice. Sci. Rep. 2017, 7, 6082. [Google Scholar] [CrossRef]
- Ragot, A.; Pietropaolo, S.; Vincent, J.; Delage, P.; Zhang, H.; Allinquant, B.; Leinekugel, X.; Fischer, A.; Cho, Y.H. Genetic deletion of the Histone Deacetylase 6 exacerbates selected behavioral deficits in the R6/1 mouse model for Huntington’s disease. Brain Behav. 2015, 5, e00361. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Tsuji, J.; Labadorf, A.; Roussos, P.; Chen, J.-F.; Myers, R.H.; Akbarian, S.; Weng, Z. The Role of H3K4me3 in Transcriptional Regulation Is Altered in Huntington’s Disease. PLoS ONE 2015, 10, e0144398. [Google Scholar] [CrossRef]
- Song, W.; Zsindely, N.; Faragó, A.; Marsh, J.L.; Bodai, L. Systematic genetic interaction studies identify histone demethylase Utx as potential target for ameliorating Huntington’s disease. Hum. Mol. Genet. 2017, 27, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hwang, Y.J.; Kim, Y.; Lee, M.Y.; Hyeon, S.J.; Lee, S.; Kim, D.H.; Jang, S.; Im, H.; Min, S.-J.; et al. Remodeling of heterochromatin structure slows neuropathological progression and prolongs survival in an animal model of Huntington’s disease. Acta Neuropathol. 2017, 134, 729–748. [Google Scholar] [CrossRef] [PubMed]
- March, Z.M.; King, O.D.; Shorter, J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016, 1647, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.-Y.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef]
- Guo, L.; Shorter, J. Biology and Pathobiology of TDP-43 and Emergent Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2016, 7, a024554. [Google Scholar] [CrossRef]
- Harrison, A.F.; Shorter, J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017, 474, 1417–1438. [Google Scholar] [CrossRef]
- Murakami, K.; Ono, K. Interactions of amyloid coaggregates with biomolecules and its relevance to neurodegeneration. FASEB J. 2022, 36, e22493. [Google Scholar] [CrossRef]
- Henriques, A.D.; Machado-Silva, W.; Leite, R.E.P.; Suemoto, C.K.; Leite, K.R.M.; Srougi, M.; Pereira, A.C.; Jacob-Filho, W.; Nóbrega, O.T. Genome-wide profiling and predicted significance of post-mortem brain microRNA in Alzheimer’s disease. Mech. Ageing Dev. 2020, 191, 111352. [Google Scholar] [CrossRef] [PubMed]
- Moradifard, S.; Hoseinbeyki, M.; Ganji, S.M.; Minuchehr, Z. Analysis of microRNA and Gene Expression Profiles in Alzheimer’s Disease: A Meta-Analysis Approach. Sci. Rep. 2018, 8, 4767. [Google Scholar] [CrossRef] [PubMed]
- Nunez-Iglesias, J.; Liu, C.-C.; Morgan, T.E.; Finch, C.E.; Zhou, X.J. Joint Genome-Wide Profiling of miRNA and mRNA Expression in Alzheimer’s Disease Cortex Reveals Altered miRNA Regulation. PLoS ONE 2010, 5, e8898. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.N.; Begum, G.; Ashman, E.; Thorn, D.; Yakoub, K.M.; Al Hariri, M.; Nehme, A.; Mondello, S.; Kobeissy, F.; Belli, A.; et al. Co-Expression Analysis of microRNAs and Proteins in Brain of Alzheimer’s Disease Patients. Cells 2022, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.J.; Zhang, Y.F.; Dammer, E.B.; Zhou, Y.; Wang, L.L.; Liu, X.H.; Feng, B.L.; Jiang, G.X.; Chen, S.D.; Wang, G.; et al. Peripheral Blood MicroRNA Expression Profiles in Alzheimer’s Disease: Screening, Validation, Association with Clinical Phenotype and Implications for Molecular Mechanism. Mol. Neurobiol. 2016, 53, 5772–5781. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, L.; Wang, Y.; Huang, S. Integrated analysis of miRNA and mRNA expression in the blood of patients with Alzheimer’s disease. Mol. Med. Rep. 2020, 22, 1053–1062. [Google Scholar] [CrossRef]
- Dong, H.; Li, J.; Huang, L.; Chen, X.; Li, D.; Wang, T.; Hu, C.; Xu, J.; Zhang, C.; Zen, K.; et al. Serum MicroRNA Profiles Serve as Novel Biomarkers for the Diagnosis of Alzheimer’s Disease. Dis. Markers 2015, 2015, 625659. [Google Scholar] [CrossRef]
- Lu, L.; Dai, W.-Z.; Zhu, X.-C.; Ma, T. Analysis of Serum miRNAs in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dementias 2021, 36. [Google Scholar] [CrossRef]
- Dangla-Valls, A.; Molinuevo, J.L.; Altirriba, J.; Sánchez-Valle, R.; Alcolea, D.; Fortea, J.; Rami, L.; Balasa, M.; Muñoz-García, C.; Ezquerra, M.; et al. CSF microRNA Profiling in Alzheimer’s Disease: A Screening and Validation Study. Mol. Neurobiol. 2016, 54, 6647–6654. [Google Scholar] [CrossRef]
- Dong, Z.; Gu, H.; Guo, Q.; Liang, S.; Xue, J.; Yao, F.; Liu, X.; Li, F.; Liu, H.; Sun, L.; et al. Profiling of Serum Exosome MiRNA Reveals the Potential of a MiRNA Panel as Diagnostic Biomarker for Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3084–3094. [Google Scholar] [CrossRef]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [PubMed]
- Serpente, M.; Fenoglio, C.; D’Anca, M.; Arcaro, M.; Sorrentino, F.; Visconte, C.; Arighi, A.; Fumagalli, G.G.; Porretti, L.; Cattaneo, A.; et al. MiRNA Profiling in Plasma Neural-Derived Small Extracellular Vesicles from Patients with Alzheimer’s Disease. Cells 2020, 9, 1443. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Tie, C.; Yu, B.; Zhang, W.; Wan, J. Identifying lncRNA-miRNA-mRNA networks to investigate Alzheimer’s disease pathogenesis and therapy strategy. Aging 2020, 12, 2897–2920. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Qu, Y.; Xu, Y.; Zhang, L.; Zhou, L.; Han, Y.; Zhao, W.; Yu, P.; Zhang, Y.; Li, X.; et al. Microarray microRNA profiling of urinary exosomes in a 5XFAD mouse model of Alzheimer’s disease. Anim. Model. Exp. Med. 2021, 4, 233–242. [Google Scholar] [CrossRef]
- Li, Z.-R.; Liu, R.; Zeng, L.; Jiang, H.-L.; Ashraf, G.M. MicroRNA and mRNA profiling of cerebral cortex in a transgenic mouse model of Alzheimer’s disease by RNA sequencing. Neural Regen. Res. 2021, 16, 2099–2108. [Google Scholar] [CrossRef]
- Cardo, L.F.; Coto, E.; Ribacoba, R.; Menéndez, M.; Moris, G.; Suárez, E.; Alvarez, V. MiRNA Profile in the Substantia Nigra of Parkinson’s Disease and Healthy Subjects. J. Mol. Neurosci. 2014, 54, 830–836. [Google Scholar] [CrossRef]
- Hoss, A.G.; Labadorf, A.; Beach, T.G.; Latourelle, J.C.; Myers, R.H. microRNA Profiles in Parkinson’s Disease Pre-frontal Cortex. Front. Aging Neurosci. 2016, 8, 36. [Google Scholar] [CrossRef]
- Schulz, J.; Takousis, P.; Wohlers, I.; Itua, I.O.; Dobricic, V.; Rücker, G.; Binder, H.; Middleton, L.; Ioannidis, J.P.; Perneczky, R.; et al. Meta-analyses identify differentially expressed microRNAs in Parkinson’s disease. Ann. Neurol. 2019, 85, 835–851. [Google Scholar] [CrossRef]
- Tatura, R.; Kraus, T.; Giese, A.; Arzberger, T.; Buchholz, M.; Höglinger, G.; Müller, U. Parkinson’s disease: SNCA-, PARK2-, and LRRK2- targeting microRNAs elevated in cingulate gyrus. Park. Relat. Disord. 2016, 33, 115–121. [Google Scholar] [CrossRef]
- Kurz, A.; Kumar, R.; Northoff, B.H.; Wenk, C.; Schirra, J.; Donakonda, S.; Höglinger, G.U.; Schwarz, J.; Rozanski, V.; Hübner, R.; et al. Differential expression of gut miRNAs in idiopathic Parkinson’s disease. Park. Relat. Disord. 2021, 88, 46–50. [Google Scholar] [CrossRef]
- Cai, M.; Chai, S.; Xiong, T.; Wei, J.; Mao, W.; Zhu, Y.; Li, X.; Wei, W.; Dai, X.; Yang, B.; et al. Aberrant Expression of Circulating MicroRNA Leads to the Dysregulation of Alpha-Synuclein and Other Pathogenic Genes in Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 695007. [Google Scholar] [CrossRef]
- Cardo, L.F.; Coto, E.; de Mena, L.; Ribacoba, R.; Moris, G.; Menéndez, M.; Álvarez, L.D.M. Profile of microRNAs in the plasma of Parkinson’s disease patients and healthy controls. J. Neurol. 2013, 260, 1420–1422. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Freischmidt, A.; Grozdanov, V.; Roth, V.; Brockmann, S.; Mollenhauer, B.; Martin, D.; Haslinger, B.; Fundel-Clemens, K.; Otto, M.; et al. Protein Binding Partners of Dysregulated miRNAs in Parkinson’s Disease Serum. Cells 2021, 10, 791. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, M.C.T.; Barreto-Sanz, M.A.; Correia, B.R.S.; Bell, R.; Widnall, C.; Perez, L.T.; Berteau, C.; Schulte, C.; Scheller, D.; Berg, D.; et al. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget 2018, 9, 17455–17465. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Xiao, Y.; Huang, S.; Cen, L.; Chen, X.; Zhang, L.; Luo, Q.; Li, S.; Yang, X.; Lin, X.; et al. MicroRNA expressing profiles in A53T mutant alpha-synuclein transgenic mice and Parkinsonian. Oncotarget 2016, 8, 15–28. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, J.; Sun, Y.; Li, F.; Wei, L.; Sun, W.; Deng, J.; Yuan, Y.; Wang, Z. Profiling of Differentially Expressed MicroRNAs in Saliva of Parkinson’s Disease Patients. Front. Neurol. 2021, 12, 738530. [Google Scholar] [CrossRef]
- He, S.; Huang, L.; Shao, C.; Nie, T.; Xia, L.; Cui, B.; Lu, F.; Zhu, L.; Chen, B.; Yang, Q. Several miRNAs derived from serum extracellular vesicles are potential biomarkers for early diagnosis and progression of Parkinson’s disease. Transl. Neurodegener. 2021, 10, 25. [Google Scholar] [CrossRef]
- Yadav, S.K.; Pandey, A.; Sarkar, S.; Yadav, S.S.; Parmar, D.; Yadav, S. Identification of Altered Blood MicroRNAs and Plasma Proteins in a Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2022, 59, 1781–1798. [Google Scholar] [CrossRef]
- Tolosa, E.; Botta-Orfila, T.; Morató, X.; Calatayud, C.; Ferrer-Lorente, R.; Martí, M.J.; Fernández, M.; Gaig, C.; Raya, A.; Consiglio, A.; et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol. Aging 2018, 69, 283–291. [Google Scholar] [CrossRef]
- Campos-Melo, D.; Droppelmann, C.A.; He, Z.; Volkening, K.; Strong, M.J. Altered microRNA expression profile in amyotrophic lateral sclerosis: A role in the regulation of NFL mRNA levels. Mol. Brain 2013, 6, 26. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Mori, F.; Kakita, A.; Takahashi, H.; Utsumi, J.; Sasaki, H. Analysis of microRNA from archived formalin-fixed paraffin-embedded specimens of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2014, 2, 173. [Google Scholar] [CrossRef] [PubMed]
- Aksu-Menges, E.; Balci-Hayta, B.; Bekircan-Kurt, C.E.; Aydinoglu, A.T.; Erdem-Ozdamar, S.; Tan, E. Two distinct skeletal muscle microRNA signatures revealing the complex mechanism of sporadic ALS. Acta Neurol. Belg. 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Manfellotto, F.; Fiorentino, G.; Annunziata, A.; Biffali, E.; Pannone, R.; Federico, A. Wide-Ranging Analysis of MicroRNA Profiles in Sporadic Amyotrophic Lateral Sclerosis Using Next-Generation Sequencing. Front. Genet. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, Q.; Chen, X.; Li, C.; Cao, B.; Ou, R.; Hadano, S.; Shang, H.-F. Aberration of miRNAs Expression in Leukocytes from Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2016, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; de Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- Liguori, M.; Nuzziello, N.; Introna, A.; Consiglio, A.; Licciulli, F.; D’Errico, E.; Scarafino, A.; Distaso, E.; Simone, I.L. Dysregulation of MicroRNAs and Target Genes Networks in Peripheral Blood of Patients With Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2018, 11, 288. [Google Scholar] [CrossRef]
- Takahashi, I.; Hama, Y.; Matsushima, M.; Hirotani, M.; Kano, T.; Hohzen, H.; Yabe, I.; Utsumi, J.; Sasaki, H. Identification of plasma microRNAs as a biomarker of sporadic Amyotrophic Lateral Sclerosis. Mol. Brain 2015, 8, 67. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Zondler, L.; Weydt, P.; Volk, A.E.; Božič, A.L.; Walter, M.; Bonin, M.; Mayer, B.; von Arnim, C.A.F.; et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers. Brain 2014, 137, 2938–2950. [Google Scholar] [CrossRef]
- Joilin, G.; Gray, E.; Thompson, A.G.; Bobeva, Y.; Talbot, K.; Weishaupt, J.; Ludolph, A.; Malaspina, A.; Leigh, P.N.; Newbury, S.F.; et al. Identification of a potential non-coding RNA biomarker signature for amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa053. [Google Scholar] [CrossRef]
- Taguchi, Y.-H.; Wang, H. Exploring microRNA Biomarker for Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 1318. [Google Scholar] [CrossRef]
- Benigni, M.; Ricci, C.; Jones, A.R.; Giannini, F.; Al-Chalabi, A.; Battistini, S. Identification of miRNAs as Potential Biomarkers in Cerebrospinal Fluid from Amyotrophic Lateral Sclerosis Patients. NeuroMolecular Med. 2016, 18, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Katsu, M.; Hama, Y.; Utsumi, J.; Takashina, K.; Yasumatsu, H.; Mori, F.; Wakabayashi, K.; Shoji, M.; Sasaki, H. MicroRNA expression profiles of neuron-derived extracellular vesicles in plasma from patients with am-yotrophic lateral sclerosis. Neurosci. Lett. 2019, 708, 134176. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.-W.; Figueroa-Romero, C.; Hur, J.; Pacut, C.; Stoll, E.; Spring, C.; Lewis, R.; Nair, A.; Goutman, S.A.; Sakowski, S.A.; et al. Extracellular Vesicles in Serum and Central Nervous System Tissues Contain microRNA Signatures in Sporadic Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2021, 14, 739016. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, M.; Melzi, V.; Gagliardi, D.; Resnati, D.; Meneri, M.; Dioni, L.; Masrori, P.; Hersmus, N.; Poesen, K.; Locatelli, M.; et al. Insights into the identification of a molecular signature for amyotrophic lateral sclerosis exploiting integrated microRNA profiling of iPSC-derived motor neurons and exosomes. Cell. Mol. Life Sci. 2022, 79, 189. [Google Scholar] [CrossRef]
- Saucier, D.; Wajnberg, G.; Roy, J.; Beauregard, A.P.; Chacko, S.; Crapoulet, N.; Fournier, S.; Ghosh, A.; Lewis, S.M.; Marrero, A.; et al. Identification of a circulating miRNA signature in extracellular vesicles collected from amyotrophic lateral sclerosis patients. Brain Res. 2019, 1708, 100–108. [Google Scholar] [CrossRef]
- Toivonen, J.M.; Manzano, R.; Oliván, S.; Zaragoza, P.; García-Redondo, A.; Osta, R. MicroRNA-206: A Potential Circulating Biomarker Candidate for Amyotrophic Lateral Sclerosis. PLoS ONE 2014, 9, e89065. [Google Scholar] [CrossRef]
- Matamala, J.M.; Arias-Carrasco, R.; Sanchez, C.; Uhrig, M.; Bargsted, L.; Matus, S.; Maracaja-Coutinho, V.; Abarzua, S.; van Zundert, B.; Verdugo, R.; et al. Genome-wide circulating microRNA expression profiling reveals potential biomarkers for amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 64, 123–138. [Google Scholar] [CrossRef]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.P.; Barabino, S.; Nizzardo, M.; et al. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 2018, 8, 10105. [Google Scholar] [CrossRef]
- Dong, X.; Cong, S. Bioinformatic analysis of microRNA expression in Huntington’s disease. Mol. Med. Rep. 2018, 18, 2857–2865. [Google Scholar] [CrossRef]
- Hoss, A.G.; Kartha, V.K.; Dong, X.; Latourelle, J.C.; Dumitriu, A.; Hadzi, T.C.; Macdonald, M.E.; Gusella, J.F.; Akbarian, S.; Chen, J.-F.; et al. MicroRNAs Located in the Hox Gene Clusters Are Implicated in Huntington’s Disease Pathogenesis. PLoS Genet. 2014, 10, e1004188. [Google Scholar] [CrossRef]
- Wang, Z.M.; Dong, X.Y.; Cong, S.Y. Bioinformatic analysis of a microRNA regulatory network in Huntington’s disease. J. Integr. Neurosci. 2020, 19, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Díez-Planelles, C.; Sánchez-Lozano, P.; Crespo, M.; Gil-Zamorano, J.; Ribacoba, R.; González, N.; Suárez, E.; Martínez-Descals, A.; Martínez-Camblor, P.; Álvarez, V.; et al. Circulating microRNAs in Huntington’s disease: Emerging mediators in metabolic impairment. Pharmacol. Res. 2016, 108, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Reed, E.R.; Latourelle, J.C.; Bockholt, J.H.; Bregu, J.; Smock, J.; Paulsen, J.S.; Myers, R.H. PREDICT-HD CSF ancillary study investigators MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology 2017, 90, e264–e272. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Kong, G.; Tran, H.; Li, S.; Pang, T.Y.; Hannan, A.J.; Renoir, T. Small Non-coding RNAs Are Dysregulated in Huntington’s Disease Transgenic Mice Independently of the Therapeutic Effects of an Environmental Intervention. Mol. Neurobiol. 2021, 58, 3308–3318. [Google Scholar] [CrossRef]
- Langfelder, P.; Gao, F.; Wang, N.; Howland, D.; Kwak, S.; Vogt, T.F.; Aaronson, J.S.; Rosinski, J.; Coppola, G.; Horvath, S.; et al. MicroRNA signatures of endogenous Huntingtin CAG repeat expansion in mice. PLoS ONE 2018, 13, e0190550. [Google Scholar] [CrossRef]
- Olmo, I.G.; Olmo, R.P.; Gonçalves, A.N.A.; Pires, R.G.W.; Marques, J.T.; Ribeiro, F.M. High-Throughput Sequencing of BACHD Mice Reveals Upregulation of Neuroprotective miRNAs at the Pre-Symptomatic Stage of Huntington’s Disease. ASN Neuro 2021, 13. [Google Scholar] [CrossRef]
- Pircs, K.; Petri, R.; Madsen, S.; Brattås, P.L.; Vuono, R.; Ottosson, D.R.; St-Amour, I.; Hersbach, B.; Matusiak-Brückner, M.; Lundh, S.H.; et al. Huntingtin Aggregation Impairs Autophagy, Leading to Argonaute-2 Accumulation and Global MicroRNA Dysregulation. Cell Rep. 2018, 24, 1397–1406. [Google Scholar] [CrossRef]
- Li, K.; Zhang, J.; Ji, C.; Wang, L. MiR-144-3p and Its Target Gene β-Amyloid Precursor Protein Regulate 1-Methyl-4-Phenyl-1,2-3,6-Tetrahydropyridine-Induced Mitochondrial Dysfunction. Mol. Cells 2016, 39, 543–549. [Google Scholar] [CrossRef]
- Jovicic, A.; Zaldivar Jolissaint, J.F.; Moser, R.; de Fatima Silva Santos, M.; Luthi-Carter, R. MicroRNA-22 (miR-22) Overexpression Is Neuroprotective via General Anti-Apoptotic Effects and May also Target Specific Huntington’s Disease-Related Mechanisms. PLoS ONE 2013, 8, e54222. [Google Scholar]
- Cao, F.; Liu, Z.; Sun, G. Diagnostic value of miR-193a-3p in Alzheimer’s disease and miR-193a-3p attenuates amyloid-β induced neurotoxicity by targeting PTEN. Exp. Gerontol. 2019, 130, 110814. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, Q.; Yin, Y. miR-133b is a potential diagnostic biomarker for Alzheimer’s disease and has a neuroprotective role. Exp. Ther. Med. 2019, 18, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fei, Z.; Luo, S.; Wang, H. MiR-335-5p Inhibits β-Amyloid (Aβ) Accumulation to Attenuate Cognitive Deficits Through Targeting c-jun-N-terminal Kinase 3 in Alzheimer’s Disease. Curr. Neurovasc. Res. 2020, 17, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, D.; Zhang, B.; Lu, H. MiR-361-3p inhibits β-amyloid accumulation and attenuates cognitive deficits through targeting BACE1 in Alzheimer’s disease. J. Integr. Neurosci. 2019, 18, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p Mediates Neuroprotection in Alzheimer-Related Deficits and Attenuates Amyloid-Beta Overproduction and Tau Hyperphosphorylation via Coregulating BACE1 and PRKACB. Front. Pharmacol. 2019, 10, 806. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, K.; Fan, Q.; Zong, B.; Han, L. Knockdown of long non-coding RNA SOX21-AS1 attenuates amyloid-β-induced neuronal damage by sponging miR-107. Biosci. Rep. 2020, 40, BSR20194295. [Google Scholar] [CrossRef]
- Tan, X.; Luo, Y.; Pi, D.; Xia, L.; Li, Z.; Tu, Q. miR-340 reduces accumulation of amyloid-β through targeting BACE1 (β-site amyloid precursor protein cleaving enzyme 1) in Alzheimer’ s disease. Curr. Neurovasc. Res. 2020, 17, 86–92. [Google Scholar] [CrossRef]
- Baby, N.; Alagappan, N.; Dheen, S.T.; Sajikumar, S. MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1–42)-induced model of Alzheimer’s disease. Aging Cell 2020, 19, e13046. [Google Scholar] [CrossRef]
- Hou, T.Y.; Zhou, Y.; Zhu, L.S.; Wang, X.; Pang, P.; Wang, D.Q.; Liuyang, Z.Y.; Man, H.; Lu, Y.; Zhu, L.Q.; et al. Correcting abnormalities in miR-124/PTPN1 signaling rescues tau pathology in Alzheimer’s disease. J. Neurochem. 2020, 154, 441–457. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, K.; Zhou, H.; Jiang, L.; Xie, B.; Wang, R.; Xia, W.; Yin, Y.; Gao, Z.; Cui, D.; et al. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s Disease. Aging Cell 2020, 19, e13125. [Google Scholar] [CrossRef]
- Wang, X.; Liu, D.; Huang, H.Z.; Wang, Z.H.; Hou, T.Y.; Yang, X.; Pang, P.; Wei, N.; Zhou, Y.F.; Dupras, M.J.; et al. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 395–405. [Google Scholar] [CrossRef]
- Ansari, A.; Maffioletti, E.; Milanesi, E.; Marizzoni, M.; Frisoni, G.B.; Blin, O.; Richardson, J.C.; Bordet, R.; Forloni, G.; Gennarelli, M.; et al. miR-146a and miR-181a are involved in the progression of mild cognitive impairment to Alzheimer’s disease. Neurobiol. Aging 2019, 82, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Ghanbari, M.; Munshi, S.T.; Ma, B.; Lendemeijer, B.; Bansal, S.; Adams, H.H.; Wang, W.; Goth, K.; Slump, D.E.; van den Hout, M.C.G.N.; et al. A functional variant in the miR-142 promoter modulating its expression and conferring risk of Alzheimer disease. Hum. Mutat. 2019, 40, 2131–2145. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yu, L.; Li, M.; Chen, X.; Tian, Q.; Jiang, Y.; Li, N. MicroRNA-150 serves as a diagnostic biomarker and is involved in the inflammatory pathogenesis of Parkinson’s disease. Mol. Genet. Genom. Med. 2020, 8, e1189. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, D.; Zhang, Z.; Qu, X.; Bao, K.; Lu, G.; Duan, J. miR-let-7a suppresses α-Synuclein-induced microglia inflammation through targeting STAT3 in Parkinson’s disease. Biochem. Biophys. Res. Commun. 2019, 519, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, S.; Chen, J.; Cai, H.; Huang, W.; Zhang, Y.; Wang, L.; Xing, Y. MicroRNA-190 alleviates neuronal damage and inhibits neuroinflammation via Nlrp3 in MPTP-induced Parkinson’s disease mouse model. J. Cell. Physiol. 2019, 234, 23379–23387. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Luo, D.-X.; Li, H.-P.; Zhang, Q.-S.; Lei, S.-S.; Chen, J.-H. MicroRNA-135b alleviates MPP+-mediated Parkinson’s disease in in vitro model through suppressing FoxO1-induced NLRP3 inflammasome and pyroptosis. J. Clin. Neurosci. 2019, 65, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.J.; Tu, L.; Li, T.; Yang, X.L.; Ren, Y.P.; Gu, R.; Zhang, Q.; Yao, H.; Qu, X.; Wang, Q.; et al. Up-regulation of microRNA-375 ameliorates the damage of dopaminergic neurons, reduces oxidative stress and inflammation in Parkinson’s disease by inhibiting SP1. Aging 2020, 12, 672–689. [Google Scholar] [CrossRef]
- Wang, R.; Yang, Y.; Wang, H.; He, Y.; Li, C. MiR-29c protects against inflammation and apoptosis in Parkinson’s disease model in vivo and in vitro by targeting SP1. Clin. Exp. Pharmacol. Physiol. 2019, 47, 372–382. [Google Scholar] [CrossRef]
- Liu, W.; Li, L.; Liu, S.; Wang, Z.; Kuang, H.; Xia, Y.; Tang, C.; Yin, D. MicroRNA Expression Profiling Screen miR-3557/324-Targeted CaMK/mTOR in the Rat Striatum of Parkinson’s Disease in Regular Aerobic Exercise. Biomed Res. Int. 2019, 2019, 7654798. [Google Scholar] [CrossRef]
- Ge, H.; Yan, Z.; Zhu, H.; Zhao, H. MiR-410 exerts neuroprotective effects in a cellular model of Parkinson’s disease induced by 6-hydroxydopamine via inhibiting the PTEN/AKT/mTOR signaling pathway. Exp. Mol. Pathol. 2019, 109, 16–24. [Google Scholar] [CrossRef]
- Zhao, X.H.; Wang, Y.B.; Yang, J.; Liu, H.Q.; Wang, L.L. MicroRNA-326 suppresses iNOS expression and promotes autophagy of dopaminergic neurons through the JNK signaling by targeting XBP1 in a mouse model of Parkinson’s disease. J. Cell. Biochem. 2019, 120, 14995–15006. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, H.; Xie, W.; Wei, N.; Liu, M. MicroRNA-195 triggers neuroinflammation in Parkinson’s disease in a Rho-associated kinase 1-dependent manner. Mol. Med. Rep. 2019, 19, 5153–5161. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jiang, Y.; Xu, Y.; Li, Y.; Li, B. Identification of miRNA-7 as a regulator of brain-derived neurotrophic fac-tor/A-synuclein axis in atrazine-induced Parkinson’s disease by peripheral blood and brain microRNA profiling. Chemosphere 2019, 233, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Baghi, M.; Delavar, M.R.; Yadegari, E.; Peymani, M.; Pozo, D.; Nasr-Esfahani, M.H.; Ghaedi, K. Modified level of miR-376a is associated with Parkinson’s disease. J. Cell. Mol. Med. 2020, 24, 2622–2634. [Google Scholar] [CrossRef] [PubMed]
- De Luna, N.; Turon-Sans, J.; Cortes-Vicente, E.; Carrasco-Rozas, A.; Illán-Gala, I.; Dols-Icardo, O.; Clarimón, J.; Lleó, A.; Gallardo, E.; Illa, I.; et al. Downregulation of miR-335-5P in Amyotrophic Lateral Sclerosis Can Contribute to Neuronal Mitochondrial Dysfunction and Apoptosis. Sci. Rep. 2020, 10, 4308. [Google Scholar] [CrossRef]
- Rohm, M.; May, C.; Marcus, K.; Steinbach, S.; Theis, V.; Theiss, C.; Matschke, V. The microRNA miR-375-3p and the tumor suppressor NDRG2 are involved in sporadic amyotrophic lateral sclerosis. Cell. Physiol. Biochem. 2019, 52, 1412–1426. [Google Scholar]
- Klatt, C.L.; Theis, V.; Hahn, S.; Theiss, C.; Matschke, V. Deregulated miR-29b-3p Correlates with Tissue-Specific Activation of Intrinsic Apoptosis in An Animal Model of Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1077. [Google Scholar] [CrossRef]
- Maimon, R.; Ionescu, A.; Bonnie, A.; Sweetat, S.; Wald-Altman, S.; Inbar, S.; Gradus, T.; Trotti, D.; Weil, M.; Behar, O.; et al. miR126-5p Downregulation Facilitates Axon Degeneration and NMJ Disruption via a Non-Cell-Autonomous Mechanism in ALS. J. Neurosci. 2018, 38, 5478–5494. [Google Scholar] [CrossRef]
- Varcianna, A.; Myszczynska, M.A.; Castelli, L.M.; O’Neill, B.; Kim, Y.; Talbot, J.; Nyberg, S.; Nyamali, I.; Heath, P.R.; Stopford, M.J.; et al. Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. eBioMedicine 2019, 40, 626–635. [Google Scholar] [CrossRef]
- Helferich, A.M.; Brockmann, S.J.; Reinders, J.; Deshpande, D.; Holzmann, K.; Brenner, D.; Andersen, P.M.; Petri, S.; Thal, D.R.; Michaelis, J.; et al. Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell. Mol. Life Sci. 2018, 75, 4301–4319. [Google Scholar] [CrossRef]
- Li, C.; Wei, Q.; Gu, X.; Chen, Y.; Chen, X.; Cao, B.; Ou, R.; Shang, H. Decreased Glycogenolysis by miR-338-3p Promotes Regional Glycogen Accumulation Within the Spinal Cord of Amyotrophic Lateral Sclerosis Mice. Front. Mol. Neurosci. 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Hawley, Z.C.E.; Campos-Melo, D.; Strong, M.J. MiR-105 and miR-9 regulate the mRNA stability of neuronal intermediate filaments. Implications for the pathogenesis of amyotrophic lateral sclerosis (ALS). Brain Res. 2019, 1706, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hoye, M.L.; Regan, M.R.; A Jensen, L.; Lake, A.M.; Reddy, L.V.; Vidensky, S.; Richard, J.-P.; Maragakis, N.J.; Rothstein, J.D.; Dougherty, J.D.; et al. Motor neuron-derived microRNAs cause astrocyte dysfunction in amyotrophic lateral sclerosis. Brain 2018, 141, 2561–2575. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 Delays ALS Progression and Promotes Regeneration of Neuromuscular Synapses in Mice. Science 2009, 326, 1549–1554. [Google Scholar] [CrossRef]
- Tung, Y.-T.; Peng, K.-C.; Chen, Y.-C.; Yen, Y.-P.; Chang, M.; Thams, S.; Chen, J.-A. Mir-17~92 Confers Motor Neuron Subtype Differential Resistance to ALS-Associated Degeneration. Cell Stem Cell 2019, 25, 193–209.e7. [Google Scholar] [CrossRef]
- Mégret, L.; Nair, S.S.; Dancourt, J.; Aaronson, J.; Rosinski, J.; Neri, C. Combining feature selection and shape analysis uncovers precise rules for miRNA regulation in Huntington’s disease mice. BMC Bioinform. 2020, 21, 75. [Google Scholar] [CrossRef]
- Fukuoka, M.; Takahashi, M.; Fujita, H.; Chiyo, T.; Popiel, H.A.; Watanabe, S.; Furuya, H.; Murata, M.; Wada, K.; Okada, T.; et al. Supplemental Treatment for Huntington’s Disease with miR-132 that Is Deficient in Huntington’s Disease Brain. Mol. Ther. Nucleic Acids 2018, 11, 79–90. [Google Scholar] [CrossRef]
- Kunkanjanawan, T.; Carter, R.L.; Prucha, M.S.; Yang, J.; Parnpai, R.; Chan, A.W.S. miR-196a Ameliorates Cytotoxicity and Cellular Phenotype in Transgenic Huntington’s Disease Monkey Neural Cells. PLoS ONE 2016, 11, e0162788. [Google Scholar] [CrossRef]
- Her, L.-S.; Mao, S.-H.; Chang, C.-Y.; Cheng, P.-H.; Chang, Y.-F.; Yang, H.-I.; Chen, C.-M.; Yang, S.-H. miR-196a Enhances Neuronal Morphology through Suppressing RANBP10 to Provide Neuroprotection in Huntington’s Disease. Theranostics 2017, 7, 2452–2462. [Google Scholar] [CrossRef]
- Ban, J.-J.; Chung, J.-Y.; Lee, M.; Im, W.; Kim, M. MicroRNA-27a reduces mutant hutingtin aggregation in an in vitro model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 488, 316–321. [Google Scholar] [CrossRef]
- Pfister, E.L.; DiNardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial miRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington’s Disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2019, 48, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Macedo, B.; Silva, J.L.; Gomes, M.P.B. Pathological implications of nucleic acid interactions with proteins associated with neurodegenerative diseases. Biophys. Rev. 2014, 6, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, Y.; Silva, J.L. The Hypothesis of the Catalytic Action of Nucleic Acid on the Conversion of Prion Protein. Protein Pept. Lett. 2005, 12, 251–255. [Google Scholar] [CrossRef]
- Macedo, B.; Millen, T.A.; Braga, C.A.C.A.; Gomes, M.P.B.; Ferreira, P.S.; Kraineva, J.; Winter, R.; Silva, J.; Cordeiro, Y. Nonspecific Prion Protein–Nucleic Acid Interactions Lead to Different Aggregates and Cytotoxic Species. Biochemistry 2012, 51, 5402–5413. [Google Scholar] [CrossRef]
- Gomes, M.P.B.; Millen, T.A.; Ferreira, P.S.; e Silva, N.L.C.; Vieira, T.C.R.G.; Almeida, M.; Silva, J.L.; Cordeiro, Y. Prion Protein Complexed to N2a Cellular RNAs through Its N-terminal Domain Forms Aggregates and Is Toxic to Murine Neuroblastoma Cells. J. Biol. Chem. 2008, 283, 19616–19625. [Google Scholar] [CrossRef]
- Tandon, A.; Subramani, V.K.; Kim, K.K.; Park, S.H. Interaction of Prion Peptides with DNA Structures. ACS Omega 2021, 7, 176–186. [Google Scholar] [CrossRef]
- Adler, V.; Zeiler, B.; Kryukov, V.; Kascsak, R.; Rubenstein, R.; Grossman, A. Small, highly structured RNAs participate in the conversion of human recombinant PrPSen to PrPRes in vitro. J. Mol. Biol. 2003, 332, 47–57. [Google Scholar] [CrossRef]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef]
- Mashima, T.; Nishikawa, F.; Kamatari, Y.O.; Fujiwara, H.; Saimura, M.; Nagata, T.; Kodaki, T.; Nishikawa, S.; Kuwata, K.; Katahira, M. Anti-prion activity of an RNA aptamer and its structural basis. Nucleic Acids Res. 2012, 41, 1355–1362. [Google Scholar] [CrossRef]
- Dalai, W.; Matsuo, E.; Takeyama, N.; Kawano, J.; Saeki, K. CpG site DNA methylation patterns reveal a novel regulatory element in the mouse prion protein gene. J. Vet. Med. Sci. 2017, 79, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Dalai, W.; Matsuo, E.; Takeyama, N.; Kawano, J.; Saeki, K. Increased expression of prion protein gene is accompanied by demethylation of CpG sites in a mouse embryonal carcinoma cell line, P19C6. J. Vet. Med. Sci. 2017, 79, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Dabin, L.C.; Guntoro, F.; Campbell, T.; Bélicard, T.; Smith, A.R.; Smith, R.G.; Raybould, R.; Schott, J.M.; Lunnon, K.; Sarkies, P.; et al. Altered DNA methylation profiles in blood from patients with sporadic Creutzfeldt–Jakob disease. Acta Neuropathol. 2020, 140, 863–879. [Google Scholar] [CrossRef] [PubMed]
- Hernaiz, A.; Sanz, A.; Sentre, S.; Ranera, B.; Lopez-Pérez, O.; Zaragoza, P.; Badiola, J.J.; Filali, H.; Bolea, R.; Toivonen, J.M.; et al. Genome-Wide Methylation Profiling in the Thalamus of Scrapie Sheep. Front. Vet. Sci. 2022, 9, 824677. [Google Scholar] [CrossRef]
- Guntoro, F.; Viré, E.; Giordani, C.; Darwent, L.; Hummerich, H.; Linehan, J.; Sinka, K.; Jaunmuktane, Z.; Brandner, S.; Collinge, J.; et al. DNA methylation analysis of archival lymphoreticular tissues in Creutzfeldt–Jakob disease. Acta Neuropathol. 2022, 144, 785–787. [Google Scholar] [CrossRef]
- Harvey, Z.H.; Chakravarty, A.K.; Futia, R.A.; Jarosz, D.F. A Prion Epigenetic Switch Establishes an Active Chromatin State. Cell 2020, 180, 928–940.e14. [Google Scholar] [CrossRef]
- Zhu, T.; Zhao, D.; Song, Z.; Yuan, Z.; Li, C.; Wang, Y.; Zhou, X.; Yin, X.; Hassan, M.F.; Yang, L. HDAC6 alleviates prion peptide-mediated neuronal death via modulating PI3K-Akt-mTOR pathway. Neurobiol. Aging 2016, 37, 91–102. [Google Scholar] [CrossRef]
- Seo, J.-S.; Moon, M.-H.; Jeong, J.-K.; Seol, J.-W.; Lee, Y.-J.; Park, B.-H.; Park, S.-Y. SIRT1, a histone deacetylase, regulates prion protein-induced neuronal cell death. Neurobiol. Aging 2012, 33, 1110–1120. [Google Scholar] [CrossRef]
- Jeong, J.-K.; Moon, M.-H.; Lee, Y.-J.; Seol, J.-W.; Park, S.-Y. Autophagy induced by the class III histone deacetylase Sirt1 prevents prion peptide neurotoxicity. Neurobiol. Aging 2012, 34, 146–156. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, J.; Shi, Q.; Zhang, B.-Y.; Chen, C.; Chen, L.-N.; Sun, J.; Wang, H.; Xiao, K.; Dong, X.-P. Scrapie Infection in Experimental Rodents and SMB-S15 Cells Decreased the Brain Endogenous Levels and Activities of Sirt1. J. Mol. Neurosci. 2014, 55, 1022–1030. [Google Scholar] [CrossRef]
- Boese, A.S.; Saba, R.; Campbell, K.; Majer, A.; Medina, S.; Burton, L.; Booth, T.F.; Chong, P.; Westmacott, G.; Dutta, S.M.; et al. MicroRNA abundance is altered in synaptoneurosomes during prion disease. Mol. Cell. Neurosci. 2016, 71, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wei, J.; Zhang, B.-Y.; Shi, Q.; Chen, C.; Wang, J.; Shi, Q.; Dong, X.-P. MiRNA expression profiles in the brains of mice infected with scrapie agents 139A, ME7 and S15. Emerg. Microbes Infect. 2016, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Toivonen, J.M.; Sanz-Rubio, D.; López-Pérez, Ó.; Marín-Moreno, A.; Bolea, R.; Osta, R.; Badiola, J.J.; Zaragoza, P.; Espinosa, J.-C.; Torres, J.-M.; et al. MicroRNA Alterations in a Tg501 Mouse Model of Prion Disease. Biomolecules 2020, 10, 908. [Google Scholar] [CrossRef] [PubMed]
- Slota, J.; Medina, S.J.; Klassen, M.; Gorski, D.; Mesa, C.M.; Robertson, C.; Mitchell, G.; Coulthart, M.B.; Pritzkow, S.; Soto, C.; et al. Identification of circulating microRNA signatures as potential biomarkers in the serum of elk infected with chronic wasting disease. Sci. Rep. 2019, 9, 19705. [Google Scholar] [CrossRef]
- Rubio, D.S.; López-Pérez, Ó.; de Andrés Pablo, A.; Bolea, R.; Osta, R.; Badiola, J.J.; Zaragoza, P.; Martín-Burriel, I.; Toivonen, J.M. Increased circulating microRNAs miR-342-3p and miR-21-5p in natural sheep prion disease. J. Gen. Virol. 2017, 98, 305–310. [Google Scholar] [CrossRef]
- Pease, D.; Scheckel, C.; Schaper, E.; Eckhardt, V.; Emmenegger, M.; Xenarios, I.; Aguzzi, A. Genome-wide identification of microRNAs regulating the human prion protein. Brain Pathol. 2018, 29, 232–244. [Google Scholar] [CrossRef]
- Burak, K.; Lamoureux, L.; Boese, A.; Majer, A.; Saba, R.; Niu, Y.; Frost, K.; Booth, S.A. MicroRNA-16 targets mRNA involved in neurite extension and branching in hippocampal neurons during presymptomatic prion disease. Neurobiol. Dis. 2018, 112, 1–13. [Google Scholar] [CrossRef]
- Gao, C.; Shi, Q.; Wei, J.; Zhou, W.; Xiao, K.; Wang, J.; Shi, Q.; Dong, X.-P. The associations of two SNPs in miRNA-146a and one SNP in ZBTB38-RASA2 with the disease susceptibility and the clinical features of the Chinese patients of sCJD and FFI. Prion 2018, 12, 34–41. [Google Scholar] [CrossRef]
- Kang, S.-G.; Kim, C.; Aiken, J.; Yoo, H.S.; McKenzie, D. Dual MicroRNA to Cellular Prion Protein Inhibits Propagation of Pathogenic Prion Protein in Cultured Cells. Mol. Neurobiol. 2017, 55, 2384–2396. [Google Scholar] [CrossRef]


| miRNA | Prion Diseases | AD | PD | ALS | HD | Upregulated (+)/Downregulated (−) |
|---|---|---|---|---|---|---|
| miR-5100 | + | + | + | Upregulated | ||
| miR-342-3p | +/− | − | +/− | +/− | Upregulated/Downregulated | |
| let-7f-5p | + | + | + | Upregulated | ||
| miR-146b-5p | + | + | + | + | Upregulated | |
| let-7a-5p | + | + | + | Upregulated | ||
| miR-378c | + | + | + | Upregulated | ||
| miR-27a-3p | + | + | + | Upregulated | ||
| miR-339-3p | + | + | + | Upregulated | ||
| miR-142-5p | + | + | + | + | Upregulated | |
| miR-146a-5p | + | + | Upregulated | |||
| miR-320a-3p | +/− | + | + | − | Upregulated/Downregulated | |
| miR-10a-5p | + | + | Upregulated | |||
| miR-326 | + | + | Upregulated | |||
| miR-21-5p | + | + | Upregulated | |||
| miR-324-5p | + | + | Upregulated | |||
| miR-103a-3p | + | + | Upregulated | |||
| miR-331-3p | +/− | − | +/− | Upregulated/Downregulated | ||
| miR-107 | + | + | Upregulated | |||
| miR-142-3p | + | + | Upregulated | |||
| miR-129-5p | − | − | − | Downregulated | ||
| miR-423-5p | − | − | − | Downregulated | ||
| miR-125a-5p | − | − | − | Downregulated | ||
| miR-148a-3p | − | − | − | Downregulated | ||
| miR-186-5p | − | − | − | Downregulated | ||
| miR-141-3p | − | − | − | Downregulated | ||
| miR-149-5p | − | − | Downregulated | |||
| miR-200a-3p | − | − | Downregulated | |||
| miR-200b | − | − | Downregulated | |||
| miR-323-3p | − | − | Downregulated | |||
| miR-338-3p | − | − | − | Downregulated | ||
| miR-342-5p | − | − | Downregulated | |||
| miR-382 | − | − | Downregulated | |||
| miR-383 | − | − | Downregulated | |||
| miR-433 | − | − | Downregulated | |||
| miR-455-5p | − | − | Downregulated | |||
| let-7b | − | − | Downregulated | |||
| let-7c | − | − | Downregulated | |||
| miR-486-3p | − | − | Downregulated | |||
| miR-183-5p | − | − | Downregulated | |||
| miR-100-5p | − | − | Downregulated | |||
| miR-125b-5p | − | − | Downregulated | |||
| miR-99a-5p | − | − | Downregulated | |||
| miR-145-3p | − | − | Downregulated | |||
| miR-410-3p | − | − | Downregulated | |||
| miR-181d-5p | − | − | Downregulated | |||
| miR-375 | − | − | Downregulated | |||
| miR-99b-5p | − | − | Downregulated | |||
| miR-30e-3p | − | − | − | − | Downregulated | |
| miR-129-2-3p | − | − | − | − | Downregulated | |
| miR-223-3p | − | − | − | − | Downregulated | |
| miR-493-3p | − | − | Downregulated | |||
| miR-182-3p | − | − | Downregulated | |||
| miR-877-5p | − | − | − | Downregulated | ||
| miR-182-5p | − | − | − | Downregulated | ||
| miR-144-5p | − | − | − | Downregulated | ||
| miR-181c-5p | − | − | − | Downregulated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernaiz, A.; Toivonen, J.M.; Bolea, R.; Martín-Burriel, I. Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 12609. https://doi.org/10.3390/ijms232012609
Hernaiz A, Toivonen JM, Bolea R, Martín-Burriel I. Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives. International Journal of Molecular Sciences. 2022; 23(20):12609. https://doi.org/10.3390/ijms232012609
Chicago/Turabian StyleHernaiz, Adelaida, Janne Markus Toivonen, Rosa Bolea, and Inmaculada Martín-Burriel. 2022. "Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives" International Journal of Molecular Sciences 23, no. 20: 12609. https://doi.org/10.3390/ijms232012609
APA StyleHernaiz, A., Toivonen, J. M., Bolea, R., & Martín-Burriel, I. (2022). Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives. International Journal of Molecular Sciences, 23(20), 12609. https://doi.org/10.3390/ijms232012609