
The Brain Protein Acylation System Responds to Seizures in the Rat Model of PTZ-Induced Epilepsy

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
2.1. Seizures-Induced Alteration in the Brain Protein Acylation Levels Involves Enzymes of Central Energy Metabolism, with More Acylation Sites Responding to Single Than Chronic Seizures
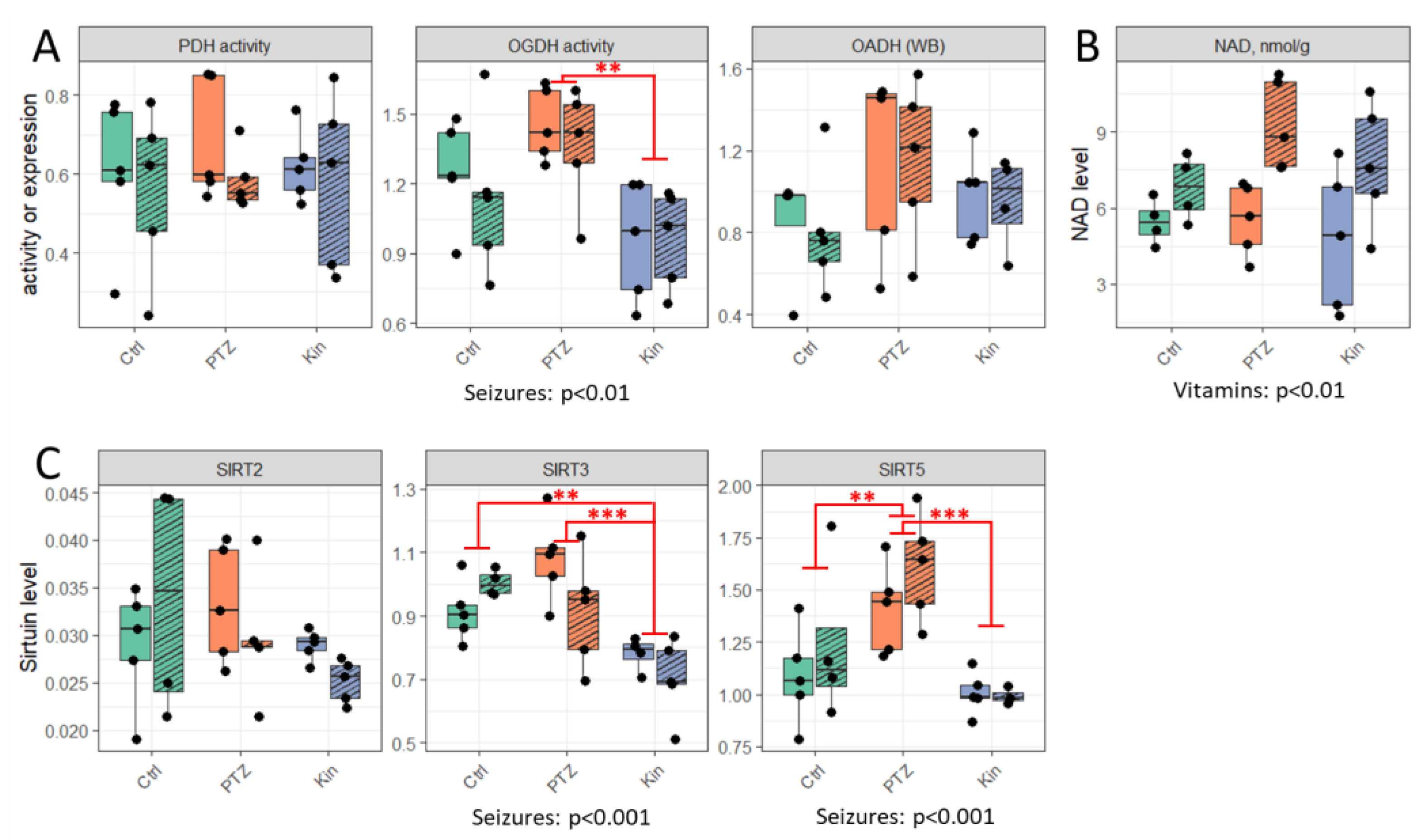
2.2. Activity of OGDH Complex and Expression of SIRT3 and SIRT5 Are Lower in Chronic vs. Single Seizures, While Activity of PDH Complex and Expression of SIRT2 Remain Unchanged
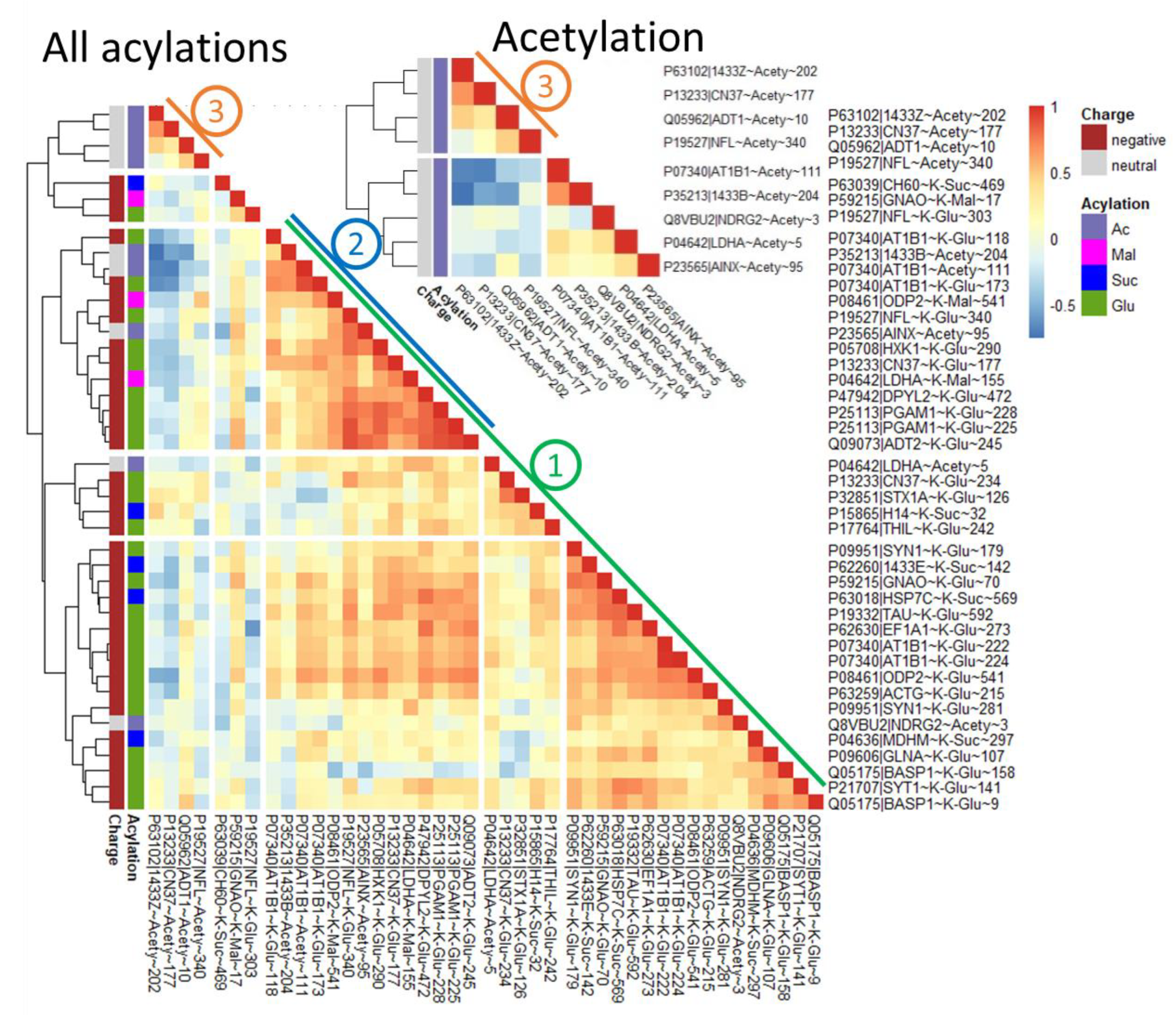
2.3. Correlations between Different Types of Protein Acylation and acyl-CoA Producers, Acyl-Specific Sirtuins and NAD+
2.4. Dependent on the Acylation Site/Protein, Both Negative and Positive Correlations May Be Observed with Acylation of Other Sites, Even for the Same Type of Acylation
3. Discussion
3.1. Altered Acylation System of the Brain after Seizures
3.2. Regulation of Acylation in the Rat Model of PTZ-Induced Epilepsy
3.3. Involvement of Perturbed Acylation in Neurological Disorders
3.4. Regulation of Energy Metabolism by Acylation upon Seizures
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Modelling Seizures by Single or Repeated Administration of Pentylenetetrazole
4.4. Homogenization and Extraction of Rat Tissues
4.5. Mass-Spectrometric Detection and Quantification of Acylations and Sirtuin 2 Level
- Cleavage sites (the ends of the peptides) occur only after lysine and arginine or methylated lysine and arginine, which correspond to the cleavage sites of the trypsin used to obtain the peptides,
- Removal of starting methionine is allowed,
- The real charge of the peptide does not exceed that theoretically calculated from the sequence, assuming N-terminus, Lys, Arg and His to potentially be charged in the positive MS mode,
- The peptide is unique (i.e., occurs in only one protein according to the assignment of the peptides during their identification).
4.6. Enzymatic Assays
4.7. Western-Blotting Quantification of the Protein Levels of 2-Oxoadipate Dehydrogenase, Sirtuin 3, and Sirtuin 5
4.8. NAD+ Quantification
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PTZ | pentylenetetrazole |
| kin | kindling |
| MS | mass-spectrometry |
| OGDH | 2-oxoglutarate dehydrogenase |
| OADH | 2-oxoadipate dehydrogenase |
| PDH | pyruvate dehydrogenase |
References
- Treiman, D.M. GABAergic Mechanisms in Epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.Y.; Baik, E.J. Glutamate Dehydrogenase as a Neuroprotective Target Against Neurodegeneration. Neurochem. Res. 2018, 44, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Eid, T.; Tu, N.; Lee, T.-S.W.; Lai, J.C.K. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem. Int. 2013, 63, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Storici, P.; Capitani, G.; De Biase, D.; Moser, M.; John, R.A.; Jansonius, J.N.; Schirmer, T. Crystal Structure of GABA-Aminotransferase, a Target for Antiepileptic Drug Therapy. Biochemistry 1999, 38, 8628–8634. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Liang, X.-J.; Li, W.-J.; Wu, D.; Liu, M.; Cao, B.-Y.; Chen, J.-J.; Qin, M.; Meng, X.; Gong, C.-X. Clinical and Molecular Spectrum of Glutamate Dehydrogenase Gene Defects in 26 Chinese Congenital Hyperinsulinemia Patients. J. Diabetes Res. 2018, 2018, 2802540. [Google Scholar] [CrossRef]
- Wasterlain, C.G.; Fujikawa, D.G.; Penix, L.; Sankar, R. Pathophysiological Mechanisms of Brain Damage from Status Epilepticus. Epilepsia 1993, 34, S37–S53. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J.; Guo, R.; Peng, Y.; Zheng, W.; Liu, D.; Song, Z. Glycolysis in energy metabolism during seizures. Neural Regen. Res. 2013, 8, 1316–1326. [Google Scholar] [CrossRef]
- Rheims, S.; Alvarez, B.M.; Alexandre, V.; Curot, J.; Maillard, L.; Bartolomei, F.; Derambure, P.; Hirsch, E.; Michel, V.; Chassoux, F.; et al. Hypoxemia following generalized convulsive seizures: Risk factors and effect of oxygen therapy. Neurology 2019, 92, e183–e193. [Google Scholar] [CrossRef]
- Bond, S.T.; Howlett, K.F.; Kowalski, G.M.; Mason, S.; Connor, T.; Cooper, A.; Streltsov, V.; Bruce, C.R.; Walder, K.R.; McGee, S.L. Lysine post-translational modification of glyceraldehyde-3-phosphate dehydrogenase regulates hepatic and systemic metabolism. FASEB J. 2017, 31, 2592–2602. [Google Scholar] [CrossRef]
- Hallows, W.C.; Yu, W.; Denu, J.M. Regulation of glycolytic enzyme phosphoglycerate mutase-1 by Sirt1 protein-mediated deacetylation. J. Biol. Chem. 2012, 287, 3850–3858. [Google Scholar] [CrossRef]
- Zhao, D.; Xiong, Y.; Lei, Q.Y.; Guan, K.L. LDH-A acetylation: Implication in cancer. Oncotarget 2013, 4, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Shan, C.; Kang, H.B.; Elf, S.; Xie, J.; Tucker, M.; Gu, T.L.; Aguiar, M.; Lonning, S.; Chen, H.; et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 2014, 53, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Assiri, M.A.; Roy, S.R.; Harris, P.S.; Ali, H.; Liang, Y.; Shearn, C.T.; Orlicky, D.J.; Roede, J.R.; Hirschey, M.D.; Backos, D.S.; et al. Chronic Ethanol Metabolism Inhibits Hepatic Mitochondrial Superoxide Dismutase via Lysine Acetylation. Alcohol. Clin. Exp. Res. 2017, 41, 1705–1714. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Graf, A.V.; Bunik, V.I. Daytime Dependence of the Activity of the Rat Brain Pyruvate Dehydrogenase Corresponds to the Mitochondrial Sirtuin 3 Level and Acetylation of Brain Proteins, All Regulated by Thiamine Administration Decreasing Phosphorylation of PDHA Ser293. Int. J. Mol. Sci. 2021, 22, 8006. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Z.; Bao, L. An Expanding Repertoire of Protein Acylations. Mol. Cell. Proteom. 2022, 21, 100193. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Mkrtchyan, G.V.; Kaehne, T.; Graf, A.V.; Maslova, M.V.; Bunik, V.I. Diurnal regulation of the function of the rat brain glutamate dehydrogenase by acetylation and its dependence on thiamine administration. J. Neurochem. 2020, 153, 80–102. [Google Scholar] [CrossRef]
- Graf, A.V.; Maslova, M.V.; Artiukhov, A.V.; Ksenofontov, A.L.; Aleshin, V.A.; Bunik, V.I. Acute Prenatal Hypoxia in Rats Affects Physiology and Brain Metabolism in the Offspring, Dependent on Sex and Gestational Age. Int. J. Mol. Sci. 2022, 23, 2579. [Google Scholar] [CrossRef]
- Yang, Y.; Gibson, G.E. Succinylation Links Metabolism to Protein Functions. Neurochem. Res. 2019, 44, 2346–2359. [Google Scholar] [CrossRef]
- Boyko, A.I.; Karlina, I.S.; Zavileyskiy, L.G.; Aleshin, V.A.; Artiukhov, A.V.; Kaehne, T.; Ksenofontov, A.L.; Ryabov, S.I.; Graf, A.V.; Tramonti, A.; et al. Delayed Impact of 2-Oxoadipate Dehydrogenase Inhibition on the Rat Brain Metabolism Is Linked to Protein Glutarylation. Front. Med. 2022, 9, 896263. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef]
- Guedouari, H.; Daigle, T.; Scorrano, L.; Hebert-Chatelain, E. Sirtuin 5 protects mitochondria from fragmentation and degradation during starvation. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 169–176. [Google Scholar] [CrossRef]
- Aleshin, V.A.; Graf, A.V.; Artiukhov, A.V.; Boyko, A.I.; Ksenofontov, A.L.; Maslova, M.V.; Nogues, I.; di Salvo, M.L.; Bunik, V.I. Physiological and Biochemical Markers of the Sex-Specific Sensitivity to Epileptogenic Factors, Delayed Consequences of Seizures and Their Response to Vitamins B1 and B6 in a Rat Model. Pharmaceuticals 2021, 14, 737. [Google Scholar] [CrossRef]
- Cole, A.J.; Koh, S.; Zheng, Y. Are seizures harmful: What can we learn from animal models? In Do Seizures Damage the Brain; Elsevier: Amsterdam, The Netherlands, 2002; pp. 13–23. [Google Scholar] [CrossRef]
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and How Do Seizures Kill Neurons, and Is Cell Death Relevant to Epileptogenesis? In Issues in Clinical Epileptology: A View from the Bench; Springer: Dordrecht, The Netherlands, 2014; pp. 109–122. [Google Scholar] [CrossRef]
- Shimada, T.; Yamagata, K. Pentylenetetrazole-Induced Kindling Mouse Model. J. Vis. Exp. 2018, 136, e56573. [Google Scholar] [CrossRef]
- Dhir, A. Pentylenetetrazol (PTZ) Kindling Model of Epilepsy. Curr. Protoc. Neurosci. 2012, 58, 9–37. [Google Scholar] [CrossRef]
- Li, F.; Liu, L. SIRT5 Deficiency Enhances Susceptibility to Kainate-Induced Seizures and Exacerbates Hippocampal Neurodegeneration not through Mitochondrial Antioxidant Enzyme SOD2. Front. Cell. Neurosci. 2016, 10, 171. [Google Scholar] [CrossRef]
- Gano, L.B.; Liang, L.P.; Ryan, K.; Michel, C.R.; Gomez, J.; Vassilopoulos, A.; Reisdorph, N.; Fritz, K.S.; Patel, M. Altered mitochondrial acetylation profiles in a kainic acid model of temporal lobe epilepsy. Free Radic. Biol. Med. 2018, 123, 116–124. [Google Scholar] [CrossRef]
- Wu, G.; Liu, J.; Li, S.; Gao, W.; Qiu, M.; Yang, C.; Ma, Y.; Song, X. Glycyrrhizic acid protects juvenile epileptic rats against hippocampal damage through activation of Sirtuin3. Brain Res. Bull. 2020, 164, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zeng, X.; Mai, Q.; Bai, X.; Jiang, Y.; Li, J.; Fan, S.; Ding, H. Insulin injections inhibits PTZ-induced mitochondrial dysfunction, oxidative stress and neurological deficits via the SIRT1/PGC-1α/SIRT3 pathway. Biochim. Et Biophys. Acta (BBA)—Mol. Basis Dis. 2021, 1867, 166124. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.; Hodson, M.P.; Bederman, I.; Puchowicz, M.; Borges, K. Triheptanoin alters [U-13C6]-glucose incorporation into glycolytic intermediates and increases TCA cycling by normalizing the activities of pyruvate dehydrogenase and oxoglutarate dehydrogenase in a chronic epilepsy mouse model. J. Cereb. Blood Flow Metab. 2019, 40, 678–691. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef]
- Pougovkina, O.; te Brinke, H.; Ofman, R.; van Cruchten, A.G.; Kulik, W.; Wanders, R.J.; Houten, S.M.; de Boer, V.C. Mitochondrial protein acetylation is driven by acetyl-CoA from fatty acid oxidation. Hum. Mol. Genet. 2014, 23, 3513–3522. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef]
- Barnes, J.A.; Gomes, A.V. PEST sequences in calmodulin-binding proteins. In Signal Transduction Mechanisms; Springer: Boston, MA, USA, 1995; pp. 17–27. [Google Scholar] [CrossRef]
- Mosevitsky, M.I.; Capony, J.P.; Skladchikova, G.Y.; Novitskaya, V.A.; Plekhanov, A.Y.; Zakharov, V.V. The BASP1 family of myristoylated proteins abundant in axonal termini. Primary structure analysis and physico-chemical properties. Biochimie 1997, 79, 373–384. [Google Scholar] [CrossRef]
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Akaaboune, M.; Cohen, L.D.; Zuchman, R.; Sorokina, O.; Müller, A.; Dieterich, D.C.; Armstrong, J.D.; Ziv, T.; Ziv, N.E. Metabolic Turnover of Synaptic Proteins: Kinetics, Interdependencies and Implications for Synaptic Maintenance. PLoS ONE 2013, 8, e63191. [Google Scholar] [CrossRef]
- Tsepkova, P.M.; Artiukhov, A.V.; Boyko, A.I.; Aleshin, V.A.; Mkrtchyan, G.V.; Zvyagintseva, M.A.; Ryabov, S.I.; Ksenofontov, A.L.; Baratova, L.A.; Graf, A.V.; et al. Thiamine Induces Long-Term Changes in Amino Acid Profiles and Activities of 2-Oxoglutarate and 2-Oxoadipate Dehydrogenases in Rat Brain. Biochemistry 2017, 82, 723–736. [Google Scholar] [CrossRef]
- Feng, H.; Larrivee, C.L.; Demireva, E.Y.; Xie, H.; Leipprandt, J.R.; Neubig, R.R. Mouse models of GNAO1-associated movement disorder: Allele- and sex-specific differences in phenotypes. PLoS ONE 2019, 14, e0211066. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Patry, L.; Congia, S.; Onofri, F.; Piton, A.; Gauthier, J.; Pozzi, D.; Messa, M.; Defranchi, E.; Fadda, M.; et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 2011, 20, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.E.; Affleck, A.J.; Bahar, A.Y.; Carew-Jones, F.; Halliday, G.M. Intracellular and secreted forms of clusterin are elevated early in Alzheimer’s disease and associate with both Abeta and tau pathology. Neurobiol. Aging 2020, 89, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, J.C.; Breuillaud, L.; Do Carmo, S.; Hall, H.; Welikovitch, L.A.; Macdonald, J.A.; Goedert, M.; Cuello, A.C. Neuropathological changes and cognitive deficits in rats transgenic for human mutant tau recapitulate human tauopathy. Neurobiol. Dis. 2019, 127, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.J.; Uchida, A.; Brown, A. Charcot-Marie-Tooth disease Type 2E/1F mutant neurofilament proteins assemble into neurofilaments. Cytoskeleton 2019, 76, 423–439. [Google Scholar] [CrossRef]
- Gabrikova, D.; Mistrik, M.; Bernasovska, J.; Bozikova, A.; Behulova, R.; Tothova, I.; Macekova, S. Founder mutations in NDRG1 and HK1 genes are common causes of inherited neuropathies among Roma/Gypsies in Slovakia. J. Appl. Genet. 2013, 54, 455–460. [Google Scholar] [CrossRef]
- Xu, W.-y.; Gu, M.-m.; Sun, L.-h.; Guo, W.-t.; Zhu, H.-b.; Ma, J.-f.; Yuan, W.-t.; Kuang, Y.; Ji, B.-j.; Wu, X.-l.; et al. A Nonsense Mutation in DHTKD1 Causes Charcot-Marie-Tooth Disease Type 2 in a Large Chinese Pedigree. Am. J. Hum. Genet. 2012, 91, 1088–1094. [Google Scholar] [CrossRef]
- Xu, W.-Y.; Zhu, H.; Shen, Y.; Wan, Y.-H.; Tu, X.-D.; Wu, W.-T.; Tang, L.; Zhang, H.-X.; Lu, S.-Y.; Jin, X.-L.; et al. DHTKD1 Deficiency Causes Charcot-Marie-Tooth Disease in Mice. Mol. Cell. Biol. 2018, 38, e00085-18. [Google Scholar] [CrossRef]
- Prasad, C.; Rupar, T.; Prasad, A.N. Pyruvate dehydrogenase deficiency and epilepsy. Brain Dev. 2011, 33, 856–865. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.; Trojanowski, J.Q. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tapias, V.; Acosta, D.; Xu, H.; Chen, H.; Bhawal, R.; Anderson, E.T.; Ivanova, E.; Lin, H.; Sagdullaev, B.T.; et al. Altered succinylation of mitochondrial proteins, APP and tau in Alzheimer’s disease. Nat. Commun. 2022, 13, 159. [Google Scholar] [CrossRef] [PubMed]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD(+) in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Pallàs, M. Resveratrol in epilepsy: Preventive or treatment opportunities? Front. Biosci. 2014, 19, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, T.; Xia, J.; Yao, W.; Huang, F. Enzymatic and nonenzymatic protein acetylations control glycolysis process in liver diseases. FASEB J. 2019, 33, 11640–11654. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [CrossRef]
- Wardell, S.E.; Ilkayeva, O.R.; Wieman, H.L.; Frigo, D.E.; Rathmell, J.C.; Newgard, C.B.; McDonnell, D.P. Glucose metabolism as a target of histone deacetylase inhibitors. Mol. Endocrinol. 2009, 23, 388–401. [Google Scholar] [CrossRef]
- Li, F.L.; Liu, J.P.; Bao, R.X.; Yan, G.; Feng, X.; Xu, Y.P.; Sun, Y.P.; Yan, W.; Ling, Z.Q.; Xiong, Y.; et al. Acetylation accumulates PFKFB3 in cytoplasm to promote glycolysis and protects cells from cisplatin-induced apoptosis. Nat. Commun. 2018, 9, 508. [Google Scholar] [CrossRef]
- Li, T.; Liu, M.; Feng, X.; Wang, Z.; Das, I.; Xu, Y.; Zhou, X.; Sun, Y.; Guan, K.L.; Xiong, Y.; et al. Glyceraldehyde-3-phosphate dehydrogenase is activated by lysine 254 acetylation in response to glucose signal. J. Biol. Chem. 2014, 289, 3775–3785. [Google Scholar] [CrossRef]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef]
- Lv, L.; Xu, Y.P.; Zhao, D.; Li, F.L.; Wang, W.; Sasaki, N.; Jiang, Y.; Zhou, X.; Li, T.T.; Guan, K.L.; et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 2013, 52, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Kim, W.K.; Kang, H.J.; Kim, J.H.; Chung, S.J.; Seo, Y.S.; Park, S.G.; Lee, S.C.; Bae, K.H. Acetylation of malate dehydrogenase 1 promotes adipogenic differentiation via activating its enzymatic activity. J. Lipid Res. 2012, 53, 1864–1876. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhou, L.; Shi, Q.; Zhao, Y.; Lin, H.; Zhang, M.; Zhao, S.; Yang, Y.; Ling, Z.Q.; Guan, K.L.; et al. SIRT3-dependent GOT2 acetylation status affects the malate-aspartate NADH shuttle activity and pancreatic tumor growth. EMBO J. 2015, 34, 1110–1125. [Google Scholar] [CrossRef] [PubMed]
- Corda, M.G.; Orlandi, M.; Lecca, D.; Carboni, G.; Frau, V.; Giorgi, O. Pentylenetetrazol-induced kindling in rats: Effect of GABA function inhibitors. Pharmacol. Biochem. Behav. 1991, 40, 329–333. [Google Scholar] [CrossRef]
- Schmoll, H.; Badan, I.; Grecksch, G.; Walker, L.; Kessler, C.; Popa-Wagner, A. Kindling Status in Sprague-Dawley Rats Induced by Pentylenetetrazole. Am. J. Pathol. 2003, 162, 1027–1034. [Google Scholar] [CrossRef]
- Hoeller, A.; de Carvalho, C.; Franco, P.; Formolo, D.; Imthon, A.; dos Santos, H.; Eidt, I.; Souza, G.; Constantino, L.; Ferreira, C.; et al. Behavioral and Neurochemical Consequences of Pentylenetetrazol-Induced Kindling in Young and Middle-Aged Rats. Pharmaceuticals 2017, 10, 75. [Google Scholar] [CrossRef]
- Pierozan, P.; Jerneren, F.; Ransome, Y.; Karlsson, O. The Choice of Euthanasia Method Affects Metabolic Serum Biomarkers. Basic Clin. Pharmacol. Toxicol. 2017, 121, 113–118. [Google Scholar] [CrossRef]
- Suckow, M.A.; Stevens, K.A.; Wilson, R.P. The laboratory rabbit, guinea pig, hamster, and other rodents. In American College of Laboratory Animal Medicine Series, 1st ed.; Elsevier: London, UK, 2012. [Google Scholar]
- Leary, S.L.; Underwood, W.; Anthony, R.; Cartner, S.; Grandin, T.; Greenacre, C.; Gwaltney-Brant, S.; McCrackin, M.; Meyer, R.; Miller, D.; et al. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition. Available online: https://www.spandidos-publications.com/var/AVMA_euthanasia_guidelines_2020.pdf (accessed on 1 July 2022).
- Araujo, W.L.; Trofimova, L.; Mkrtchyan, G.; Steinhauser, D.; Krall, L.; Graf, A.; Fernie, A.R.; Bunik, V.I. On the role of the mitochondrial 2-oxoglutarate dehydrogenase complex in amino acid metabolism. Amino Acids 2013, 44, 683–700. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Boyko, A.; Tsepkova, P.; Aleshin, V.; Artiukhov, A.; Mkrtchyan, G.; Ksenofontov, A.; Baratova, L.; Ryabov, S.; Graf, A.; Bunik, V. Severe Spinal Cord Injury in Rats Induces Chronic Changes in the Spinal Cord and Cerebral Cortex Metabolism, Adjusted by Thiamine That Improves Locomotor Performance. Front. Mol. Neurosci. 2021, 14, 620593. [Google Scholar] [CrossRef]
- Schwab, M.A.; Kolker, S.; van den Heuvel, L.P.; Sauer, S.; Wolf, N.I.; Rating, D.; Hoffmann, G.F.; Smeitink, J.A.; Okun, J.G. Optimized spectrophotometric assay for the completely activated pyruvate dehydrogenase complex in fibroblasts. Clin. Chem. 2005, 51, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Ladner, C.L.; Yang, J.; Turner, R.J.; Edwards, R.A. Visible fluorescent detection of proteins in polyacrylamide gels without staining. Anal. Biochem. 2004, 326, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Artiukhov, A.V.; Pometun, A.A.; Zubanova, S.A.; Tishkov, V.I.; Bunik, V.I. Advantages of formate dehydrogenase reaction for efficient NAD(+) quantification in biological samples. Anal. Biochem. 2020, 603, 113797. [Google Scholar] [CrossRef] [PubMed]
- Glass, G.V.; Peckham, P.D.; Sanders, J.R. Consequences of Failure to Meet Assumptions Underlying the Fixed Effects Analyses of Variance and Covariance. Rev. Educ. Res. 2016, 42, 237–288. [Google Scholar] [CrossRef]
- Harwell, M.R.; Rubinstein, E.N.; Hayes, W.S.; Olds, C.C. Summarizing Monte Carlo Results in Methodological Research: The One- and Two-Factor Fixed Effects ANOVA Cases. J. Educ. Stat. 1992, 17, 315–339. [Google Scholar] [CrossRef]
- Lix, L.M.; Keselman, J.C.; Keselman, H.J. Consequences of Assumption Violations Revisited: A Quantitative Review of Alternatives to the One-Way Analysis of Variance F Test. Rev. Educ. Res. 2016, 66, 579–619. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulator | Brain Region | Model | Direction of Changes | Reference | |
|---|---|---|---|---|---|
| Published | This Work | ||||
| sirtuin 5 | hippocampus | kainate, single | increase | increase | [31] |
| sirtuin 3 | hippocampus | kainate, chronic | decrease | decrease | [32] |
| sirtuin 3 | hippocampus | pilocarpine, single, in juvenile rats | decrease | n.d. | [33] |
| sirtuin 3 | hippocampus | PTZ, chronic | decrease | decrease | [34] |
| PDHC | hippocampus | PTZ, chronic | decrease | n.d. | [34] |
| OGDHC | hippocampus | PTZ, chronic | decrease | n.d. | [34] |
| PDHC | hippocampus | pilocarpine, chronic | decrease | n.d. | [35] |
| OGDHC | hippocampus | pilocarpine, chronic | decrease | n.d. | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zavileyskiy, L.G.; Aleshin, V.A.; Kaehne, T.; Karlina, I.S.; Artiukhov, A.V.; Maslova, M.V.; Graf, A.V.; Bunik, V.I. The Brain Protein Acylation System Responds to Seizures in the Rat Model of PTZ-Induced Epilepsy. Int. J. Mol. Sci. 2022, 23, 12302. https://doi.org/10.3390/ijms232012302
Zavileyskiy LG, Aleshin VA, Kaehne T, Karlina IS, Artiukhov AV, Maslova MV, Graf AV, Bunik VI. The Brain Protein Acylation System Responds to Seizures in the Rat Model of PTZ-Induced Epilepsy. International Journal of Molecular Sciences. 2022; 23(20):12302. https://doi.org/10.3390/ijms232012302
Chicago/Turabian StyleZavileyskiy, Lev G., Vasily A. Aleshin, Thilo Kaehne, Irina S. Karlina, Artem V. Artiukhov, Maria V. Maslova, Anastasia V. Graf, and Victoria I. Bunik. 2022. "The Brain Protein Acylation System Responds to Seizures in the Rat Model of PTZ-Induced Epilepsy" International Journal of Molecular Sciences 23, no. 20: 12302. https://doi.org/10.3390/ijms232012302
APA StyleZavileyskiy, L. G., Aleshin, V. A., Kaehne, T., Karlina, I. S., Artiukhov, A. V., Maslova, M. V., Graf, A. V., & Bunik, V. I. (2022). The Brain Protein Acylation System Responds to Seizures in the Rat Model of PTZ-Induced Epilepsy. International Journal of Molecular Sciences, 23(20), 12302. https://doi.org/10.3390/ijms232012302