
mTOR as a Potential Target for the Treatment of Microbial Infections, Inflammatory Bowel Diseases, and Colorectal Cancer

 , , , , and
, , , , and
Abstract
1. Introduction
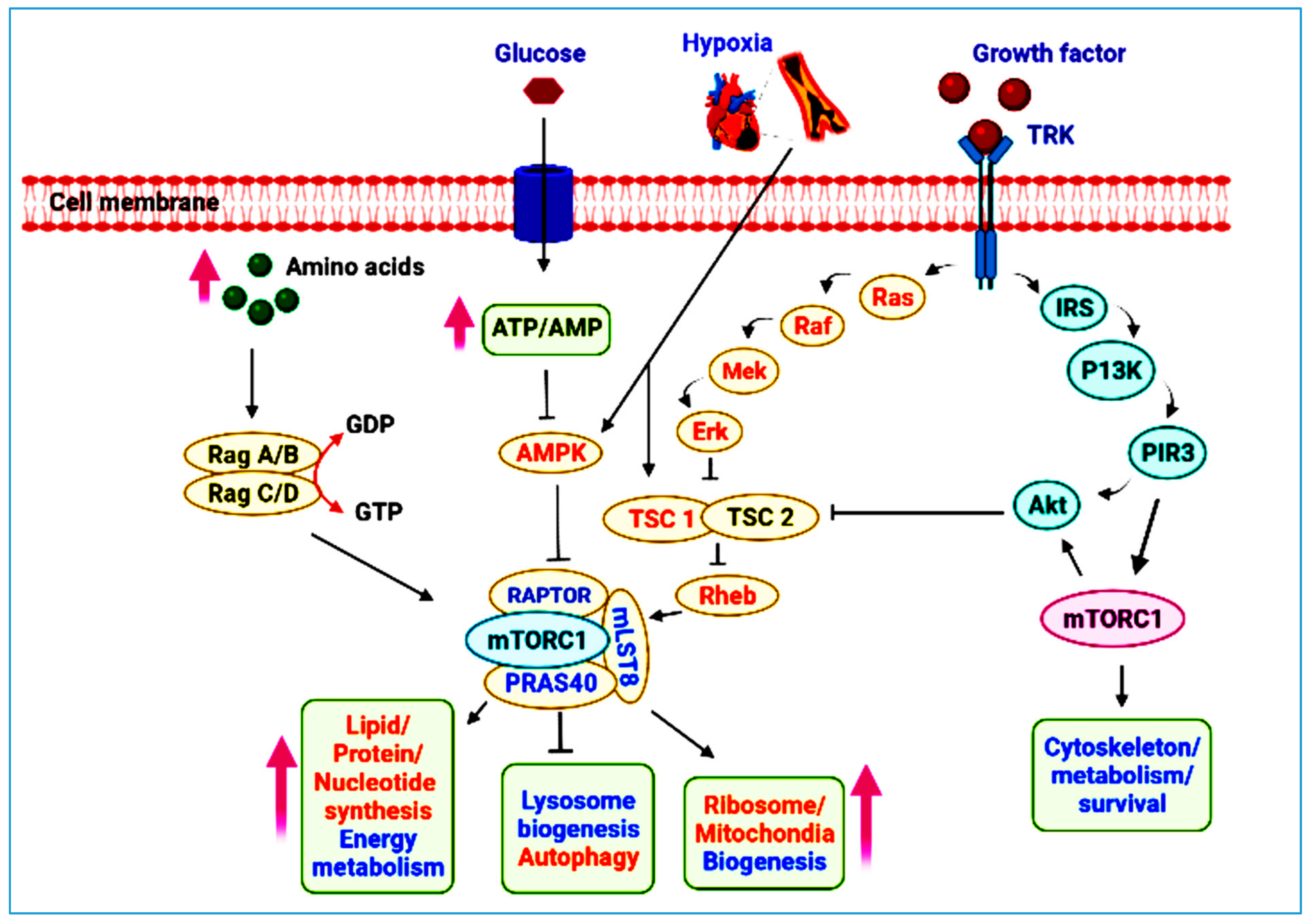
2. mTOR Pathway at a Glance
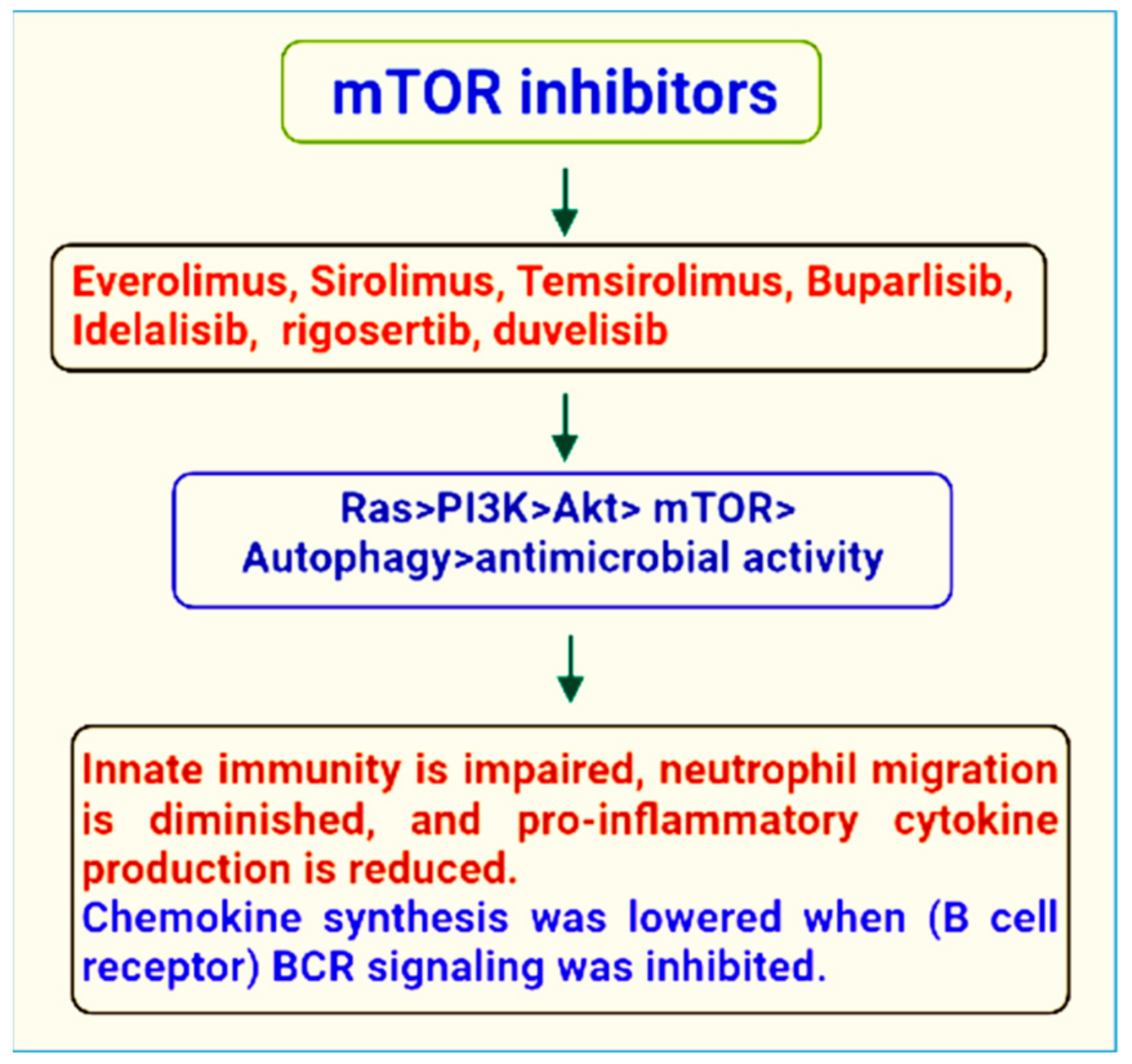
3. mTOR Inhibitors
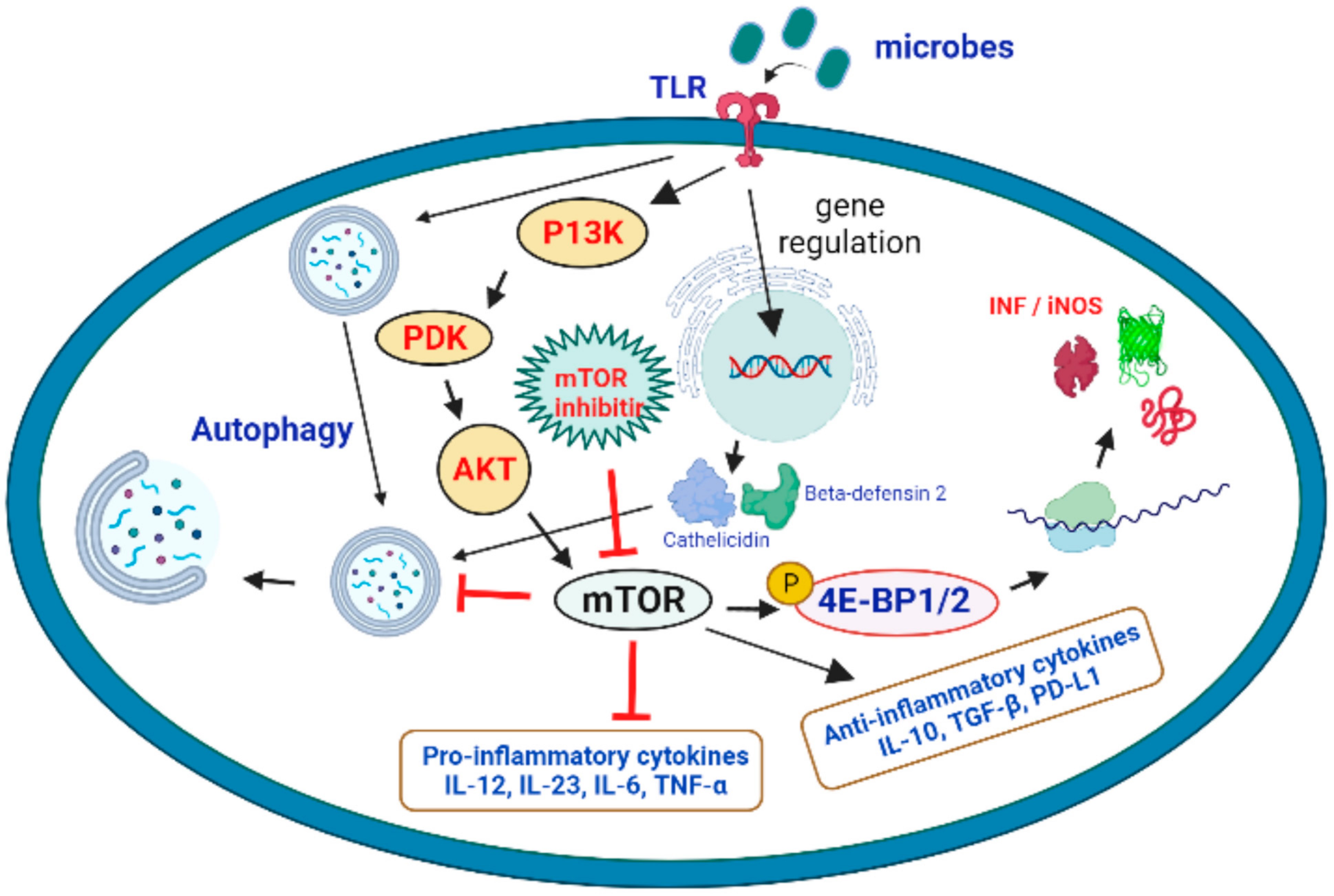
4. Therapeutic Role of mTOR Signaling in the Treatment of Microbial Infections
5. mTOR in Inflammatory Bowel Diseases
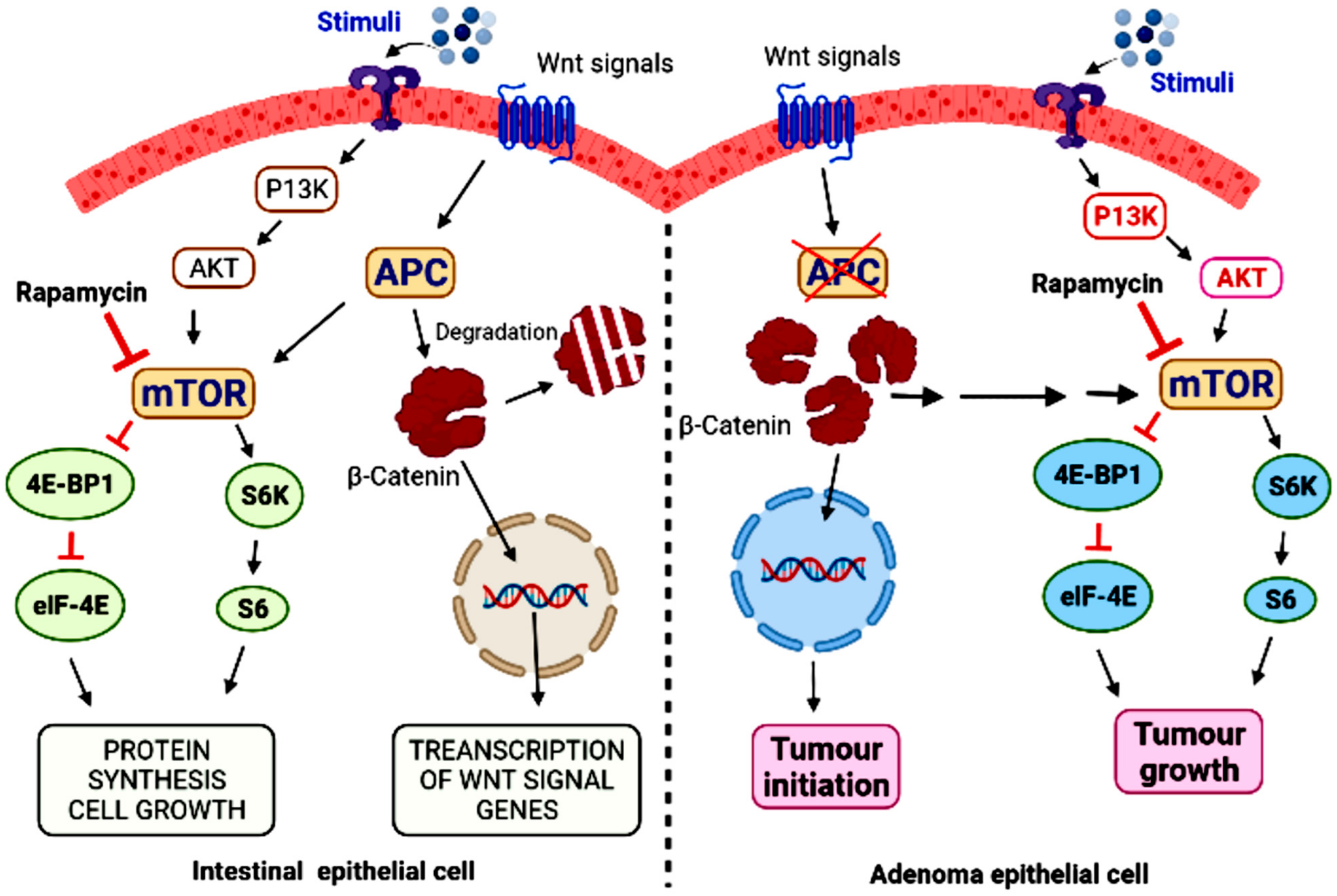
6. mTOR and Colorectal Cancer
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pignataro, G.; Capone, D.; Polichetti, G.; Vinciguerra, A.; Gentile, A.; Di Renzo, G.; Annunziato, L. Neuroprotective, immunosuppressant and antineoplastic properties of mTOR inhibitors: Current and emerging therapeutic options. Curr. Opin. Pharmacol. 2011, 11, 378–394. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garrido, J.; Shenoy, A.R. Regulation and repurposing of nutrient sensing and autophagy in innate immunity. Autophagy 2021, 17, 1571–1591. [Google Scholar] [CrossRef] [PubMed]
- Jastrzebski, K.; Hannan, K.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 2007, 25, 209–226. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Robichaud, N.; Hulea, L.; Sonenberg, N.; Pelletier, J.; Topisirovic, I. Targeting the translation machinery in cancer. Nat. Rev. Drug Discov. 2015, 14, 261–278. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef]
- Giguere, V. Canonical signaling and nuclear activity of mTOR—A teamwork effort to regulate metabolism and cell growth. FEBS J. 2018, 285, 1572–1588. [Google Scholar] [CrossRef]
- Holmes, B.; Benavides-Serrato, A.; Saunders, J.T.; Kumar, S.; Nishimura, R.N.; Gera, J. mTORC2-mediated direct phosphorylation regulates YAP activity promoting glioblastoma growth and invasive characteristics. Neoplasia 2021, 23, 951–965. [Google Scholar] [CrossRef]
- Rajala, R.V. Phosphoinositide 3-kinase signaling in the vertebrate retina. J. Lipid Res. 2010, 51, 4–22. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2008, 1784, 116–132. [Google Scholar] [CrossRef]
- Wu, X.; Kihara, T.; Akaike, A.; Niidome, T.; Sugimoto, H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem. Biophys. Res. Commun. 2010, 393, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Kwon, C.-H.; Zhu, X.; Zhang, J.; Baker, S.J. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 12923–12928. [Google Scholar] [CrossRef] [PubMed]
- Avruch, J.; Lin, Y.; Long, X.; Murthy, S.; Ortiz-Vega, S. Recent advances in the regulation of the TOR pathway by insulin and nutrients. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Phosphorylation and Signal Transduction Pathways in Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a033050. [Google Scholar] [CrossRef]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.-K.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed]
- Giovanelli, P.; Sandoval, T.A.; Cubillos-Ruiz, J.R. Dendritic Cell Metabolism and Function in Tumors. Trends Immunol. 2019, 40, 699–718. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, I.; Rubio, J.; Piedra-Quintero, Z.; Lopez-Mendez, O.; Serrano, C.; Reyes-Maldonado, E.; Salinas-Lara, C.; Betanzos, A.; Shibayama, M.; Silva-Olivares, A.; et al. mTORC1 Prevents Epithelial Damage During Inflammation and Inhibits Colitis-Associated Colorectal Cancer Development. Transl. Oncol. 2019, 12, 24–35. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Sridhar, S.; Botbol, Y.; Macian, F.; Cuervo, A.M. Autophagy and disease: Always two sides to a problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Schulze, A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp. Mol. Med. 2018, 50, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Keating, R.; McGargill, M.A. mTOR regulation of lymphoid cells in immunity to pathogens. Front. Immunol. 2016, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting autophagy in cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Subramani, S. The Roles of Ubiquitin-Binding Protein Shuttles in the Degradative Fate of Ubiquitinated Proteins in the Ubiquitin-Proteasome System and Autophagy. Cells 2019, 8, 40. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Cassidy, K.; Zhao, H. Redefining the Scope of Targeted Protein Degradation: Translational Opportunities in Hijacking the Autophagy–Lysosome Pathway. Biochemistry 2021. [Google Scholar] [CrossRef]
- Sarkar, S.; Vaccaro, M.I.; Nezis, I.P. Editorial: Autophagy: From Big Data to Physiological Significance. Front. Cell Dev. Biol. 2020, 7, 376. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Vaidya, A.; Jain, S.; Sahu, S.; Jain, P.K.; Pathak, K.; Pathak, D.; Kumar, R.; Jain, S.K. Anticancer Agents Based on Vulnerable Components in a Signalling Pathway. Mini-Rev. Med. Chem. 2020, 20, 886–907. [Google Scholar] [CrossRef]
- Wang, C.-H.; Chung, F.-T.; Lin, S.-M.; Huang, S.-Y.; Chou, C.-L.; Lee, K.-Y.; Lin, T.-Y.; Kuo, H.-P. Adjuvant Treatment With a Mammalian Target of Rapamycin Inhibitor, Sirolimus, and Steroids Improves Outcomes in Patients With Severe H1N1 Pneumonia and Acute Respiratory Failure*. Crit. Care Med. 2014, 42, 313–321. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Ding, J.; Huang, Y.; Liu, J.; Liu, N.; Ao, Y.; Hong, Y.; Wang, L.; Zhang, L.; et al. Targeting mTOR suppressed colon cancer growth through 4EBP1/eIF4E/PUMA pathway. Cancer Gene Ther. 2020, 27, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.; Sun, R.C.; Aladegbami, B.; Erwin, C.R.; Warner, B.W.; Guo, J. Intestinal Epithelial-Specific mTORC1 Activation Enhances Intestinal Adaptation After Small Bowel Resection. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Sampson, L.L.; Davis, A.K.; Grogg, M.W.; Zheng, Y. mTOR disruption causes intestinal epithelial cell defects and intestinal atrophy postinjury in mice. FASEB J. 2016, 30, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.P.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef]
- Alcaide, A.; Dominguez, C. Pharmacological Control of Autophagy: Therapeutic Perspectives in Inflammatory Bowel Disease and Colorectal Cancer. Curr. Pharm. Des. 2012, 18, 3853–3873. [Google Scholar] [CrossRef]
- Tian, T.; Li, X.; Zhang, J. mTOR Signaling in Cancer and mTOR Inhibitors in Solid Tumor Targeting Therapy. Int. J. Mol. Sci. 2019, 20, 755. [Google Scholar] [CrossRef]
- Yang, M.; Huang, Q.; Li, C.; Jiang, Z.; Sun, J.; Wang, Z.; Liang, R.; Li, D.; Li, B.; Zhao, H. TOX Acts as a Tumor Suppressor by Inhibiting mTOR Signaling in Colorectal Cancer. Front. Immunol. 2021, 12, 647540. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82. [Google Scholar] [CrossRef]
- Ramasamy, T.S.; Ayob, A.Z.; Myint, H.H.L.; Thiagarajah, S.; Amini, F. Targeting colorectal cancer stem cells using curcumin and curcumin analogues: Insights into the mechanism of the therapeutic efficacy. Cancer Cell Int. 2015, 15, 1–15. [Google Scholar] [CrossRef]
- Radovanović, M. Autophagy and renal cell carcinoma: What do we know so far? Med. Podml. 2021, 72, 43–49. [Google Scholar] [CrossRef]
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mTOR pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Jiang, Y.; Su, S.; Zhang, Y.; Qian, J.; Liu, P. Control of mTOR signaling by ubiquitin. Oncogene 2019, 38, 3989–4001. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J.; Staruch, M.J.; Koprak, S.L.; Melino, M.R.; Sigal, N.H. Distinct mechanisms of suppression of murine T cell activation by the related macrolides FK-506 and rapamycin. J. Immunol. 1990, 144, 251–258. [Google Scholar]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.; Schreiber, S.L. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef]
- Lempiäinen, H.; Halazonetis, T.D. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009, 28, 3067–3073. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Jewell, J.L.; Guan, K.-L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Lampaki, S.; Turner, J.F.; Huang, H.; Kakolyris, S.; Syrigos, K.; Zarogoulidis, K. mTOR pathway: A current, up-to-date mini-review (Review). Oncol. Lett. 2014, 8, 2367–2370. [Google Scholar] [CrossRef] [PubMed]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef]
- Kim, D.-H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a Positive Regulator of the Rapamycin-Sensitive Pathway Required for the Nutrient-Sensitive Interaction between Raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef]
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse signaling mechanisms of mTOR complexes: mTORC1 and mTORC2 in forming a formidable relationship. Adv. Biol. Regul. 2019, 72, 51–62. [Google Scholar] [CrossRef]
- Hong-Brown, L.Q.; Brown, C.R.; Navaratnarajah, M.; Huber, D.S.; Lang, C.H. Alcohol-Induced Modulation of Rictor and mTORC2 Activity in C2C12 Myoblasts. Alcohol. Clin. Exp. Res. 2011, 35, 1445–1453. [Google Scholar] [CrossRef]
- Gleason, C.E.; Oses-Prieto, J.A.; Li, K.H.; Saha, B.; Situ, G.; Burlingame, A.L.; Pearce, D. Phosphorylation at distinct subcellular locations underlies specificity in mTORC2-mediated activation of SGK1 and Akt. J. Cell Sci. 2019, 132, jcs224931. [Google Scholar] [CrossRef]
- Dancey, J.; Sausville, E.A. Issues and progress with protein kinase inhibitors for cancer treatment. Nat. Rev. Drug Discov. 2003, 2, 296–313. [Google Scholar] [CrossRef] [PubMed]
- Montor, W.R.; Salas, A.R.O.S.E.; De Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Arif, M.; Senapati, P.; Shandilya, J.; Kundu, T.K. Protein lysine acetylation in cellular function and its role in cancer manifestation. Biochim. et Biophys. Acta 2010, 1799, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Chauhan, P.; Saha, B.; Kubatzky, K.F. Conceptual evolution of cell signaling. Int. J. Mol. Sci. 2019, 20, 3292. [Google Scholar] [CrossRef]
- Gross, A.S.; Graef, M. Mechanisms of Autophagy in Metabolic Stress Response. J. Mol. Biol. 2020, 432, 28–52. [Google Scholar] [CrossRef]
- Wiegering, A.; Rüther, U.; Gerhardt, C. The Role of Primary Cilia in the Crosstalk between the Ubiquitin–Proteasome System and Autophagy. Cells 2019, 8, 241. [Google Scholar] [CrossRef]
- Finley, D. Recognition and Processing of Ubiquitin-Protein Conjugates by the Proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Schwartz, A.L.; Ciechanover, A. Targeting Proteins for Destruction by the Ubiquitin System: Implications for Human Pathobiology. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 73–96. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2017, 14, 207–215. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef]
- Pieri, M.; Miraglia, N.; Polichetti, G.; Tarantino, G.; Acampora, A.; Capone, D. Analytical and Pharmacological Aspects of Therapeutic Drug Monitoring of mTOR Inhibitors. Curr. Drug Metab. 2011, 12, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Bullock, K.E.; Petros, W.P.; Younis, I.; Uronis, H.E.; Morse, M.A.; Blobe, G.C.; Zafar, S.Y.; Gockerman, J.P.; Lager, J.J.; Truax, R.; et al. A phase I study of bevacizumab (B) in combination with everolimus (E) and erlotinib (E) in advanced cancer (BEE). Cancer Chemother. Pharmacol. 2011, 67, 465–474. [Google Scholar] [CrossRef][Green Version]
- Capone, D.; Palmiero, G.; Gentile, A.; Basile, V.; Federico, S.; Sabbatini, M.; Potenza, M.; Perfetti, A.; Pieri, M.; Tarantino, G. A Pharmacokinetic Interaction Between Clarithromycin and Sirolimus in Kidney Transplant Recipient. Curr. Drug Metab. 2007, 8, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Palavra, F.; Robalo, C.; Reis, F. Recent Advances and Challenges of mTOR Inhibitors Use in the Treatment of Patients with Tuberous Sclerosis Complex. Oxidative Med. Cell. Longev. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-Y.; Huang, S.-L. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin. J. Cancer 2013, 32, 8–18. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; De Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef]
- Powers, R.W.; Kaeberlein, M.; Caldwell, S.D.; Kennedy, B.K.; Fields, S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006, 20, 174–184. [Google Scholar] [CrossRef]
- Granata, S.; Gassa, A.D.; Carraro, A.; Brunelli, M.; Stallone, G.; Lupo, A.; Zaza, G. Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects. Int. J. Mol. Sci. 2016, 17, 735. [Google Scholar] [CrossRef]
- Pal, S.K.; Quinn, D.I. Differentiating mTOR inhibitors in renal cell carcinoma. Cancer Treat. Rev. 2013, 39, 709–719. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro andIn Vivo Models of Parkinson’s Disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.L.; Ruan, X.Z.; Powis, S.H.; Moorhead, J.F.; Varghese, Z. Anti-atherosclerotic effects of sirolimus on human vascular smooth muscle cells. Am. J. Physiol. Circ. Physiol. 2007, 292, H2721–H2728. [Google Scholar] [CrossRef] [PubMed]
- Goudar, R.K.; Shi, Q.; Hjelmeland, M.D.; Keir, S.T.; McLendon, R.E.; Wikstrand, C.J.; Reese, E.D.; Conrad, C.A.; Traxler, P.; Lane, H.A.; et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol. Cancer Ther. 2005, 4, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, G.I.; Meier-Wiedenbach, I.; Manns, M.P. Clinical Pharmacokinetics of Everolimus. Clin. Pharmacokinet. 2004, 43, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, M.; Everolimus, A. Proliferation signal inhibitor targeting primary causes of allograft dysfunction. Drugs Today 2004, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; de Vries, E.G.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Sessa, C.; Tosi, D.; Viganò, L.; Albanell, J.; Hess, D.; Maur, M.; Cresta, S.; Locatelli, A.; Angst, R.; Rojo, F.; et al. Phase Ib study of weekly mammalian target of rapamycin inhibitor ridaforolimus (AP23573; MK-8669) with weekly paclitaxel. Ann. Oncol. 2009, 21, 1315–1322. [Google Scholar] [CrossRef]
- Chawla, S.P.; Staddon, A.P.; Baker, L.H.; Schuetze, S.M.; Tolcher, A.W.; D’Amato, G.Z.; Blay, J.-Y.; Mita, M.M.; Sankhala, K.K.; Berk, L.; et al. Phase II Study of the Mammalian Target of Rapamycin Inhibitor Ridaforolimus in Patients with Advanced Bone and Soft Tissue Sarcomas. J. Clin. Oncol. 2020, 30, 78–84, Coriggendum in 2017, 35, 2722. [Google Scholar] [CrossRef]
- Serruys, P.W.; Silber, S.; Garg, S.; van Geuns, R.J.; Richardt, G.; Buszman, P.E.; Kelbæk, H.; van Boven, A.J.; Hofma, S.H.; Linke, A.; et al. Comparison of Zotarolimus-Eluting and Everolimus-Eluting Coronary Stents. N. Engl. J. Med. 2010, 363, 136–146. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. mTOR pathway in colorectal cancer: An update. Oncotarget 2014, 5, 49–66. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Hur, W.; Ahmed, T.; Sabatini, D.M.; Gray, N.S. Discovery of 9-(6-Aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h]1,6naphthyridin-2(1H)-one (Torin2) as a Potent, Selective, and Orally Available Mammalian Target of Rapamycin (mTOR) Inhibitor for Treatment of Cancer. J. Med. Chem. 2011, 54, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-L.; Nokin, M.-J.; Egorov, M.; Tomé, M.; Bodineau, C.; Di Primo, C.; Minder, L.; Wdzieczak-Bakala, J.; Garcia-Alvarez, M.C.; Bignon, J.; et al. mTOR Inhibition via Displacement of Phosphatidic Acid Induces Enhanced Cytotoxicity Specifically in Cancer Cells. Cancer Res. 2018, 78, 5384–5397. [Google Scholar] [CrossRef]
- Bendell, J.C.; Varghese, A.M.; Hyman, D.M.; Bauer, T.M.; Pant, S.; Callies, S.; Lin, J.; Martinez, R.; Wickremsinhe, E.R.; Fink, A.; et al. A First-in-Human Phase 1 Study of LY3023414, an Oral PI3K/mTOR Dual Inhibitor, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Plews, R.L.; Yusof, A.M.; Wang, C.; Saji, M.; Zhang, X.; Chen, C.-S.; Ringel, M.D.; Phay, J.E. A Novel Dual AMPK Activator/mTOR Inhibitor Inhibits Thyroid Cancer Cell Growth. J. Clin. Endocrinol. Metab. 2015, 100, E748–E756. [Google Scholar] [CrossRef]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C.; et al. IKKβ Suppression of TSC1 Links Inflammation and Tumor Angiogenesis via the mTOR Pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef]
- von Manteuffel, S.R.; Gingras, A.C.; Ming, X.F.; Sonenberg, N.; Thomas, G. 4E-BP1 phosphorylation is mediated by the FRAP-p70s6k pathway and is independent of mitogen-activated protein kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 4076–4080. [Google Scholar] [CrossRef]
- Yip, C.K.; Murata, K.; Walz, T.; Sabatini, D.M.; Kang, S.A. Structure of the Human mTOR Complex I and Its Implications for Rapamycin Inhibition. Mol. Cell 2010, 38, 768–774. [Google Scholar] [CrossRef]
- Saucedo, L.J.; Gao, X.; Chiarelli, D.A.; Li, L.; Pan, D.; Edgar, B.A. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat. Cell Biol. 2003, 5, 566–571. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk: Implications for Tuberous Sclerosisand Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, D.; Roux, P.; Mieulet, V.; Cohen, M.S.; Raught, B.; Taunton, J.; Hershey, J.W.B.; Blenis, J.; Pende, M.; Sonenberg, N. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 2006, 25, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Avruch, J. Glutamatergic Regulation of the p70S6 Kinase in Primary Mouse Neurons. J. Biol. Chem. 2005, 280, 38121–38124. [Google Scholar] [CrossRef] [PubMed]
- Parate, S.; Kumar, V.; Lee, G.; Rampogu, S.; Hong, J.; Lee, K. Marine-Derived Natural Products as ATP-Competitive mTOR Kinase Inhibitors for Cancer Therapeutics. Pharmaceuticals 2021, 14, 282. [Google Scholar] [CrossRef]
- Yordy, B.; Iwasaki, A. Autophagy in the control and pathogenesis of viral infection. Curr. Opin. Virol. 2011, 1, 196–203. [Google Scholar] [CrossRef]
- Siqueira, M.d.S.; Ribeiro, R.d.M.; Travassos, L.H. Autophagy and its interaction with intracellular bacterial pathogens. Front. Immunol. 2018, 9, 935. [Google Scholar] [CrossRef]
- Bahia, D.; Satoskar, A.R.; Dussurget, O. Cell signaling in host–pathogen interactions: The host point of view. Front. Immunol. 2018, 9, 221. [Google Scholar] [CrossRef]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Axtell, R.C.; De Jong, B.A.; Boniface, K.; Van Der Voort, L.F.; Bhat, R.; De Sarno, P.; Naves, R.; Han, M.; Zhong, F.; Castellanos, J.G.; et al. T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 2010, 16, 406–412. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Powell, J.D. Regulation of T cells by mTOR: The known knowns and the known unknowns. Trends Immunol. 2014, 36, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Limon, J.J.; Fruman, D.A. Akt and mTOR in B Cell Activation and Differentiation. Front. Immunol. 2012, 3, 228. [Google Scholar] [CrossRef] [PubMed]
- Babiński, S.; Giermaziak, H. Influenza epidemic in 1971 in diabetics treated with 1-butyl-biguanidine hydrochloride (Silubin retard) and 1-phenylethyl-biguanidine hydrochloride (Phenformin). Polski Tyg. Lek. 1973, 28, 1815–1817. [Google Scholar]
- Chuang, Y.-C.; Ruan, S.-Y. Compelling Results of Adjuvant Therapy with Sirolimus for Severe H1N1 Pneumonia. Crit. Care Med. 2014, 42, e687–e688. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Das, S.; Losert, W.; Parent, C.A. mTORC2 regulates neutrophil chemotaxis in a cAMP-and RhoA-dependent fashion. Dev. Cell 2010, 19, 845–857. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.; Zhang, Y.; Liu, H.; Liao, J.; Shao, K.; Chu, Y.; Liu, G. Modulation of TSC-mTOR signaling on immune cells in immunity and autoimmunity. J. Cell. Physiol. 2014, 229, 17–26. [Google Scholar] [CrossRef]
- Squarize, C.; Castilho, R.M.; Bugge, T.H.; Gutkind, J.S. Accelerated Wound Healing by mTOR Activation in Genetically Defined Mouse Models. PLoS ONE 2010, 5, e10643. [Google Scholar] [CrossRef]
- Katholnig, K.; Linke, M.; Pham, H.; Hengstschläger, M.; Weichhart, T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem. Soc. Trans. 2013, 41, 927–933. [Google Scholar] [CrossRef]
- Kufer, T.A.; Banks, D.J.; Philpott, D.J. Innate Immune Sensing of Microbes by Nod Proteins. Ann. N. Y. Acad. Sci. 2006, 1072, 19–27. [Google Scholar] [CrossRef]
- Abdel-Nour, M.; Tsalikis, J.; Kleinman, D.; Girardin, S.E. The emerging role of mTOR signalling in antibacterial immunity. Immunol. Cell Biol. 2014, 92, 346–353. [Google Scholar] [CrossRef]
- Rampersad, S.; Tennant, P. Replication and Expression Strategies of Viruses. Viruses 2018, 55–82. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- Terrazzano, G.; Rubino, V.; Palatucci, A.T.; Giovazzino, A.; Carriero, F.; Ruggiero, G. An open question: Is it rational to inhibit the mTor-dependent pathway as COVID-19 therapy? Front. Pharmacol. 2020, 11, 856. [Google Scholar] [CrossRef]
- Castle, B.T.; Dock, C.; Hemmat, M.; Kline, S.; Tignanelli, C.; Rajasingham, R.; Masopust, D.; Provenzano, P.; Langlois, R.; Schacker, T.; et al. Biophysical modeling of the SARS-CoV-2 viral cycle reveals ideal antiviral targets. Biorxiv 2020. [Google Scholar] [CrossRef]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef]
- Cheng, F.; da Silva, S.R.; Huang, I.-C.; Jung, J.U.; Gao, S.-J. Suppression of Zika Virus Infection and Replication in Endothelial Cells and Astrocytes by PKA Inhibitor PKI 14-22. J. Virol. 2018, 92, e02019-17. [Google Scholar] [CrossRef]
- Nakashima, K.; Takeuchi, K.; Chihara, K.; Hotta, H.; Sada, K. Inhibition of hepatitis C virus replication through adenosine monophosphate-activated protein kinase-dependent and-independent pathways. Microbiol. Immunol. 2011, 55, 774–782. [Google Scholar] [CrossRef]
- Del Campo, J.A.; Garcia-Valdecasas, M.; Gil-Gomez, A.; Rojas, A.; Gallego, P.; Ampuero, J.; Gallego-Durán, R.; Pastor, H.; Grande, L.; Padillo, F.J.; et al. Simvastatin and metformin inhibit cell growth in hepatitis C virus infected cells via mTOR increasing PTEN and autophagy. PLoS ONE 2018, 13, e0191805. [Google Scholar] [CrossRef]
- Denys, A.; Bocian, J. Effect of Silubin-retard (1-butyl-biguanide hydrochloride) on the course of influenza-virus infection in mice. Polski Tyg. Lek. 1970, 25, 332–334. [Google Scholar]
- Rightsel, W.A.; Dice, J.R.; McAlpine, R.J.; Timm, E.A.; McLean, I.W.; Dixon, G.J.; Schabel, F.M. Antiviral Effect of Guanidine. Science 1961, 134, 558–559. [Google Scholar] [CrossRef]
- Ueda, T.; Toyoshima, S.; Tsuji, T.; Seto, Y.; Nomoto, J. Antiviral effect of guanidine and its derivatives part 1. the inhibitory effect of guanidine on the multiplication of poliomyelitis virus in tissue culture. Keio J. Med. 1961, 10, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Loddo, B. Inhibition of the multiplication in vitro of poliomyelitis viruses by guanidine. VIII. Elective inhibitory activity of guanidine on the PCE of enteroviruses. Boll. Della Soc. Ital. Di Biol. Sper. 1962, 38, 8–10. [Google Scholar]
- Loddo, B.; Ferrari, W.; Brotzu, G.; Spanedda, A. Inhibitory action of guanidine on the multiplication of enteroviruses and especially of polioviruses. Boll. Dell’Istituto Sieroter. Milan. 1962, 41, 111–120. [Google Scholar]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral Potential of ERK/MAPK and PI3K/AKT/mTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef]
- Garcia, G., Jr.; Sharma, A.; Ramaiah, A.; Sen, C.; Kohn, D.; Gomperts, B.; Svendsen, C.N.; Damoiseaux, R.D.; Arumugaswami, V. Antiviral drug screen of kinase inhibitors identifies cellular signaling pathways critical for SARS-CoV-2 replication. BioRxiv 2020. [Google Scholar]
- Appelberg, S.; Gupta, S.; Svensson Akusjärvi, S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Végvári, Á.; Benfeitas, R.; Sperk, M.; Ståhlberg, M. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef]
- Luo, P.; Qiu, L.; Liu, Y.; Liu, X.-L.; Zheng, J.-L.; Xue, H.-Y.; Liu, W.-H.; Liu, D.; Li, J. Metformin Treatment Was Associated with Decreased Mortality in COVID-19 Patients with Diabetes in a Retrospective Analysis. Am. J. Trop. Med. Hyg. 2020, 103, 69–72. [Google Scholar] [CrossRef]
- Karam, B.S.; Morris, R.S.; Bramante, C.T.; Puskarich, M.; Ms, E.J.Z.; Lotfi-Emran, S.; Ingraham, N.E.; Charles, A.; Odde, D.J.; Tignanelli, C.J. mTOR inhibition in COVID-19: A commentary and review of efficacy in RNA viruses. J. Med. Virol. 2021, 93, 1843–1846. [Google Scholar] [CrossRef]
- Martini, E.; Krug, S.M.; Siegmund, B.; Neurath, M.F.; Becker, C. Mend your fences: The epithelial barrier and its relationship with mucosal immunity in inflammatory bowel disease. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 33–46. [Google Scholar] [CrossRef]
- Wijnands, A.M.; Mahmoud, R.; Lutgens, M.W.; Oldenburg, B. Surveillance and management of colorectal dysplasia and cancer in inflammatory bowel disease: Current practice and future perspectives. Eur. J. Intern. Med. 2021, 93, 35–41. [Google Scholar] [CrossRef]
- Zenlea, T.; Peppercorn, M.A. Immunosuppressive therapies for inflammatory bowel disease. World J. Gastroenterol. WJG 2014, 20, 3146. [Google Scholar] [CrossRef] [PubMed]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of response to anti-TNFs: Definition, epidemiology, and management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, V.Z.; Jones, F.; Doherty, G.; Ryan, E.J. Targeting Immune Cell Metabolism in the Treatment of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2021, 27, 1684–1693. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Saber, S.; Youssef, M.E.; Sharaf, H.; Amin, N.A.; El-Shedody, R.; Aboutouk, F.H.; AbdEl-Galeel, Y.; El-Hefnawy, A.; Shabaka, D.; Khalifa, A.; et al. BBG enhances OLT1177-induced NLRP3 inflammasome inactivation by targeting P2X7R/NLRP3 and MyD88/NF-κB signaling in DSS-induced colitis in rats. Life Sci. 2021, 270, 119123. [Google Scholar] [CrossRef]
- Saber, S.; El-Fattah, E.A.; Yahya, G.; Gobba, N.; Maghmomeh, A.; Khodir, A.; Mourad, A.; Saad, A.; Mohammed, H.; Nouh, N.; et al. A Novel Combination Therapy Using Rosuvastatin and Lactobacillus Combats Dextran Sodium Sulfate-Induced Colitis in High-Fat Diet-Fed Rats by Targeting the TXNIP/NLRP3 Interaction and Influencing Gut Microbiome Composition. Pharmaceuticals 2021, 14, 341. [Google Scholar] [CrossRef]
- El-Rous, M.A.; Saber, S.; Raafat, E.M.; Ahmed, A.A. Dapagliflozin, an SGLT2 inhibitor, ameliorates acetic acid-induced colitis in rats by targeting NFκB/AMPK/NLRP3 axis. Inflammopharmacology 2021, 29, 1169–1185. [Google Scholar] [CrossRef]
- Jiang, H.; YiQing, Y.A.N.; Jiang, W.; RongBin, Z.H.O.U. NLRP3 inflammasome: Activation, regulation, and role in diseases. Sci. Sin. Vitae 2017, 47, 125–131. [Google Scholar]
- Wang, Y.; Lin, Z.; Zhang, B.; Jiang, Z.; Guo, F.; Yang, T. Cichorium intybus L. extract suppresses experimental gout by inhibiting the NF-κB and NLRP3 signaling pathways. Int. J. Mol. Sci. 2019, 20, 4921. [Google Scholar] [CrossRef]
- Neumann, K.; Schiller, B.; Tiegs, G. NLRP3 Inflammasome and IL-33: Novel Players in Sterile Liver Inflammation. Int. J. Mol. Sci. 2018, 19, 2732. [Google Scholar] [CrossRef]
- Dai, W.; Zhan, X.; Peng, W.; Liu, X.; Peng, W.; Mei, Q.; Hu, X. Ficus pandurata Hance Inhibits Ulcerative Colitis and Colitis-Associated Secondary Liver Damage of Mice by Enhancing Antioxidation Activity. Oxidative Med. Cell. Longev. 2021, 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cao, S. Cellular Stress Responses and Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol. Res. Pr. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.J.; Snapper, S.B.; Blumberg, R.S. Update on biologic pathways in inflammatory bowel disease and their therapeutic relevance. J. Gastroenterol. 2012, 47, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Panganiban, R.-A.M.; Snow, A.L.; Day, R.M. Mechanisms of Radiation Toxicity in Transformed and Non-Transformed Cells. Int. J. Mol. Sci. 2013, 14, 15931–15958. [Google Scholar] [CrossRef]
- Moreno-Villanueva, M.; Wu, H. Radiation and microgravity–Associated stress factors and carcinogensis. REACH 2019, 13, 100027. [Google Scholar] [CrossRef]
- Bi, S.; Wang, H.; Kuang, W. Stem cell rejuvenation and the role of autophagy in age retardation by caloric restriction: An update. Mech. Ageing Dev. 2018, 175, 46–54. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Kaur, H.; Moreau, R. Role of mTORC1 in intestinal epithelial repair and tumorigenesis. Cell. Mol. Life Sci. 2019, 76, 2525–2546. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Salmond, R.J. mTOR Regulation of Glycolytic Metabolism in T Cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Khare, V.; Dammann, K.; Asboth, M.; Krnjic, A.; Jambrich, M.; Gasche, C. Overexpression of PAK1 Promotes Cell Survival in Inflammatory Bowel Diseases and Colitis-associated Cancer. Inflamm. Bowel Dis. 2015, 21, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y.; et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.-J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Takahara, M.; Takaki, A.; Hiraoka, S.; Adachi, T.; Shimomura, Y.; Matsushita, H.; Nguyen, T.T.T.; Koike, K.; Ikeda, A.; Takashima, S.; et al. Berberine improved experimental chronic colitis by regulating interferon-γ- and IL-17A-producing lamina propria CD4+ T cells through AMPK activation. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saber, S.; Ghanim, A.M.H.; El-Ahwany, E.; El-Kader, E.M.A. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced murine hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Marín-Aguilar, F.; Pavillard, L.E.; Giampieri, F.; Bullón, P.; Cordero, M.D. Adenosine Monophosphate (AMP)-Activated Protein Kinase: A New Target for Nutraceutical Compounds. Int. J. Mol. Sci. 2017, 18, 288. [Google Scholar] [CrossRef]
- Ruderman, N.B.; Carling, D.; Prentki, M.; Cacicedo, J.M. AMPK, insulin resistance, and the metabolic syndrome. J. Clin. Investig. 2013, 123, 2764–2772. [Google Scholar] [CrossRef]
- Wu, S.; Zou, M.-H. AMPK, mitochondrial function, and cardiovascular disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Gastroenterol. Rev./Przegląd Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Erratum: Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2020, 70, 313. [Google Scholar]
- Araghi, M.; Soerjomataram, I.; Jenkins, M.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Goulet, E.J.; Livingston, E.H. Colon cancer and apoptosis. Am. J. Surg. 2006, 191, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ruder, D.; Papadimitrakopoulou, V.; Shien, K.; Behrens, C.; Kalhor, N.; Chen, H.; Shen, L.; Lee, J.J.; Hong, W.K.; Tang, X.; et al. Concomitant targeting of the mTOR/MAPK pathways: Novel therapeutic strategy in subsets of RICTOR/KRAS-altered non-small cell lung cancer. Oncotarget 2018, 9, 33995–34008. [Google Scholar] [CrossRef][Green Version]
- Quan, C.; Xiao, J.; Liu, L.; Duan, Q.; Yuan, P.; Zhu, F. Protein Kinases as Tumor Biomarkers and Therapeutic Targets. Curr. Pharm. Des. 2017, 23, 4209–4225. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef]
- Reita, D.; Bour, C.; Benbrika, R.; Groh, A.; Pencreach, E.; Guérin, E.; Guenot, D. Synergistic Anti-Tumor Effect of mTOR Inhibitors with Irinotecan on Colon Cancer Cells. Cancers 2019, 11, 1581. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Sparks, C.A.; Guertin, D.A. Targeting mTOR: Prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 2010, 29, 3733–3744. [Google Scholar] [CrossRef]
- Fujishita, T.; Aoki, M.; Taketo, M.M. The role of mTORC1 pathway in intestinal tumorigenesis. Cell Cycle 2009, 8, 3684–3687. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roulin, D.; Cerantola, Y.; Dormond-Meuwly, A.; Demartines, N.; Dormond, O. Targeting mTORC2 inhibits colon cancer cell proliferation in vitro and tumor formation in vivo. Mol. Cancer 2010, 9, 57. [Google Scholar] [CrossRef]
- Hardiman, K.M.; Liu, J.; Feng, Y.; Greenson, J.K.; Fearon, E.R. Rapamycin Inhibition of Polyposis and Progression to Dysplasia in a Mouse Model. PLoS ONE 2014, 9, e96023. [Google Scholar] [CrossRef]
- Johnson, S.M.; Gulhati, P.; Rampy, B.A.; Han, Y.; Rychahou, P.G.; Doan, H.Q.; Weiss, H.L.; Evers, B.M. Novel Expression Patterns of PI3K/Akt/mTOR Signaling Pathway Components in Colorectal Cancer. J. Am. Coll. Surg. 2010, 210, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L. Hypothesis: Cell signalling influences age-related risk of colorectal cancer. J. Cell. Mol. Med. 2014, 19, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A. PI3K/ Akt/ mTOR pathway as a therapeutic target for colorectal cancer: A review of preclinical and clinical evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Salgado, C.M.; Bauer, B.; Khakoo, Y.; Patel, J.R.; Hoehl, R.M.; Bertolini, D.M.; Zabec, J.; Brzozowski, M.R.; Reyes-Múgica, M. The Dual PI3K/mToR Inhibitor Omipalisib/GSK2126458 Inhibits Clonogenic Growth in Oncogenically-transformed Cells from Neurocutaneous Melanocytosis. Cancer Genom.-Proteom. 2018, 15, 239–248. [Google Scholar] [CrossRef]
- Galamb, O.; Kalmár, A.; Sebestyén, A.; Dankó, T.; Kriston, C.; Fűri, I.; Hollósi, P.; Csabai, I.; Wichmann, B.; Krenács, T.; et al. Promoter Hypomethylation and Increased Expression of the Long Non-coding RNA LINC00152 Support Colorectal Carcinogenesis. Pathol. Oncol. Res. 2020, 26, 2209–2223. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-Site Inhibitors of mTOR Target Rapamycin-Resistant Outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e1000038. [Google Scholar] [CrossRef] [PubMed]
- Naruse, T.; Yanamoto, S.; Okuyama, K.; Yamashita, K.; Omori, K.; Nakao, Y.; Yamada, S.-I.; Umeda, M. Therapeutic implication of mTORC2 in oral squamous cell carcinoma. Oral Oncol. 2017, 65, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, L. Discovery and development of ATP-competitive mTOR inhibitors using computational approaches. Curr. Pharm. Des. 2017, 23, 4321–4331. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Duan, Y.; Zheng, X.S. Targeting the mTOR kinase domain: The second generation of mTOR inhibitors. Drug Discov. Today 2011, 16, 325–331. [Google Scholar] [CrossRef]
- Hsu, C.-M.; Lin, P.-M.; Tsai, Y.-T.; Tsai, M.-S.; Tseng, C.-H.; Lin, S.-F.; Yang, M.-Y. NVP-BEZ235, a dual PI3K-mTOR inhibitor, suppresses the growth of FaDu hypopharyngeal squamous cell carcinoma and has a synergistic effect with Cisplatin. Cell Death Discov. 2018, 4, 1–10. [Google Scholar] [CrossRef]
- Singh, A.R.; Joshi, S.; George, E.; Durden, D.L. Anti-tumor effect of a novel PI3-kinase inhibitor, SF1126, in (12) V-Ha-Ras transgenic mouse glioma model. Cancer Cell Int. 2014, 14, 105. [Google Scholar] [CrossRef]
- De, P.; Dey, N.; Terakedis, B.; Bergsagel, P.L.; Li, Z.H.; Mahadevan, D.; Garlich, J.R.; Trudel, S.; Makale, M.T.; Durden, D.L.; et al. An integrin-targeted, pan-isoform, phosphoinositide-3 kinase inhibitor, SF1126, has activity against multiple myeloma in vivo. Cancer Chemother. Pharmacol. 2013, 71, 867–881. [Google Scholar] [CrossRef]
- Feng, Y.; Jiang, Y.; Hao, F. GSK2126458 has the potential to inhibit the proliferation of pancreatic cancer uncovered by bioinformatics analysis and pharmacological experiments. J. Transl. Med. 2021, 19, 373. [Google Scholar] [CrossRef]
- Guo, Y.; Zhu, H.; Weng, M.; Zhang, H.; Wang, C.; Sun, L. CC-223, NSC781406, and BGT226 exerts a cytotoxic effect against pancreatic cancer cells via mTOR signaling. Front. Pharmacol. 2020, 11, 580407. [Google Scholar] [CrossRef]
- Baumann, P.; Schneider, L.; Mandl-Weber, S.; Oduncu, F.; Schmidmaier, R. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anti-Cancer Drugs 2012, 23, 131–138. [Google Scholar] [CrossRef]
- Omeljaniuk, W.J.; Krętowski, R.; Ratajczak-Wrona, W.; Jabłońska, E.; Cechowska-Pasko, M. Novel dual PI3K/mTOR inhibitor, Apitolisib (GDC-0980), inhibits growth and induces apoptosis in human glioblastoma cells. Int. J. Mol. Sci. 2021, 22, 11511. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D.; et al. Randomized open-label phase II trial of Apitolisib (GDC-0980), a novel inhibitor of the PI3K/Mammalian target of rapamycin pathway, versus Everolimus in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2016, 34, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Edgar, K.A.; Guan, J.; Berry, M.; Prior, W.W.; Lee, L.; Lesnick, J.D.; Lewis, C.; Nonomiya, J.; Pang, J.; et al. GDC-0980 Is a Novel Class I PI3K/mTOR Kinase Inhibitor with Robust Activity in Cancer Models Driven by the PI3K PathwayGDC-0980, a Class I PI3K/mTOR Inhibitor in the Clinic. Mol. Cancer Ther. 2011, 10, 2426–2436. [Google Scholar] [CrossRef]
- Rehan, M. An Anti-Cancer drug candidate OSI-027 and its analog as inhibitors of mTOR: Computational insights into the inhibitory mechanisms. J. Cell. Biochem. 2017, 118, 4558–4567. [Google Scholar] [CrossRef] [PubMed]
- Zhi, X.; Chen, W.; Xue, F.; Liang, C.; Chen, B.W.; Zhou, Y.; Wen, L.; Hu, L.; Shen, J.; Bai, X.; et al. OSI-027 inhibits pancreatic ductal adenocarcinoma cell proliferation and enhances the therapeutic effect of gemcitabine both in vitro and in vivo. Oncotarget 2015, 6, 26230. [Google Scholar] [CrossRef]
- Hayman, T.J.; Wahba, A.; Rath, B.H.; Bae, H.; Kramp, T.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. The ATP-Competitive mTOR Inhibitor INK128 Enhances In Vitro and In Vivo Radiosensitivity of Pancreatic Carcinoma Cells. Clin. Cancer Res. 2014, 20, 110–119. [Google Scholar] [CrossRef]
- Zhang, H.; Dou, J.; Yu, Y.; Zhao, Y.; Fan, Y.; Cheng, J.; Xu, X.; Liu, W.; Guan, S.; Chen, Z.; et al. mTOR ATP-competitive inhibitor INK128 inhibits neuroblastoma growth via blocking mTORC signaling. Apoptosis 2015, 20, 50–62. [Google Scholar] [CrossRef]
- Basu, B.; Dean, E.; Puglisi, M.; Greystoke, A.; Ong, M.; Burke, W.; Cavallin, M.; Bigley, G.; Womack, C.; Harrington, E.A.; et al. First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014Phase I Study of AZD2014. Clin. Cancer Res. 2015, 21, 3412–3419. [Google Scholar] [CrossRef]
- Kahn, J.; Hayman, T.J.; Jamal, M.; Rath, B.H.; Kramp, T.; Camphausen, K.; Tofilon, P.J. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro-Oncol. 2014, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Curwen, J.; Bihani, T.; D’Cruz, C.M.; Yates, J.W.; Grondine, M.; Howard, Z.; Davies, B.R.; Bigley, G.; Klinowska, T.; et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous SchedulesPharmacology of AZD2014, an mTORC1/2 Inhibitor. Mol. Cancer Ther. 2015, 14, 2508–2518. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Reineke, L.C.; Knutsen, E.; Chen, M.; Pichler, M.; Ling, H.; Calin, G.A. Metformin blocks MYC protein synthesis in colorectal cancer via m TOR-4 EBP-e IF 4E and MNK1-e IF 4G-e IF 4E signaling. Mol. Oncol. 2018, 12, 1856–1870. [Google Scholar] [CrossRef]
- Higurashi, T.; Takahashi, H.; Endo, H.; Hosono, K.; Yamada, E.; Ohkubo, H.; Sakai, E.; Uchiyama, T.; Hata, Y.; Fujisawa, N.; et al. Metformin efficacy and safety for colorectal polyps: A double-blind randomized controlled trial. BMC Cancer 2012, 12, 118. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Name of mTOR Inhibitor | Therapeutic Importance | Reference |
|---|---|---|---|
| 1 | Rapamycin (Sirolimus) |
| [80] [81] [82] [83] |
| 2 | Temsirolimus | Treat advanced Renal cancer | [77] |
| 3 | Everolimus |
| [84] [85] [86] |
| 4 | Ridaforolimus/Deforolimus |
| [87] [88] |
| 5 | Zotarolimus | Antitumor activity | [89] |
| 6 | Torin 1 | Suppress colon cancer cells | [90] |
| 7 | Torin 2 | Antitumor activity | [91] |
| 8 | MLN0128 | Advanced solid tumors | [92] |
| 9 | AZD2014 (Vistusertib) | Metastatic clear cell renal cancer | [92] |
| 10 | Voxtalisib (SAR24540; XL765) | non-Hodgkin lymphoma or chronic lymphocytic lymphoma that has relapsed or is refractory | [92] |
| 11 | Gedatolisib (PKI-587 PF05212384) | Recurrent endometrial cancer | [92] |
| 12 | Rapalink-1 |
| [77] |
| 13 | Halitulin analog ICSN3250 | It has the ability to compete with and interchange phospholipids acid in the mTOR FRB domain | [93] |
| 14 | LY3023414 |
| [94] |
| 15 | O SU-53 |
| [95] |
| 16 | OSI-027 | Anticancer | [96] |
| 17 | C C-223 | Anticancer | [97] |
| 18 | PKI-587 | Gastroenteropancreatic Neuroendocrine tumor disease | [98] |
| 19 | INK-128 | Inhibit angiogenesis and tumor growth in
| [99] |
| 20 | GSK2126458 | Robust activity in cancer models | [100] |
| 21 | XL765 | Glioblastoma development is inhibited by triggering ER stress-dependent apoptosis. | [101] |
| 22 | NVP-BEZ235 | Cancer cell proliferation is inhibited by this compound | [102] |
| 23 | P529 | Stops cancer cells from multiplying | [103] |
| 24 | JR-AB2-011 | Anti-glioblastoma multiforme properties | [104] |
| Drugs | Mode of Action | References |
|---|---|---|
| Mesalamine | Inhibition of mTORC1 signaling pathway | [160] |
| Tacrolimus, | Inhibition mTORC1 signaling pathway | [161,162,163] |
| Everolimus | Inhibition mTORC1 signaling pathway | [162,163] |
| Sirolimus | Inhibition mTORC1 signaling pathway | [142,163] |
| Methotrexate | Promotes OXPHOS (Oxidative phosphorylation) by activating AMPK and blocking mTORC1 | [142] |
| Corticosteroids | Inhibits glycolysis by blocking mTORC1 | [142] |
| Aminosalicylates | Inhibits glycolysis by blocking mTORC1 | [142] |
| Tacrolimus | Inhibits glycolysis by blocking mTORC1 | [142] |
| mTOR Kinase Inhibitors | Drugs | References |
|---|---|---|
| Dual inhibitors of mTOR/PtdIns–kinase | NVPBEZ235 | [197] |
| SF1126 | [198,199] | |
| GSK2126458 | [187,200] | |
| BGT226 | [201,202] | |
| GDC0980 | [203,204,205] | |
| mTORC1/mTORC2 dual inhibitors | OSI027 | [193,206,207] |
| INK128 | [208,209] | |
| AZD2014 | [210,211,212] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afzal, O.; Altamimi, A.S.A.; Mubeen, B.; Alzarea, S.I.; Almalki, W.H.; Al-Qahtani, S.D.; Atiya, E.M.; Al-Abbasi, F.A.; Ali, F.; Ullah, I.; et al. mTOR as a Potential Target for the Treatment of Microbial Infections, Inflammatory Bowel Diseases, and Colorectal Cancer. Int. J. Mol. Sci. 2022, 23, 12470. https://doi.org/10.3390/ijms232012470
Afzal O, Altamimi ASA, Mubeen B, Alzarea SI, Almalki WH, Al-Qahtani SD, Atiya EM, Al-Abbasi FA, Ali F, Ullah I, et al. mTOR as a Potential Target for the Treatment of Microbial Infections, Inflammatory Bowel Diseases, and Colorectal Cancer. International Journal of Molecular Sciences. 2022; 23(20):12470. https://doi.org/10.3390/ijms232012470
Chicago/Turabian StyleAfzal, Obaid, Abdulmalik S. A. Altamimi, Bismillah Mubeen, Sami I. Alzarea, Waleed Hassan Almalki, Salwa D. Al-Qahtani, Eman M. Atiya, Fahad A. Al-Abbasi, Fatima Ali, Inam Ullah, and et al. 2022. "mTOR as a Potential Target for the Treatment of Microbial Infections, Inflammatory Bowel Diseases, and Colorectal Cancer" International Journal of Molecular Sciences 23, no. 20: 12470. https://doi.org/10.3390/ijms232012470
APA StyleAfzal, O., Altamimi, A. S. A., Mubeen, B., Alzarea, S. I., Almalki, W. H., Al-Qahtani, S. D., Atiya, E. M., Al-Abbasi, F. A., Ali, F., Ullah, I., Nadeem, M. S., & Kazmi, I. (2022). mTOR as a Potential Target for the Treatment of Microbial Infections, Inflammatory Bowel Diseases, and Colorectal Cancer. International Journal of Molecular Sciences, 23(20), 12470. https://doi.org/10.3390/ijms232012470