
Human Papillomavirus 16 E2 as an Apoptosis-Inducing Protein for Cancer Treatment: A Systematic Review
 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
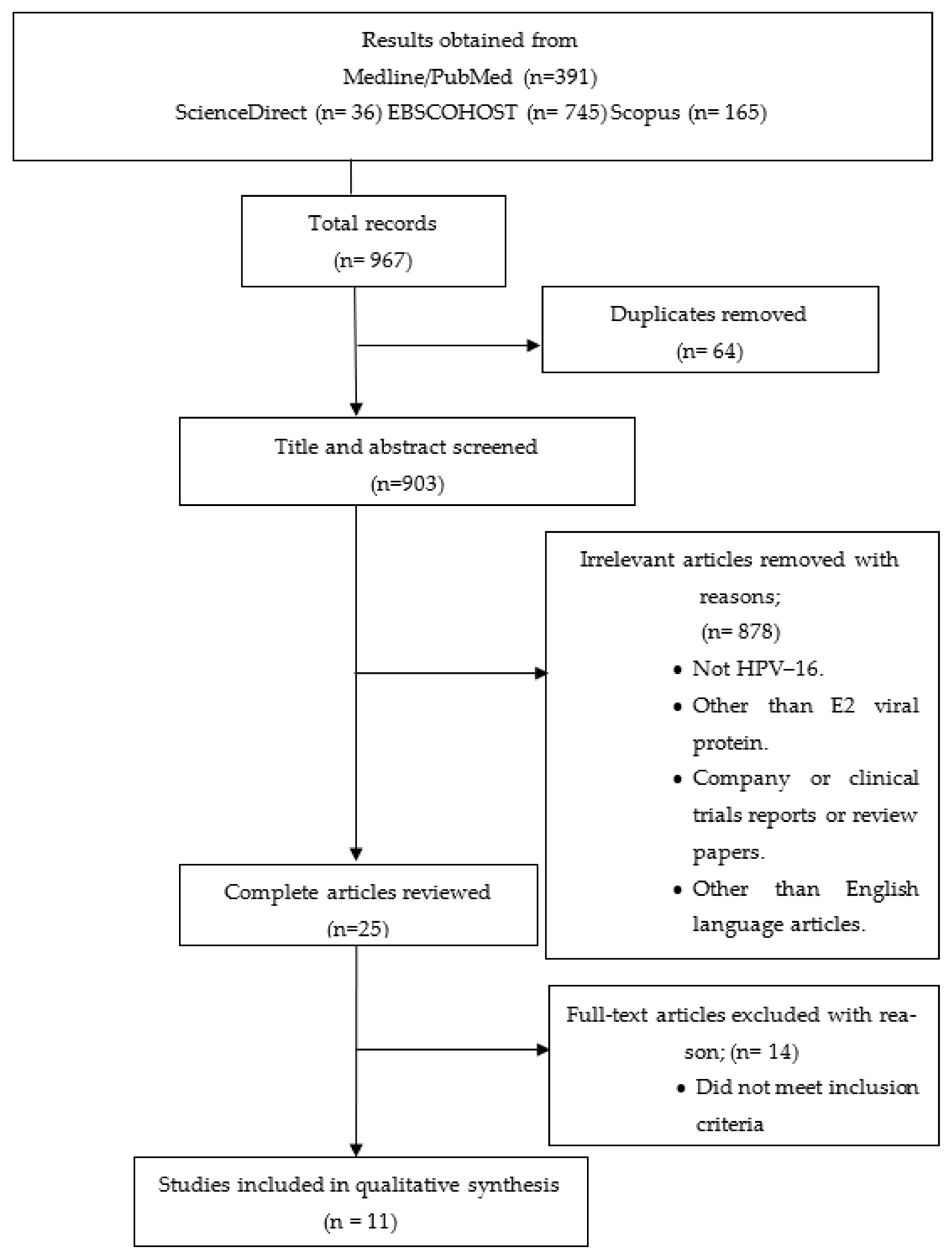
2.1. Search Methods for Selection of Studies
2.1.1. Literature Search
2.1.2. Review of Reference Lists
2.2. Data Extraction
2.3. Risk Assessment
2.4. Summary Measurement
3. Results
3.1. Methods to Determine Apoptosis and Cell Viability
3.2. Vector or Cofactor
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Estimated Number of New Cases in 2020, Worldwide, Females, All Ages. Available online: https://gco.iarc.fr/ (accessed on 20 August 2021).
- Cohen, P.A.; Jhingran, A.; Oaknin, A.; Denny, L. Cervical cancer. Lancet 2019, 393, 169–182. [Google Scholar] [CrossRef]
- Bruni, L.B.R.L.; Barrionuevo-Rosas, L.; Albero, G.; Aldea, M.; Serrano, B.; Valencia, S.; Brotons, M.; Mena, M.; Cosano, R.; Muñoz, J.; et al. Human Papillomavirus and Related Diseases in the World; Summary Report; Institut Català d’Oncologia: Barcelona, Spain, 2019. [Google Scholar]
- Robadi, I.A.; Pharaon, M.; Ducatman, B.S. The Importance of High-Risk Human Papillomavirus Types Other Than 16 and 18 in Cervical Neoplasia. Arch. Pathol. Lab. Med. 2018, 142, 693–695. [Google Scholar] [CrossRef]
- de Sanjosé, S.; Brotons, M.; Pavón, M.A. The natural history of human papillomavirus infection. Best. Pract. Res. Clin. Obstet. Gynaecol. 2018, 47, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Mac, M.; Moody, C.A. Epigenetic Regulation of the Human Papillomavirus Life Cycle. Pathogens 2020, 9, 483. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.M.; Mania-Pramanik, J. Molecular mechanisms in progression of HPV-associated cervical carcinogenesis. J. Biomed. Sci. 2019, 26, 28. [Google Scholar] [CrossRef]
- Okunade, K.S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 2020, 40, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.T.; Castellsagué, X.; Garland, S.M. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine 2012, 30 (Suppl. 5), F123–F138. [Google Scholar] [CrossRef]
- Yang, D.Y.; Bracken, K. Update on the new 9-valent vaccine for human papillomavirus prevention. Can. Fam. Physician 2016, 62, 399–402. [Google Scholar]
- Graham, S.V. Human Papillomavirus E2 Protein: Linking Replication, Transcription, and RNA Processing. J. Virol. 2016, 90, 8384–8388. [Google Scholar] [CrossRef]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef]
- Collins, S.I.; Constandinou-Williams, C.; Wen, K.; Young, L.S.; Roberts, S.; Murray, P.G.; Woodman, C.B.J. Disruption of the E2 gene is a common and early event in the natural history of cervical human papillomavirus infection: A longitudinal cohort study. Cancer Res. 2009, 69, 3828–3832. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, B.; Sengupta, S. HPV16 E2 gene disruption and polymorphisms of E2 and LCR: Some significant associations with cervical cancer in Indian women. Gynecol Oncol. 2006, 100, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Blachon, S.; Bellanger, S.; Demeret, C.; Thierry, F. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 2005, 280, 36088–36098. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Kowalczyk, A.; Chen, H.T.; Roeder, G.E.; Sessions, R.; Buckle, M.; Gaston, K. E2 proteins from high- and low-risk human papillomavirus types differ in their ability to bind p53 and induce apoptotic cell death. J. Virol. 2006, 80, 4580–4590. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.E.; Walker, H.F.; Schmitz, C.; Maitland, N.J. Phenotypic effects of HPV-16 E2 protein expression in human keratinocytes. Virology 2010, 401, 314–321. [Google Scholar] [CrossRef]
- Wang, W.; Fang, Y.; Sima, N.; Li, Y.; Li, W.; Li, L.; Han, L.; Liao, S.; Han, Z.; Gao, Q.; et al. Triggering of death receptor apoptotic signaling by human papillomavirus 16 E2 protein in cervical cancer cell lines is mediated by interaction with c-FLIP. Apoptosis 2011, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, X.; Wang, S.; Sima, N.; Li, Y.; Han, Z.; Gao, Q.; Luo, A.; Li, K.; Meng, L.; et al. Oncolytic adenovirus armed with human papillomavirus E2 gene in combination with radiation demonstrates synergistic enhancements of antitumor efficacy. Cancer Gene Ther. 2011, 18, 825–836. [Google Scholar] [CrossRef]
- Chen, Z.-l.; Su, Y.-j.; Zhang, H.-l.; Gu, P.-q.; Gao, L.-j. The role of the globular heads of the C1q receptor in HPV-16 E2-induced human cervical squamous carcinoma cell apoptosis via a mitochondria-dependent pathway. J. Transl. Med. 2014, 12, 286. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, A.M.; Soriano, S.; Clarke, A.R.; Gaston, K. Disruption of the human papillomavirus type 16 E2 gene protects cervical carcinoma cells from E2F-induced apoptosis. J. Gen. Virol. 1997, 78 Pt 11, 3009–3018. [Google Scholar] [CrossRef]
- Roeder, G.E.; Parish, J.L.; Stern, P.L.; Gaston, K. Herpes simplex virus VP22–human papillomavirus E2 fusion proteins produced in mammalian or bacterial cells enter mammalian cells and induce apoptotic cell death. Biotechnol. Appl. Biochem. 2004, 40, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.; Parish, J.; Pandya, M.; Stern, P.L.; Clarke, A.R.; Gaston, K. The human papillomavirus (HPV) 16 E2 protein induces apoptosis in the absence of other HPV proteins and via a p53-dependent pathway. J. Biol. Chem. 2000, 275, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Green, K.L.; Brown, C.; Roeder, G.E.; Southgate, T.D.; Gaston, K. A Cancer Cell-Specific Inducer of Apoptosis. Hum. Gene Ther. 2007, 18, 547–561. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Wang, P.; Yu, M.; Zhu, Y.; Teng, L.; Su, Y. The Role of the Hematopoietic Cell-Specific Protein 1-Associated Protein X-1 in Human Papillomavirus 16 E2-Induced Human Cervical Squamous Carcinoma Cell Apoptosis via a Mitochondria-Dependent Pathway. Gynecol Obstet Invest. 2021, 86, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathy, D.; Prabhakar, B.N.; Karunagaran, D. HPV16 E2-mediated potentiation of NF-κB activation induced by TNF-α involves parallel activation of STAT3 with a reduction in E2-induced apoptosis. Mol. Cell. Biochem. 2014, 394, 77–90. [Google Scholar] [CrossRef]
- Prabhavathy, D.; Subramanian, C.K.; Karunagaran, D. Re-expression of HPV16 E2 in SiHa (human cervical cancer) cells potentiates NF-κB activation induced by TNF-α concurrently increasing senescence and survival. Biosci. Rep. 2015, 35, e00175. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Morales, V.H.; Peralta-Zaragoza, O.; Guzmán-Olea, E.; García-Carrancá, A.; Bahena-Román, M.; Alcocer-González, J.M.; Madrid-Marina, V. HPV 16 E2 protein induces apoptosis in human and murine HPV 16 transformed epithelial cells and has antitumoral effects in vivo. Tumour. Biol. 2009, 30, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.; Taylor, A.; Gaston, K. Oestrogen and progesterone increase the levels of apoptosis induced by the human papillomavirus type 16 E2 and E7 proteins. J. Gen. Virol. 2001, 82, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Kowalczyk, A.M.; Taylor, E.R.; Morgan, I.M.; Gaston, K. P53 represses human papillomavirus type 16 DNA replication via the viral E2 protein. Virol J. 2008, 5, 5. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Ghosh, I.; Datta, K. Excessive reactive oxygen species induces apoptosis in fibroblasts: Role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR). Exp. Cell Res. 2008, 314, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Román-Rosales, A.A.; García-Villa, E.; Herrera, L.A.; Gariglio, P.; Díaz-Chávez, J. Mutant p53 gain of function induces HER2 over-expression in cancer cells. BMC Cancer 2018, 18, 709. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold. Spring. Harb. Protoc. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Do TUNEL and Other Apoptosis Assays Detect Cell Death in Preclinical Studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| First Author (Year) (Ref) | Study Outcome | Vector or Cofactor Used | Method to Determine the Apoptosis | Cell Viability Test | Level of Apoptosis Achieved | Cell Line Used | ||
|---|---|---|---|---|---|---|---|---|
| Flow Cytometry | Fluorescence Microscopy | TUNEL Assay | ||||||
| Wang et al., (2011) [19] |
|
| Yes | Yes | Yes | Yes |
|
|
| Wang et al., (2011)[20] |
|
| Yes | No | Yes | Yes |
|
|
| Chen et al., (2014) [21] |
|
| Yes | No | No | No |
|
|
| Sanchez-Perez et al., (1997) [22] |
|
| Yes | Yes | No | Yes |
|
|
| Roeder et al., (2004) [23] |
|
| Yes | Yes | No | No |
|
|
| Webster et al., (2000)[24] |
|
| No | Yes | No | No |
|
|
| Green et al., (2007) [25] |
|
| Yes | Yes | Yes | No |
|
|
| Gong et al., (2021) [26] |
|
| Yes | No | No | No |
|
|
| Prabhavathy et al., (2014) [27] |
|
| Yes | No | No | Yes |
|
|
| Prabhavathy et al., (2015) [28] |
|
| Yes | No | No | Yes |
| SiHa |
| Bermudez et al., (2009) [29] |
|
| No | Yes | Yes | Yes |
|
|
| Cell Line | Description | Study Involved | Apoptotic Effect |
|---|---|---|---|
| SiHa | HPV-16 transformed cervical carcinoma | [19,20,21,22,23,24,25,26,27,28,29] | Yes |
| HeLa | HPV-18 transformed cervical carcinoma | [20,23,24,25,27] | Yes |
| C33A | HPV negative from cervical carcinoma | [19,20,21], [24] *, [26], [29] * | Yes |
| HEK293 | Human embryonic kidney | [25,27] | Yes |
| COS-7 | Monkey kidney fibroblast | [23,24] | No |
| MCF-7 | Human breast adenocarcinoma | [23,25,27] | Yes |
| NIH3T3 | Mouse embryo fibroblast | [24,25] | Yes |
| BMK-16/myc | Murine c-myc-1 gene | [29] | Yes |
| 866 | HPV-16-transformed cells | [24,25] | Yes |
| B16 | Murine melanoma | [23] | Yes |
| CasKi | Cervical epidermoid carcinoma | [23,25,29] | Yes |
| 778 | HPV-18 | [25] | Yes |
| 808F | HPV negative cells, normal non-transformed cervical fibroblast cells | [24,25] | Yes |
| 873 | HPV-18 | [24,25] | Yes |
| 873F | HPV negative cells, normal non-transformed cervical fibroblast cells | [23,25] | Yes |
| 877 | HPV-18, 45 | [24] | Yes |
| 915 | HPV-16 | [24] | Yes |
| W12 | HPV-16 human cervical keratinocyte | [23] | Yes |
| Saos-2 | HPV negative | [25] | Yes |
| Transfection Reagent | Company | Author |
|---|---|---|
| Tfx-20 | Promega, Madison, WI | [25] |
| Tfx-50 | Promega | [25] |
| FuGENE 6 | Roche | [25] |
| Lipofectamine | Invitrogen | [19,26,27,28,29] |
| Lipofectamine | Life Technologies | [21] |
| Calcium phosphate | NV | [27] |
| Electroporation | NV | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jamal, D.F.; Rozaimee, Q.A.; Osman, N.H.; Mohd Sukor, A.; Elias, M.H.; Shamaan, N.A.; Das, S.; Abdul Hamid, N. Human Papillomavirus 16 E2 as an Apoptosis-Inducing Protein for Cancer Treatment: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 12554. https://doi.org/10.3390/ijms232012554
Jamal DF, Rozaimee QA, Osman NH, Mohd Sukor A, Elias MH, Shamaan NA, Das S, Abdul Hamid N. Human Papillomavirus 16 E2 as an Apoptosis-Inducing Protein for Cancer Treatment: A Systematic Review. International Journal of Molecular Sciences. 2022; 23(20):12554. https://doi.org/10.3390/ijms232012554
Chicago/Turabian StyleJamal, Dinah Farhanah, Quratul Ain Rozaimee, Nadila Haryani Osman, Atikah Mohd Sukor, Marjanu Hikmah Elias, Nor Aripin Shamaan, Srijit Das, and Nazefah Abdul Hamid. 2022. "Human Papillomavirus 16 E2 as an Apoptosis-Inducing Protein for Cancer Treatment: A Systematic Review" International Journal of Molecular Sciences 23, no. 20: 12554. https://doi.org/10.3390/ijms232012554
APA StyleJamal, D. F., Rozaimee, Q. A., Osman, N. H., Mohd Sukor, A., Elias, M. H., Shamaan, N. A., Das, S., & Abdul Hamid, N. (2022). Human Papillomavirus 16 E2 as an Apoptosis-Inducing Protein for Cancer Treatment: A Systematic Review. International Journal of Molecular Sciences, 23(20), 12554. https://doi.org/10.3390/ijms232012554