

Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression
 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Characteristics of the Patients and the Control Group
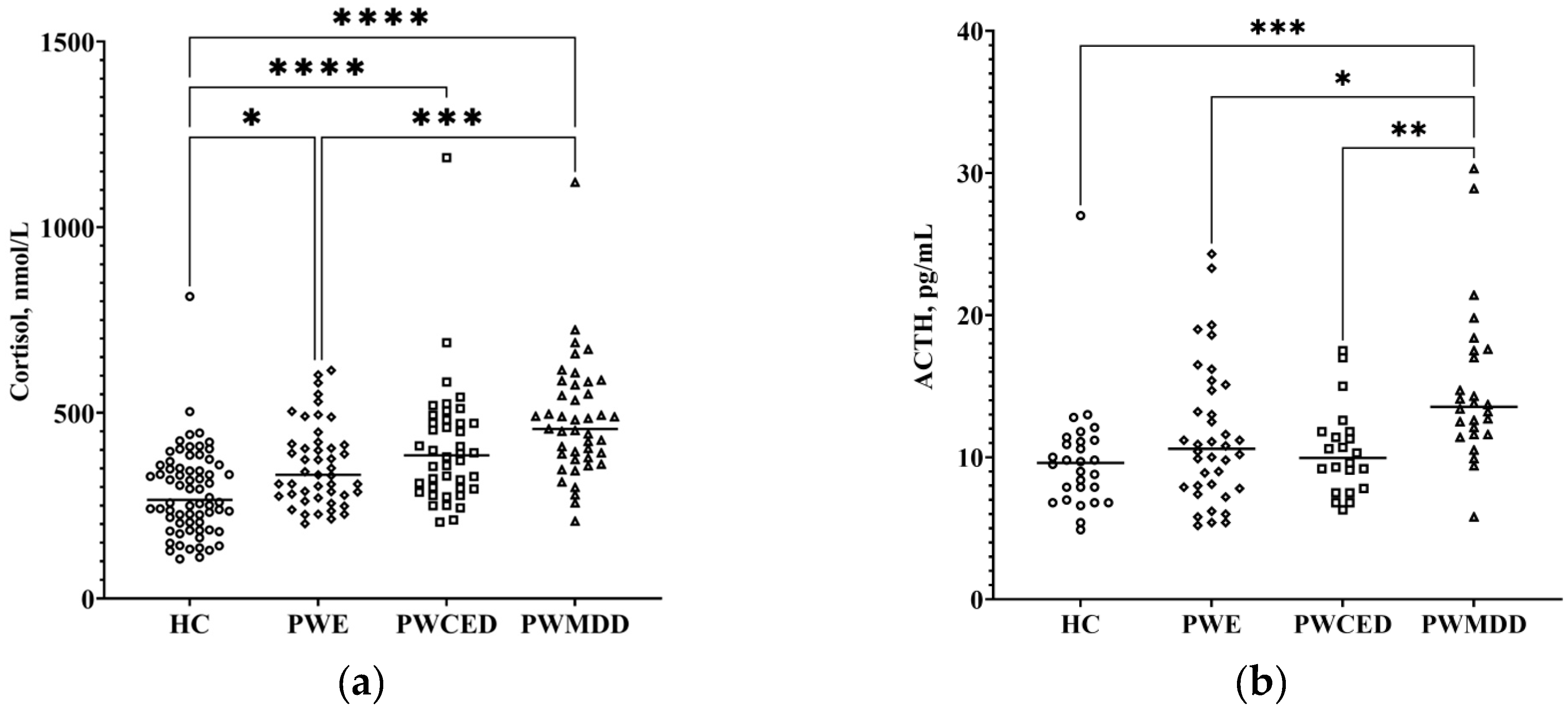
2.2. HPA Axis Indices
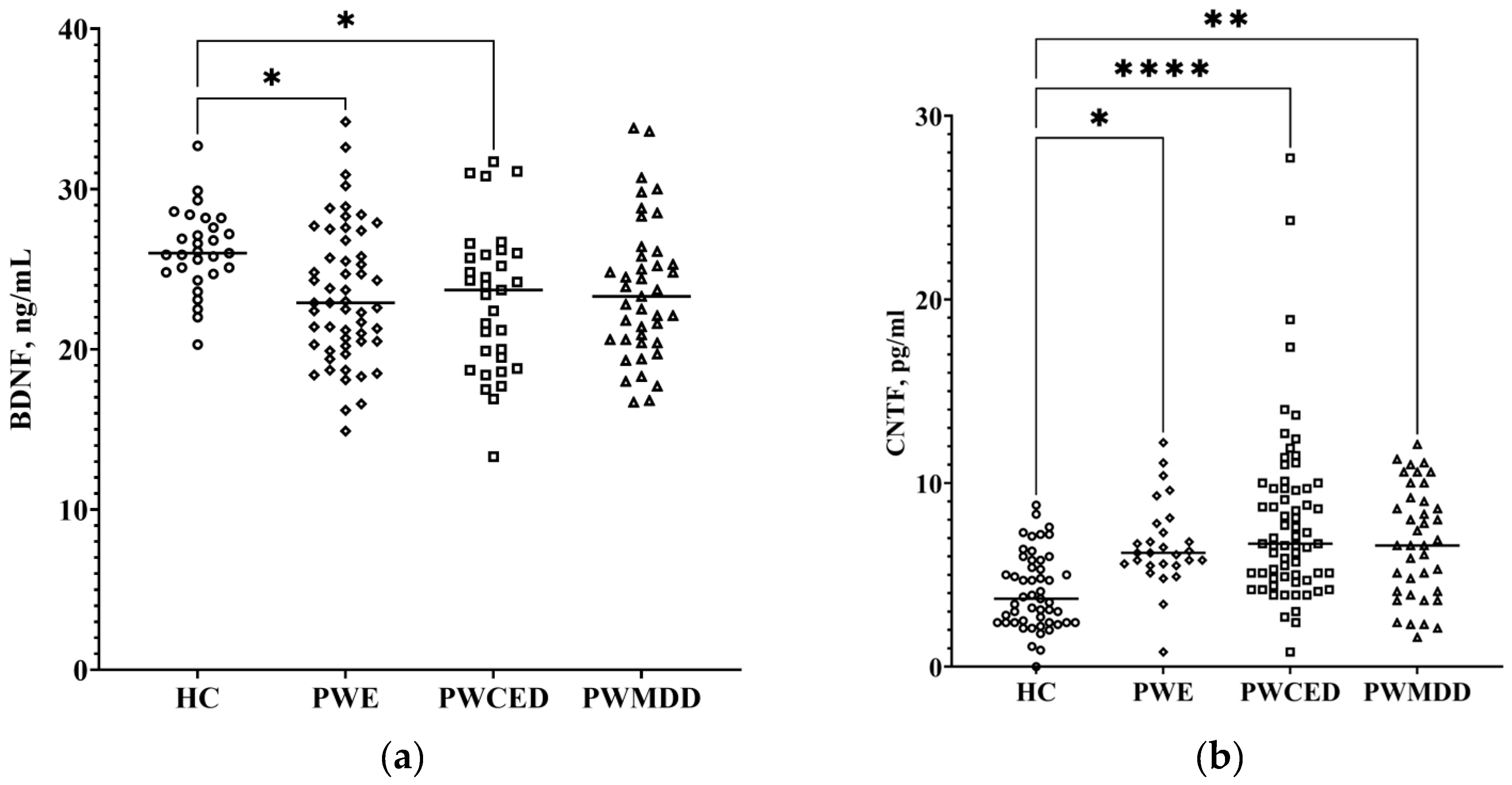
2.3. Neurotrophic Factors
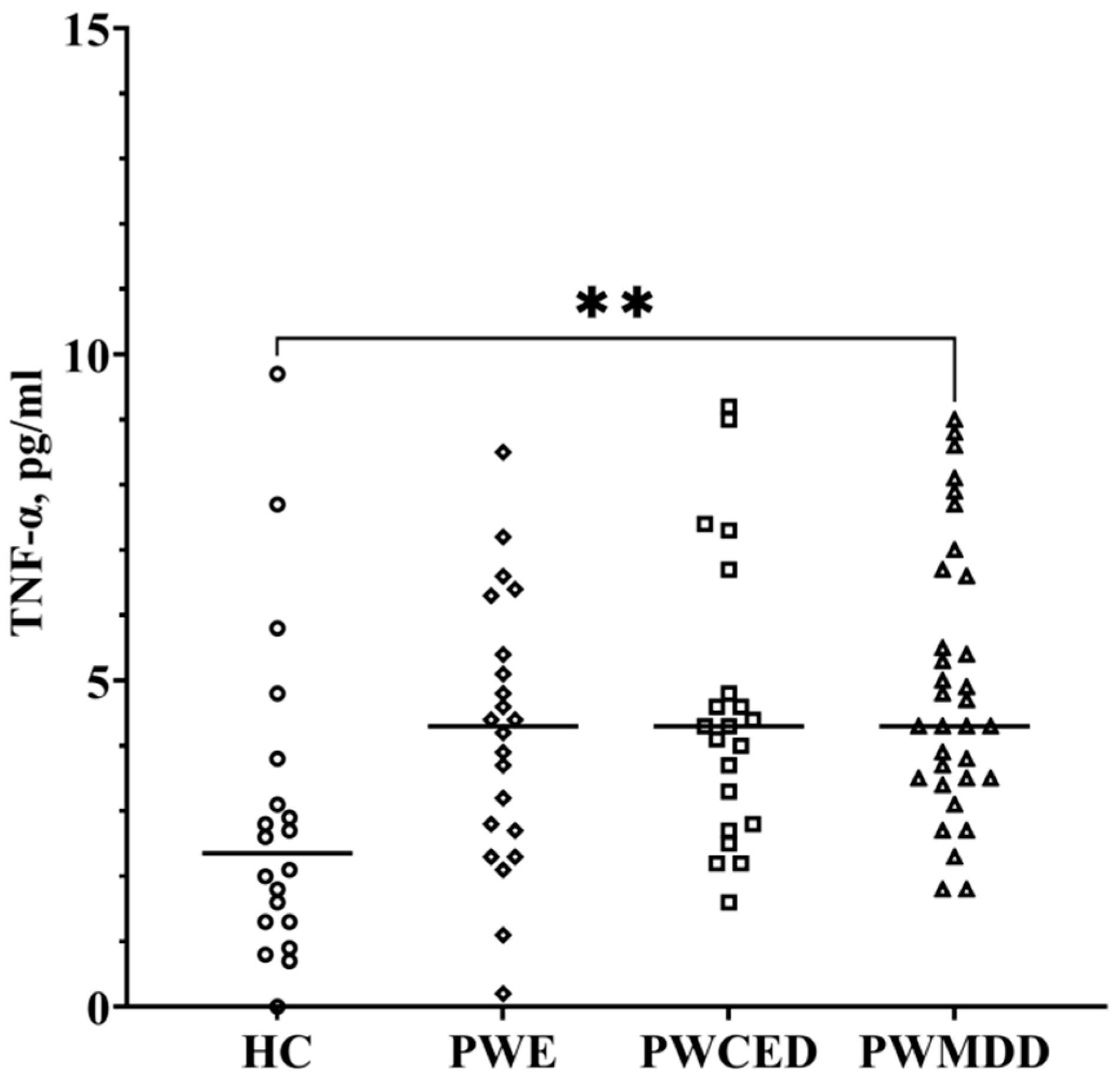
2.4. TNF-α
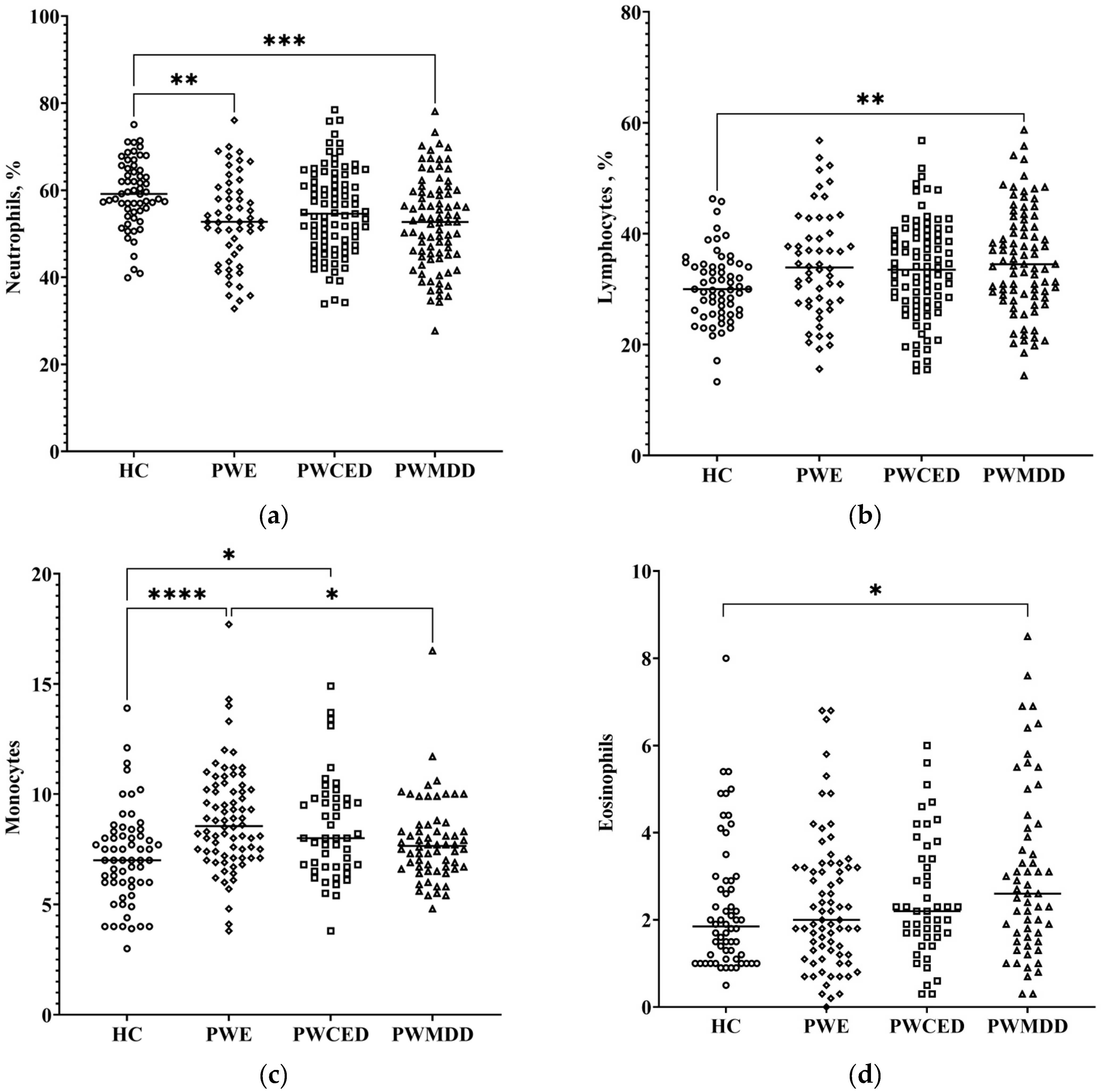
2.5. Hemogram
2.6. Correlations
3. Discussion
3.1. HPA Axis Dysfunction in Depression and Epilepsy
3.2. Inflammatory and Immune Processes in Depression and Epilepsy
3.3. Neurotrophic Factors in Depression and Epilepsy
4. Materials and Methods
4.1. Subjects
4.2. Assessment of Biochemical Indices and Hormones
4.3. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanner, A.M. Psychiatric comorbidities in new onset epilepsy: Should they be always investigated? Seizure 2017, 49, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Salpekar, J.A.; Mula, M. Common psychiatric comorbidities in epilepsy: How big of a problem is it? Epilepsy Behav. 2019, 98, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Vinti, V.; Dell’Isola, G.B.; Tascini, G.; Mencaroni, E.; Di Cara, G.; Striano, P.; Verrotti, A. Temporal Lobe Epilepsy and Psychiatric Comorbidity. Front. Neurol. 2021, 12, 775781. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Operto, F.F.; Matricardi, S.; Verrotti, A. Monitoring and Managing Depression in Adolescents with Epilepsy: Current Perspectives. Neuropsychiatr. Dis. Treat. 2019, 15, 2773–2780. [Google Scholar] [CrossRef] [PubMed]
- Mula, M.; Kaufman, K.R. Double stigma in mental health: Epilepsy and mental illness. BJPsych Open 2020, 6, e72. [Google Scholar] [CrossRef]
- Ribot, R.; Kanner, A.M. Neurobiologic properties of mood disorders may have an impact on epilepsy: Should this motivate neurologists to screen for this psychiatric comorbidity in these patients? Epilepsy Behav. 2019, 98 Pt B, 298–301. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Stress-Associated Molecular and Cellular Hippocampal Mechanisms Common for Epilepsy and Comorbid Depressive Disorders. Biochemistry 2021, 86, 641–656. [Google Scholar] [CrossRef]
- Joëls, M. Corticosteroid Actions in the Hippocampus. J. Neuroendocr. 2001, 13, 657–669. [Google Scholar] [CrossRef]
- Joëls, M.; Karst, H.; Sarabdjitsingh, R.A. The stressed brain of humans and rodents. Acta Physiol. 2018, 223, e13066. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochemistry 2019, 84, 1306–1328. [Google Scholar] [CrossRef] [PubMed]
- Wulsin, A.C.; Solomon, M.B.; Privitera, M.D.; Danzer, S.C.; Herman, J.P. Hypothalamic-pituitary-adrenocortical axis dysfunction in epilepsy. Physiol. Behav. 2016, 166, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Van Campen, J.S.; Valentijn, F.A.; Jansen, F.E.; Joëls, M.; Braun, K.P. Seizure occurrence and the circadian rhythm of cortisol: A systematic review. Epilepsy Behav. 2015, 47, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 2019, 15, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Koyama, R.; Ikegaya, Y. To BDNF or Not to BDNF: That Is the Epileptic Hippocampus. Neuroscientist 2005, 11, 282–287. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; DeSmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef]
- Mikulska, J.; Juszczyk, G.; Gawrońska-Grzywacz, M.; Herbet, M. HPA Axis in the Pathomechanism of Depression and Schizophrenia: New Therapeutic Strategies Based on Its Participation. Brain Sci. 2021, 11, 1298. [Google Scholar] [CrossRef]
- Murri, M.B.; Pariante, C.; Mondelli, V.; Masotti, M.; Atti, A.R.; Mellacqua, Z.; Antonioli, M.; Ghio, L.; Menchetti, M.; Zanetidou, S.; et al. HPA axis and aging in depression: Systematic review and meta-analysis. Psychoneuroendocrinology 2014, 41, 46–62. [Google Scholar] [CrossRef]
- Thomson, F.; Craighead, M. Innovative Approaches for the Treatment of Depression: Targeting the HPA Axis. Neurochem. Res. 2008, 33, 691–707. [Google Scholar] [CrossRef]
- Kennis, M.; Gerritsen, L.; Van Dalen, M.; Williams, A.; Cuijpers, P.; Bockting, C. Prospective biomarkers of major depressive disorder: A systematic review and meta-analysis. Mol. Psychiatry 2020, 25, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Dziurkowska, E.; Wesolowski, M. Cortisol as a Biomarker of Mental Disorder Severity. J. Clin. Med. 2021, 10, 5204. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Macare, C.; Cleare, A.J. Hypothalamic-pituitary-adrenal (HPA) axis functioning as predictor of antidepressant response–Meta-analysis. Neurosci. Biobehav. Rev. 2017, 83, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Cano-López, I.; Gonzalez-Bono, E. Cortisol levels and seizures in adults with epilepsy: A systematic review. Neurosci. Biobehav. Rev. 2019, 103, 216–229. [Google Scholar] [CrossRef] [PubMed]
- de Kloet, E.; Meijer, O.; de Nicola, A.; de Rijk, R.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocr. 2018, 49, 124–145. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Schroeter, S.; Çalışkan, G.; Salar, S.; Kobow, K.; Coras, R.; Blümcke, I.; Hamer, H.; Schwarz, M.; Buchfelder, M.; et al. Glucocorticoid modulation of synaptic plasticity in the human temporal cortex of epilepsy patients: Does chronic stress contribute to memory impairment? Epilepsia 2022, 63, 209–221. [Google Scholar] [CrossRef]
- Cano-López, I.; Hidalgo, V.; Hampel, K.G.; Garcés, M.; Salvador, A.; González-Bono, E.; Villanueva, V. Cortisol and trait anxiety as relevant factors involved in memory performance in people with drug-resistant epilepsy. Epilepsy Behav. 2019, 92, 125–134. [Google Scholar] [CrossRef]
- de Kloet, E.R.; Karst, H.; Joëls, M. Corticosteroid hormones in the central stress response: Quick-and-slow. Front. Neuroendocr. 2008, 29, 268–272. [Google Scholar] [CrossRef]
- Maggio, N.; Segal, M. Corticosteroid Regulation of Synaptic Plasticity in the Hippocampus. Sci. World J. 2010, 10, 462–469. [Google Scholar] [CrossRef]
- Joëls, M. Corticosteroid effects in the brain: U-shape it. Trends Pharmacol. Sci. 2006, 27, 244–250. [Google Scholar] [CrossRef]
- Basu, T.; Maguire, J.; Salpekar, J.A. Hypothalamic-pituitary-adrenal axis targets for the treatment of epilepsy. Neurosci. Lett. 2021, 746, 135618. [Google Scholar] [CrossRef] [PubMed]
- Salpekar, J.A.; Salpekar, J.A.; Basu, T.; Basu, T.; Thangaraj, S.; Thangaraj, S.; Maguire, J.; Maguire, J. The intersections of stress, anxiety and epilepsy. Int. Rev. Neurobiol. 2020, 152, 195–219. [Google Scholar] [CrossRef]
- Bauer, M.E.; Teixeira, A.L. Neuroinflammation in Mood Disorders: Role of Regulatory Immune Cells. Neuroimmunomodulation 2021, 28, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Nobis, A.; Zalewski, D.; Waszkiewicz, N. Peripheral Markers of Depression. J. Clin. Med. 2020, 9, 3793. [Google Scholar] [CrossRef] [PubMed]
- Pfau, M.L.; Ménard, C.; Russo, S.J. Inflammatory Mediators in Mood Disorders: Therapeutic Opportunities. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Debnath, M.; Berk, M.; Maes, M. Translational evidence for the Inflammatory Response System (IRS)/Compensatory Immune Response System (CIRS) and neuroprogression theory of major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 111, 110343. [Google Scholar] [CrossRef] [PubMed]
- Osimo, E.F.; Pillinger, T.; Rodriguez, I.M.; Khandaker, G.M.; Pariante, C.M.; Howes, O.D. Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain, Behav. Immun. 2020, 87, 901–909. [Google Scholar] [CrossRef]
- Enache, D.; Pariante, C.M.; Mondelli, V. Markers of central inflammation in major depressive disorder: A systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain, Behav. Immun. 2019, 81, 24–40. [Google Scholar] [CrossRef]
- Jenkins, B.J.; Hughes, S.T.O.; Figueras, A.C.; Jones, S.A. Unravelling the broader complexity of IL-6 involvement in health and disease. Cytokine 2021, 148, 155684. [Google Scholar] [CrossRef]
- Messaoud, A.; Rym, M.; Wahiba, D.; Neffati, F.; Najjar, M.F.; Gobbi, G.; Manchia, M.; Valtorta, F.; Lotfi, G.; Comai, S. Investigation of the relationship among cortisol, pro-inflammatory cytokines, and the degradation of tryptophan into kynurenine in patients with major depression and suicidal behavior. Curr. Top. Med. Chem. 2021, 21. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Furuyashiki, T. The impact of stress on immune systems and its relevance to mental illness. Neurosci. Res. 2021, 175, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E. Glia-neuronal interactions in ictogenesis and epileptogenesis: Role of inflammatory mediators. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.V., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. Available online: https://www.ncbi.nlm.nih.gov/books/NBK98146/ (accessed on 2 September 2022)ISBN 9780199746545.
- Vezzani, A.; Balosso, S.; Ravizza, T. Inflammation and epilepsy. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2012; Volume 107, pp. 163–175. ISBN 9780444528988. [Google Scholar]
- Barnes, S.E.; Zera, K.A.; Ivison, G.T.; Buckwalter, M.S.; Engleman, E.G. Brain profiling in murine colitis and human epilepsy reveals neutrophils and TNFα as mediators of neuronal hyperexcitability. J. Neuroinflammation 2021, 18, 199. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.B.; Tian, D.; Wu, L. Neuroimmune interaction in seizures and epilepsy: Focusing on monocyte infiltration. FEBS J. 2020, 287, 4822–4837. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, G.; Morichi, S.; Takamatsu, T.; Watanabe, Y.; Suzuki, S.; Ishida, Y.; Oana, S.; Yamazaki, T.; Takata, F.; Kawashima, H. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 4395. [Google Scholar] [CrossRef]
- Tan, T.H.; Perucca, P.; O’Brien, T.J.; Kwan, P.; Monif, M. Inflammation, ictogenesis, and epileptogenesis: An exploration through human disease. Epilepsia 2021, 62, 303–324. [Google Scholar] [CrossRef]
- Terrone, G.; Salamone, A.; Vezzani, A. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr. Pharm. Des. 2017, 23, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Miller, S.D.; Koh, S. Immune mechanisms in epileptogenesis. Front. Cell. Neurosci. 2013, 8, 195. [Google Scholar] [CrossRef]
- Khaboushan, A.S.; Yazdanpanah, N.; Rezaei, N. Neuroinflammation and Proinflammatory Cytokines in Epileptogenesis. Mol. Neurobiol. 2022, 59, 1724–1743. [Google Scholar] [CrossRef]
- de Vries, E.E.; Munckhof, B.V.D.; Braun, K.P.; van Royen-Kerkhof, A.; de Jager, W.; Jansen, F.E. Inflammatory mediators in human epilepsy: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2016, 63, 177–190. [Google Scholar] [CrossRef]
- Arulsamy, A.; Shaikh, M.F. Tumor Necrosis Factor-α, the Pathological Key to Post-Traumatic Epilepsy: A Comprehensive Systematic Review. ACS Chem. Neurosci. 2020, 11, 1900–1908. [Google Scholar] [CrossRef]
- Güneş, M.; Büyükgöl, H. Relationship between generalized epileptic seizure and neutrophil/lymphocyte ratio, platelet/lymphocyte ratio, and neutrophil mediated inflammation. Int. J. Neurosci. 2020, 130, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Alvim, M.K.M.; Morita-Sherman, M.E.; Yasuda, C.L.; Rocha, N.P.; Vieira, L.; Pimentel-Silva, L.R.; Nogueira, M.H.; Barbosa, R.; Watanabe, N.; Coan, A.C.; et al. Inflammatory and neurotrophic factor plasma levels are related to epilepsy independently of etiology. Epilepsia 2021, 62, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Shpak, A.; Guekht, A.; Druzhkova, T.; Rider, F.; Gudkova, A.; Gulyaeva, N. Increased ciliary neurotrophic factor in blood serum and lacrimal fluid as a potential biomarkers of focal epilepsy. Neurol. Sci. 2021, 43, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Iughetti, L.; Lucaccioni, L.; Fugetto, F.; Predieri, B.; Berardi, A.; Ferrari, F. Brain-derived neurotrophic factor and epilepsy: A systematic review. Neuropeptides 2018, 72, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Z.; Zhong, K. The Role of Brain-Derived Neurotrophic Factor in Epileptogenesis: An Update. Front. Pharmacol. 2021, 12, 758232. [Google Scholar] [CrossRef] [PubMed]
- Nowroozi, A.; Salehi, M.A.; Mohammadi, S. Brain-derived neurotrophic factor in patients with epilepsy: A systematic review and meta-analysis. Epilepsy Res. 2021, 178, 106794. [Google Scholar] [CrossRef]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Miyanishi, H.; Nitta, A. A Role of BDNF in the Depression Pathogenesis and a Potential Target as Antidepressant: The Modulator of Stress Sensitivity “Shati/Nat8l-BDNF System” in the Dorsal Striatum. Pharmaceuticals 2021, 14, 889. [Google Scholar] [CrossRef]
- Porter, G.A.; O’Connor, J.C. Brain-derived neurotrophic factor and inflammation in depression: Pathogenic partners in crime? World J. Psychiatry 2022, 12, 77–97. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Yuan, Y. The Combination of Serum BDNF, Cortisol and IFN-Gamma Can Assist the Diagnosis of Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2021, 17, 2819–2829. [Google Scholar] [CrossRef]
- Galvão, A.C.D.M.; Almeida, R.N.; Júnior, G.M.D.S.; Leocadio-Miguel, M.A.; Palhano-Fontes, F.; de Araujo, D.B.; Lobão-Soares, B.; Maia-De-Oliveira, J.P.; Nunes, E.A.; Hallak, J.E.C.; et al. Potential biomarkers of major depression diagnosis and chronicity. PLoS ONE 2021, 16, e0257251. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Zheng, Y.; Froud, K. Brain-Derived Neurotrophic Factor as a Clinical Biomarker in Predicting the Development of Post-Stroke Depression: A Review of Evidence. Cureus 2021, 13, e15662. [Google Scholar] [CrossRef] [PubMed]
- Peruga, I.; Hartwig, S.; Merkler, D.; Thöne, J.; Hovemann, B.; Juckel, G.; Gold, R.; Linker, R.A. Endogenous ciliary neurotrophic factor modulates anxiety and depressive-like behavior. Behav. Brain Res. 2012, 229, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Brown, R.W.; Malone, H.M.; Burgess, K.C.; Gill, W.D.; Keasey, M.P.; Hagg, T. Ciliary neurotrophic factor is a key sex-specific regulator of depressive-like behavior in mice. Psychoneuroendocrinology 2019, 100, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Druzhkova, T.; Pochigaeva, K.; Yakovlev, A.; Kazimirova, E.; Grishkina, M.; Chepelev, A.; Guekht, A.; Gulyaeva, N. Acute stress response to a cognitive task in patients with major depressive disorder: Potential metabolic and proinflammatory biomarkers. Metab. Brain Dis. 2018, 34, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Fisher, R.S.; Cross, J.H.; French, J.A.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; Peltola, J.; Roulet Perez, E.; et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 522–530. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-Mental State”. A Practical Method for Grading the Cognitive State of Patients for the Clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Beck, A.T.; Steer, R.A.; Brown, G.K. Manual for The Beck Depression Inventory, 2nd ed.; (BDI-II); Psychological Corporation: San Antonio, TX, USA, 1996. [Google Scholar]
- Lang, U.E.; Borgwardt, S. Molecular Mechanisms of Depression: Perspectives on New Treatment Strategies. Cell. Physiol. Biochem. 2013, 31, 761–777. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter/ Group | HC n = 78 | PWE n = 76 | PWCED n = 48 | PWMDD n = 62 |
|---|---|---|---|---|
| demographic data | ||||
| Age, years | 44.3 ± 13.4 | 42.2 ± 13.7 | 48.4 ± 12.5 | 44.6 ± 11.3 |
| BMI | 25.5 ± 4.6 | 26.3 ± 4.2 | 25.6 ± 5.00 | 24.1 ± 5.9 |
| gender (male/female), % | 37/63 | 50/50 | 25/75 | 30/70 |
| Education(sec./higher), % | 58/42 | 55/45 | 60/40 | 44/56 |
| Employment (−/+), % | 6/94 | 66/34 | 50/50 | 63/37 |
| clinical data | ||||
| take antipsychotics, % | 30 | 71 | 84 | |
| take antidepressants, % | 75 | 92 | 98 | |
| take tranquilizers, % | 37 | 52 | 55 | |
| take anti-seizure medication, % | 93 | 94 | 13 | |
| disease duration | 15.4 ± 12.9 | 14.9 ± 12.4 | 8.14 ± 5.6 | |
| BDI-II | 8.7 ± 6.1 | 25.8 ± 10.3 *** | 32.3 ± 8.00 ** | |
| MMSE | 27.6 ± 2.3 | 26.7 ± 2.3 *** | 28.5 ± 1.4 | |
| blood serum (plasma) indices | ||||
| TSH3, ulU/mL | 2.1 ± 1.2 | 2.1 ± 1.1 | 2.3 ± 2.2 | 2.5 ± 1.9 |
| ACTH, pg/mL | 9.8 ± 4.0 | 11.6 ± 4.9 *** | 10.4 ± 3.1 *** | 13.8 ± 5.8 * |
| cortisol, mmol/L | 288.3 ± 115.6 | 386.3 ± 135.2 * | 433.3 ± 174.3 * | 495.7 ± 142.5 *;** |
| BDNF, ng/mL | 26.1 ± 2.6 | 23.4 ± 4.3 * | 23.1 ± 4.5 * | 23.7 ± 4.3 |
| CNTF, pg/mL | 4.1 ± 2.1 | 7.9 ± 2.6 * | 8.3 ± 5.1 * | 6.4 ± 2.8 * |
| GDNF, pg/mL | 169.5 ± 53.1 | 143.9 ± 44.6 | 158.4 ± 53.9 | 152.8 ± 37.9 |
| NGF, pg/mL | 29.5 ± 10.1 | 27.7 ± 12.2 | 24.2 ± 7.3 | 24.4 ± 8.3 |
| TNF-α, pg/mL | 2.9 ± 2.4 | 4.2 ± 2.0 | 4.5 ± 1.93 | 5.1 ± 2.8 * |
| total bilirubin, µmol/L | 12.7 ± 3.7 | 12.3 ± 5.1 | 10.8 ± 4.7 | 13.9 ± 6.7 |
| glucose, mmol/L | 5.1 ± 0.6 | 5.5 ± 1.0 | 5.4 ± 0.6 | 5.4 ± 1.0 |
| creatinine, µmol/L | 85.1 ± 13.1 | 84.4 ± 13.8 | 80.5 ± 11.3 | 83.2 ± 13.5 |
| urea, mmol/L | 5.2 ± 1.0 | 4.2 ± 1.4 | 4.0 ± 1.2 | 3.9 ± 1.1 |
| cholesterol, mmol/L | 5.1 ± 0.7 | 6.0 ± 1.3 | 6.1 ± 1.6 | 5.4 ± 1.2 |
| triglycerides, mmol/L | 1.3 ± 0.54 | 1.4 ± 1.0 | 1.4 ± 1.3 | 1.3 ± 0.9 |
| hemogram | ||||
| platelets, PLT, 103/µL | 237.8 ± 54.1 | 226.4 ± 63.9 | 244.2 ± 62.5 | 243.0 ± 59.2 |
| erythrocytes, RBC, 106/µL | 4.7 ± 0.5 | 4.7 ± 0.5 | 4.5 ± 0.4 | 4.7 ± 0.6 |
| hemoglobin, Hb, g/L | 141.3 ± 12.8 | 143.3 ± 14.9 | 135.5 ± 12.0 | 141.9 ± 14.2 |
| while blood cell, WBC, 103/µL | 6.4 ± 1.7 | 5.8 ± 1.7 | 6.0 ± 1.9 | 6.4 ± 1.6 |
| neutrophils, NE, % | 59.2 ± 7.7 | 54.0 ± 10.3 * | 54.6 ± 10.3 | 53.0 ± 10.9 * |
| lymphocytes, LY, % | 30.4 ± 6.4 | 33.8 ± 9.6 | 33.6 ± 8.3 | 35.4 ± 9.9 * |
| monocytes, MO, % | 7.2 ± 2.1 | 8.8 ± 2.3 * | 8.5 ± 2.32 * | 7.9 ± 1.9 |
| eosinophils, EO, % | 2.3 ± 1.5 | 2.4 ± 1.6 | 2.5 ± 1.4 | 3.1 ± 2.0 * |
| basophils, BA, % | 0.8 ± 0.6 | 0.9 ± 0.51 | 0.9 ± 0.5 | 0.9 ± 0.5 |
| PLT/NE, PNR | 66.9 ± 21.1 | 80.1 ± 37.4 * | 83.2 ± 32.5 * | 79.1 ± 32.1 |
| PLT/LY, PLR | 131.2 ± 39.1 | 132.0 ± 65.0 | 135.9 ± 53.3 | 118.4 ± 42.7 |
| PLT/MO, PMR | 36.4 ± 13.9 | 27.5 ± 11.2 * | 31.1 ± 11.8 | 32.5 ± 11.0 |
| Parameter/ Group | Etiology of Epilepsy | ||
|---|---|---|---|
| Traumatic Brain Injury, Cerebrovascular Disorders, Including Stroke n = 38 | Tumors n = 20 | Reason Not Established or Multiple n = 66 | |
| Demographic data | |||
| Age, years | 45.4 ± 11.5 | 46.9 ± 14.9 | 42.7 ± 14.1 |
| BMI | 25.8 ± 4.5 | 25.5 ± 3.3 | 26.4 ± 4.9 |
| gender (male/female), % | 55/45 | 25/75 | 36/64 |
| Education (secondary/higher), % | 63/37 | 55/45 | 54/46 |
| Employment (−/+), % | 55/45 | 55/45 | 64/36 |
| Clinical data | |||
| take antipsychotics, % | 47 | 50 | 44 |
| take antidepressants, % | 71 | 85 | 86 |
| take tranquilizers, % | 32 | 40 | 50 |
| take anti-seizure medication, % | 92 | 95 | 94 |
| BDI-II | 14.8 ± 9.6 | 20.8 ± 13.8 | 14.1 ± 11.4 |
| MMSE | 26.6 ± 2.9 | 27.1 ± 2.2 | 27.7 ± 2.2 |
| disease duration | 13.7 ± 10.8 | 13.3 ± 10.0 | 16.6 ± 13.1 |
| seizure type | |||
| focal onset aware seizure, % | 34 | 25 | 33 |
| focal onset impaired/awareness seizure, % | 45 | 55 | 65 |
| focal to bilateral tonic-clonic seizure, % | 79 | 75 | 73 |
| seizure frequency | |||
| absence in the last year, % | 11 | 20 | 18 |
| less than one per year, % | 50 | 50 | 32 |
| 1–3 per month, % | 21 | 5 | 15 |
| more than one per week, % | 8 | 15 | 23 |
| blood serum (plasma) indices | |||
| TSH3, ulU/mL | 2.7 ± 2.2 | 2.0 ± 1.2 | 1.9 ± 1.1 * |
| ACTH, pg/mL | 12.2 ± 3.8 | 9.9 ± 2.2 | 11.0 ± 4.6 |
| cortisol, mmol/L | 392.1 ± 116.4 | 384.9 ± 99.6 | 414.5 ± 182.3 |
| BDNF, ng/mL | 23.7 ± 4.4 | 22.5 ± 2.6 | 23.2 ± 4.8 |
| CNTF, pg/mL | 8.3 ± 4.7 | 7.2 ± 2.6 | 7.0 ± 3.0 |
| GDNF, pg/mL | 201.2 ± 53.1 | 123.4 ± 42.3 * | 141.7 ± 40.8 * |
| NGF, pg/mL | 26.3 ± 4.0 | 28.7 ± 13.7 | 26.4 ± 20.4 |
| TNF-α, pg/mL | 5.7 ± 2.1 | 5.0 ± 2.0 | 4.0 ± 1.8 |
| total bilirubin, µmol/L | 12.4 ± 4.3 | 12.2 ± 5.6 | 11.1 ± 5.2 |
| glucose, mmol/L | 5.7 ± 1.3 | 5.6 ± 0.7 | 5.3 ± 0.5 |
| creatinine, µmol/L | 83.2 ± 17.7 | 81.4 ± 11.3 | 83.3 ± 16.1 |
| urea, mmol/L | 4.0 ± 1.2 | 4.3 ± 1.5 | 4.1 ± 1.3 |
| cholesterol, mmol/L | 5.8 ± 1.4 | 6.6 ± 1.7 | 6.0 ± 1.3 |
| triglycerides, mmol/L | 1.2 ± 0.8 ** | 2.2 ± 1.9 | 1.1 ± 0.4 ** |
| hemogram | |||
| platelets, PLT, 103/µL | 211.5 ± 66.4 | 243.1 ± 69.5 | 242.9 ± 57.9 * |
| erythrocytes, RBC, 106/µL | 4.7 ± 0.4 | 4.6 ± 0.4 | 4.6 ± 0.5 |
| hemoglobin, Hb, g/L | 143.7 ± 12.8 | 137.2 ± 14.1 | 139.2 ± 15.0 |
| while blood cell, WBC, 103/µL | 5.9 ± 1.7 | 5.4 ± 1.7 | 5.1 ± 1.8 |
| neutrophils, NE, % | 53.6 ± 10.7 | 53.9 ± 7.8 | 54.8 ± 10.8 |
| lymphocytes, LY, % | 34.4 ± 9.4 | 33.8 ± 7.2 | 33.4 ± 9.5 |
| monocytes, MO, % | 8.5 ± 2.5 | 8.8 ± 1.75 | 8.6 ± 2.36 |
| eosinophils, BA, % | 2.8 ± 1.6 | 2.5 ± 1.36 | 2.2 ± 1.43 |
| basophils, BA, % | 0.7 ± 0.3 | 0.8 ± 0.34 | 1.0 ± 0.60 |
| PLT/NE, PNR | 75.8 ± 40.0 | 89.4 ± 31.1 | 81.1 ± 33.9 |
| PLT/LY, PLR | 114.1 ± 40.6 | 143.8 ± 51.3 | 141.7 ± 70.1 |
| PLT/MO, PMR | 27.2 ± 12.2 | 29.0 ± 11.5 | 29.9 ± 11.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Druzhkova, T.A.; Yakovlev, A.A.; Rider, F.K.; Zinchuk, M.S.; Guekht, A.B.; Gulyaeva, N.V. Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression. Int. J. Mol. Sci. 2022, 23, 10414. https://doi.org/10.3390/ijms231810414
Druzhkova TA, Yakovlev AA, Rider FK, Zinchuk MS, Guekht AB, Gulyaeva NV. Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression. International Journal of Molecular Sciences. 2022; 23(18):10414. https://doi.org/10.3390/ijms231810414
Chicago/Turabian StyleDruzhkova, Tatyana A., Alexander A. Yakovlev, Flora K. Rider, Mikhail S. Zinchuk, Alla B. Guekht, and Natalia V. Gulyaeva. 2022. "Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression" International Journal of Molecular Sciences 23, no. 18: 10414. https://doi.org/10.3390/ijms231810414
APA StyleDruzhkova, T. A., Yakovlev, A. A., Rider, F. K., Zinchuk, M. S., Guekht, A. B., & Gulyaeva, N. V. (2022). Elevated Serum Cortisol Levels in Patients with Focal Epilepsy, Depression, and Comorbid Epilepsy and Depression. International Journal of Molecular Sciences, 23(18), 10414. https://doi.org/10.3390/ijms231810414