

Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R
 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Results
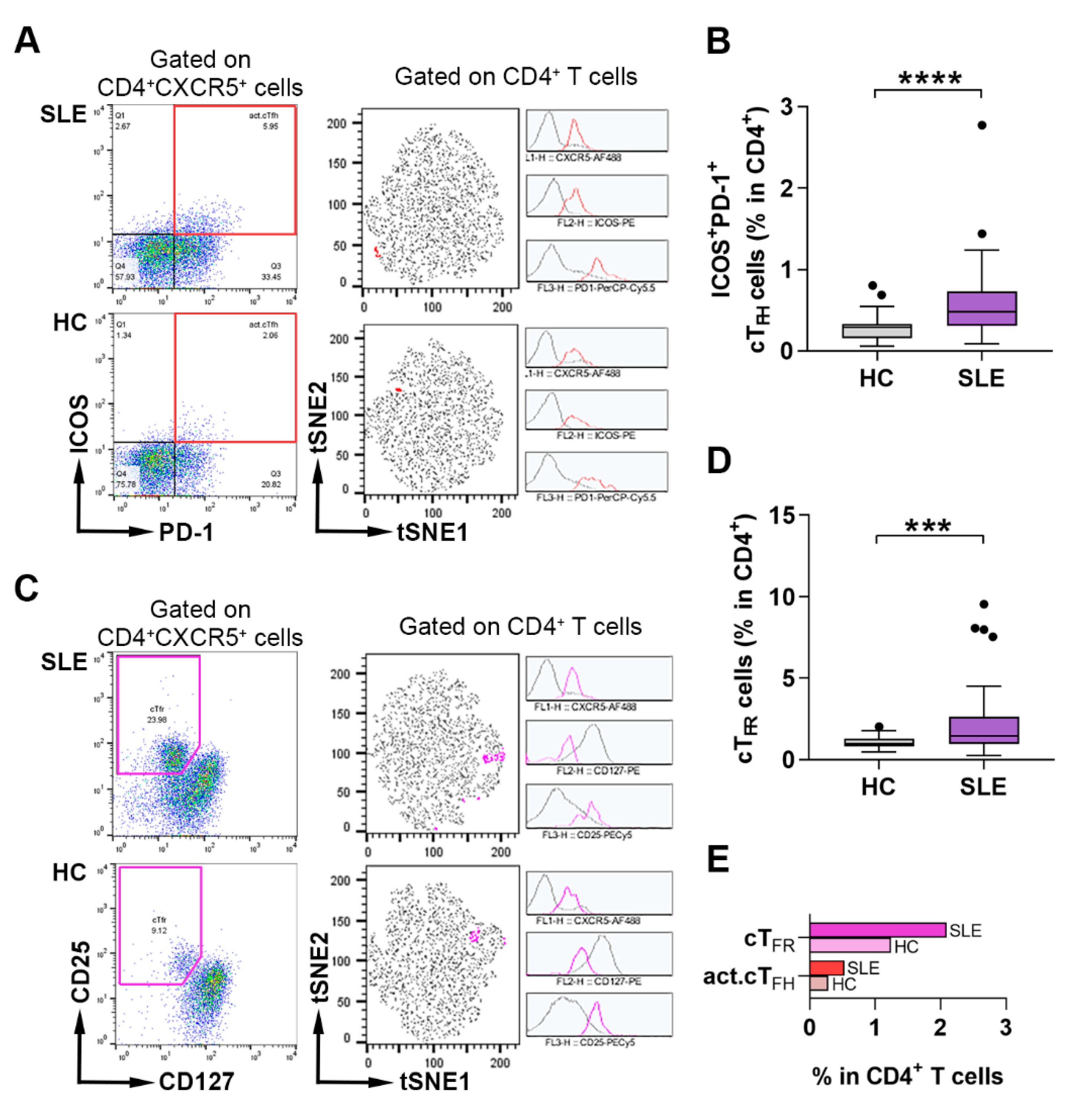
2.1. Assessment of the Frequency of Activated cTFH and cTFR Cells in Lupus Patients and Healthy Individuals
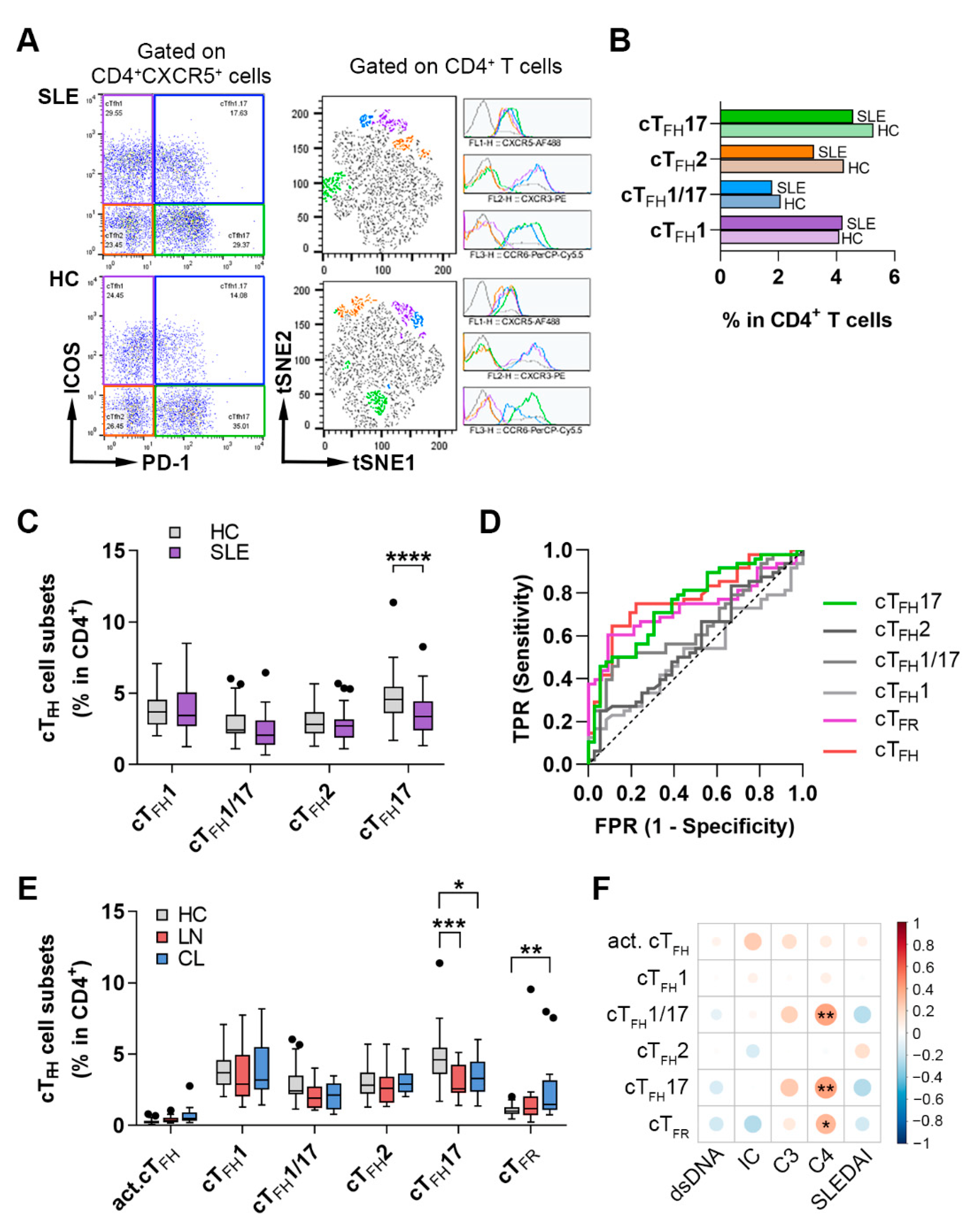
2.2. The Imbalance of the Proportions of cTFH Cell Subsets and Their Correlation with Disease Phenotype in SLE Patients
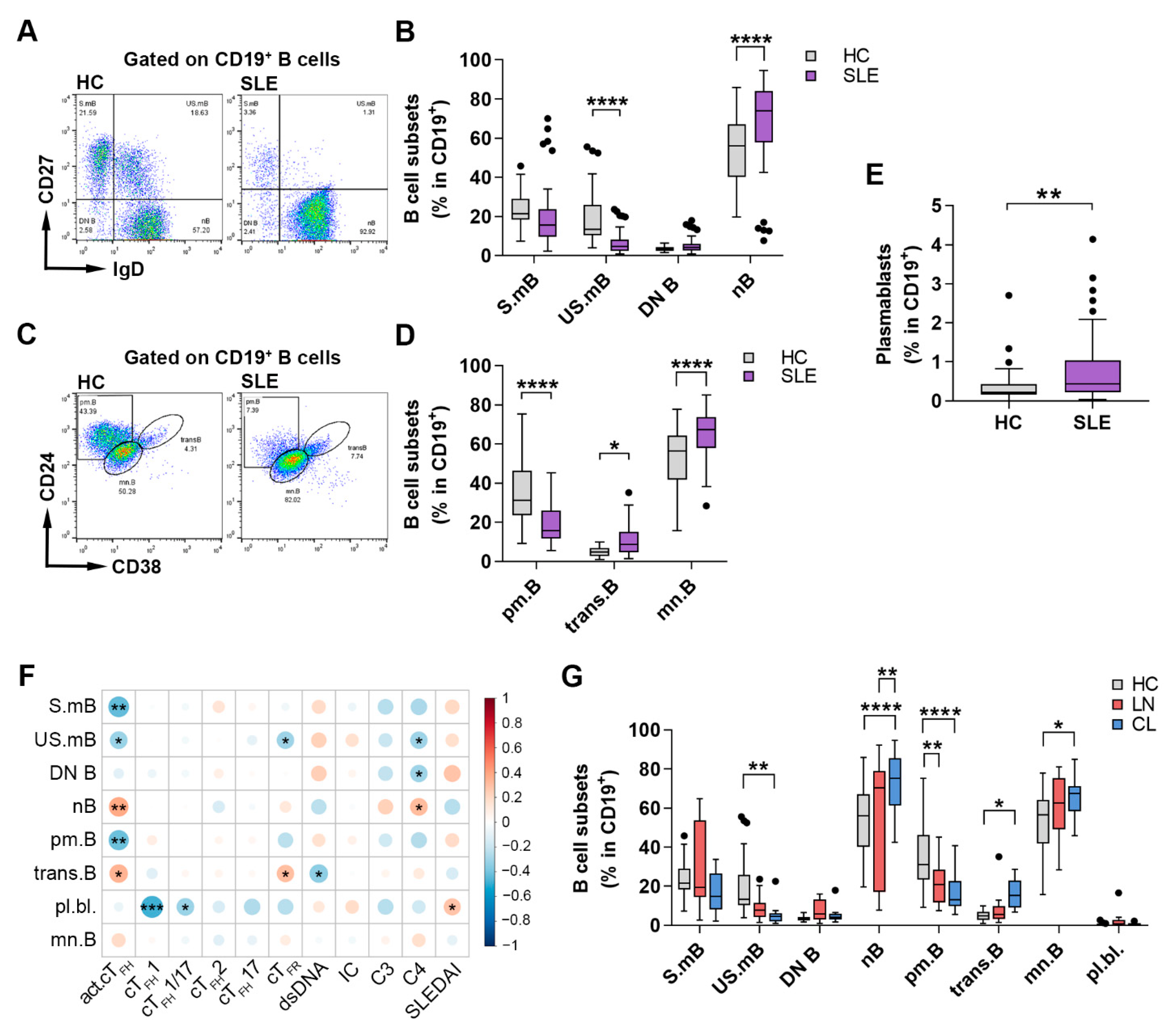
2.3. Changes in the Distribution of Circulating B Cell Subsets Is a Distinctive Characteristic of SLE
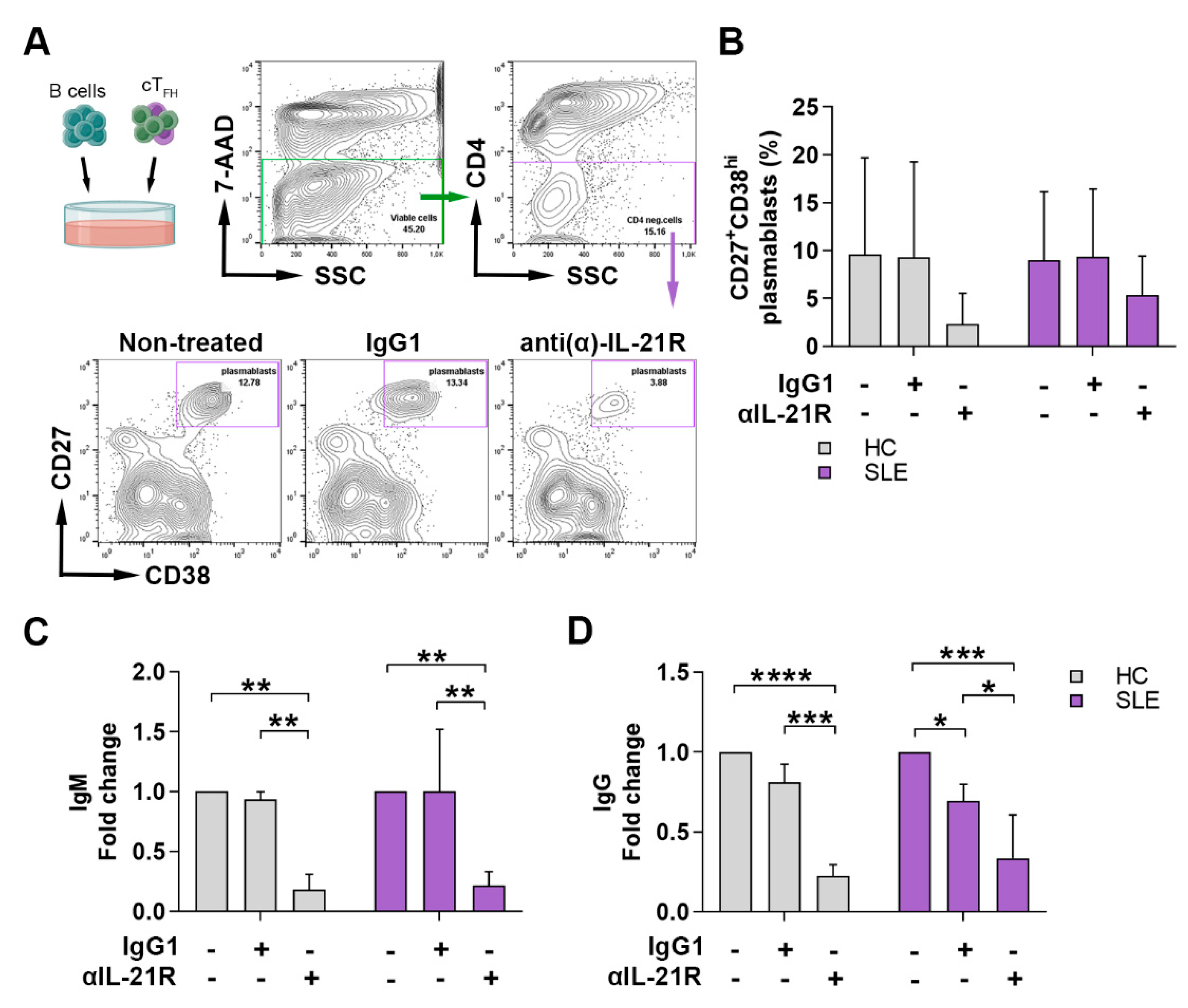
2.4. Anti-IL-21R Inhibits the TFH Cell-Dependent B Cell Differentiation in SLE Patients and Healthy Individuals
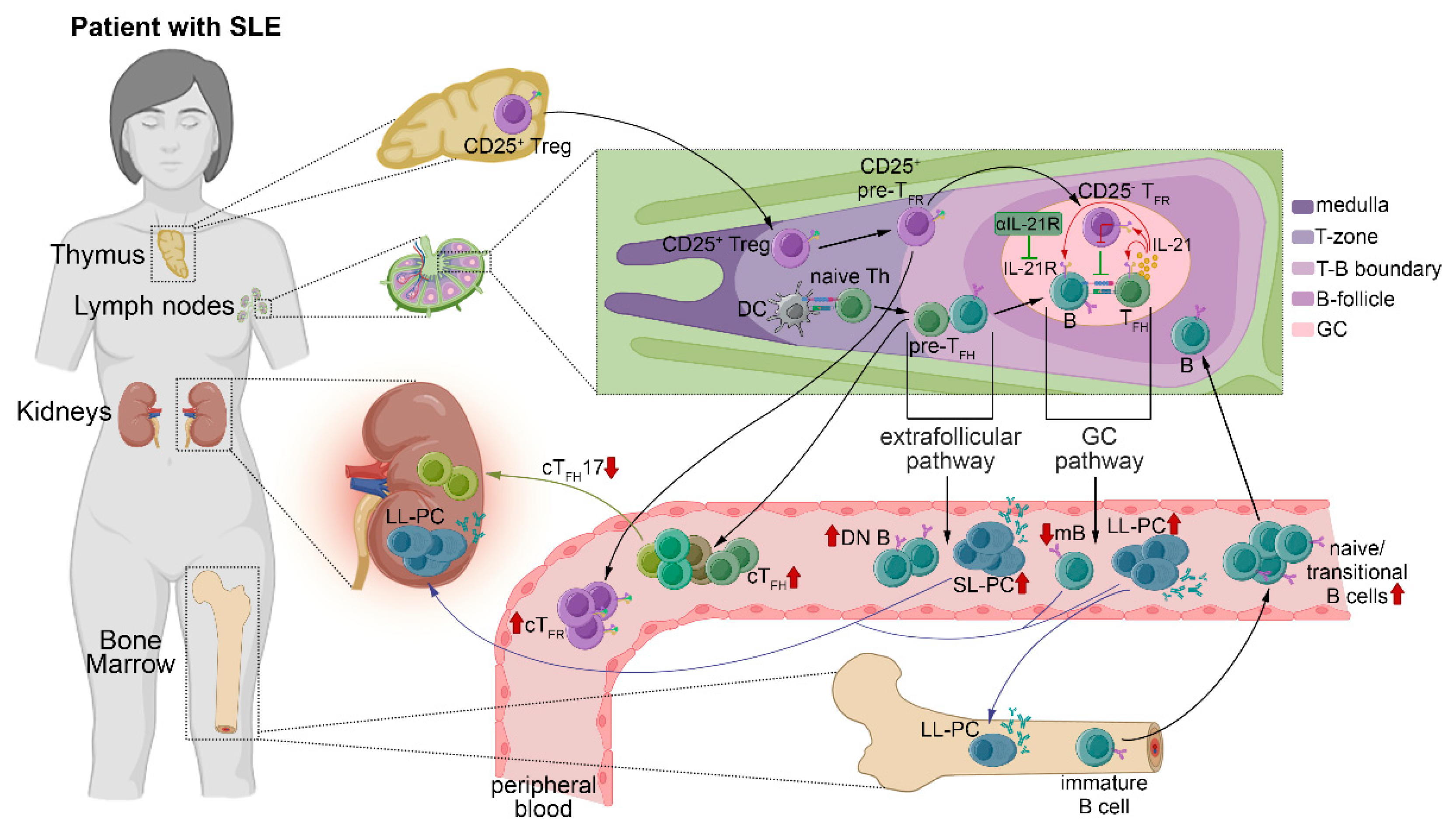
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Flow Cytometry Analysis
4.3. Cell Isolation
4.4. In Vitro cTFH-like Cell and B Cell Co-Culture Assay
4.5. ELISA to Assess B Cell Function
4.6. Assessment of Routine Laboratory Parameters
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thanou, A.; Jupe, E.; Purushothaman, M.; Niewold, T.B.; Munroe, M.E. Clinical disease activity and flare in SLE: Current concepts and novel biomarkers. J. Autoimmun. 2021, 119, 102615. [Google Scholar] [CrossRef] [PubMed]
- Tarr, T.; Papp, G.; Nagy, N.; Cserep, E.; Zeher, M. Chronic high-dose glucocorticoid therapy triggers the development of chronic organ damage and worsens disease outcome in systemic lupus erythematosus. Clin. Rheumatol. 2017, 36, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, H.; Liao, W.; Zhao, M.; Chan, V.; Li, L.; Zheng, M.; Chen, G.; Zhang, J.; Lau, C.S.; et al. A comprehensive review of immune-mediated dermatopathology in systemic lupus erythematosus. J. Autoimmun. 2018, 93, 1–15. [Google Scholar] [CrossRef]
- Anders, H.J.; Saxena, R.; Zhao, M.H.; Parodis, I.; Salmon, J.E.; Mohan, C. Lupus nephritis. Nat. Rev. Dis. Primers 2020, 6, 7. [Google Scholar] [CrossRef]
- Cashman, K.S.; Jenks, S.A.; Woodruff, M.C.; Tomar, D.; Tipton, C.M.; Scharer, C.D.; Eun-Hyung Lee, F.; Boss, J.M.; Sanz, I. Understanding and measuring human B-cell tolerance and its breakdown in autoimmune disease. Immunol. Rev. 2019, 292, 76–89. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Hsu, H.C.; Mountz, J.D. Autoreactive B cells in SLE, villains or innocent bystanders? Immunol. Rev. 2019, 292, 120–138. [Google Scholar] [CrossRef]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [Google Scholar] [CrossRef]
- Ding, T.; Su, R.; Wu, R.; Xue, H.; Wang, Y.; Su, R.; Gao, C.; Li, X.; Wang, C. Frontiers of Autoantibodies in Autoimmune Disorders: Crosstalk Between Tfh/Tfr and Regulatory B Cells. Front. Immunol. 2021, 12, 641013. [Google Scholar] [CrossRef]
- Chang, A.; Henderson, S.G.; Brandt, D.; Liu, N.; Guttikonda, R.; Hsieh, C.; Kaverina, N.; Utset, T.O.; Meehan, S.M.; Quigg, R.J.; et al. In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J. Immunol. 2011, 186, 1849–1860. [Google Scholar] [CrossRef]
- Dorraji, S.E.; Kanapathippillai, P.; Hovd, A.K.; Stenersrod, M.R.; Horvei, K.D.; Ursvik, A.; Figenschau, S.L.; Thiyagarajan, D.; Fenton, C.G.; Pedersen, H.L.; et al. Kidney Tertiary Lymphoid Structures in Lupus Nephritis Develop into Large Interconnected Networks and Resemble Lymph Nodes in Gene Signature. Am. J. Pathol. 2020, 190, 2203–2225. [Google Scholar] [CrossRef]
- Kang, S.; Fedoriw, Y.; Brenneman, E.K.; Truong, Y.K.; Kikly, K.; Vilen, B.J. BAFF Induces Tertiary Lymphoid Structures and Positions T Cells within the Glomeruli during Lupus Nephritis. J. Immunol. 2017, 198, 2602–2611. [Google Scholar] [CrossRef]
- Corsiero, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Ectopic Lymphoid Structures: Powerhouse of Autoimmunity. Front. Immunol. 2016, 7, 430. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- Kim, C.H.; Rott, L.S.; Clark-Lewis, I.; Campbell, D.J.; Wu, L.; Butcher, E.C. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J. Exp. Med. 2001, 193, 1373–1381. [Google Scholar] [CrossRef]
- Hardtke, S.; Ohl, L.; Forster, R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood 2005, 106, 1924–1931. [Google Scholar] [CrossRef]
- Nurieva, R.I.; Chung, Y.; Martinez, G.J.; Yang, X.O.; Tanaka, S.; Matskevitch, T.D.; Wang, Y.H.; Dong, C. Bcl6 mediates the development of T follicular helper cells. Science 2009, 325, 1001–1005. [Google Scholar] [CrossRef]
- Qi, H. T follicular helper cells in space-time. Nat. Rev. Immunol. 2016, 16, 612–625. [Google Scholar] [CrossRef]
- Vogelzang, A.; McGuire, H.M.; Yu, D.; Sprent, J.; Mackay, C.R.; King, C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity 2008, 29, 127–137. [Google Scholar] [CrossRef]
- Bryant, V.L.; Ma, C.S.; Avery, D.T.; Li, Y.; Good, K.L.; Corcoran, L.M.; de Waal Malefyt, R.; Tangye, S.G. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: Predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 2007, 179, 8180–8190. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef]
- Fonseca, V.R.; Ribeiro, F.; Graca, L. T follicular regulatory (Tfr) cells: Dissecting the complexity of Tfr-cell compartments. Immunol. Rev. 2019, 288, 112–127. [Google Scholar] [CrossRef]
- Deng, J.; Wei, Y.; Fonseca, V.R.; Graca, L.; Yu, D. T follicular helper cells and T follicular regulatory cells in rheumatic diseases. Nat. Rev. Rheumatol. 2019, 15, 475–490. [Google Scholar] [CrossRef]
- Vella, L.A.; Buggert, M.; Manne, S.; Herati, R.S.; Sayin, I.; Kuri-Cervantes, L.; Bukh Brody, I.; O’Boyle, K.C.; Kaprielian, H.; Giles, J.R.; et al. T follicular helper cells in human efferent lymph retain lymphoid characteristics. J. Clin. Investig. 2019, 129, 3185–3200. [Google Scholar] [CrossRef]
- Morita, R.; Schmitt, N.; Bentebibel, S.E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121. [Google Scholar] [CrossRef]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef]
- Ueno, H. T follicular helper cells in human autoimmunity. Curr. Opin. Immunol. 2016, 43, 24–31. [Google Scholar] [CrossRef]
- Bentebibel, S.E.; Lopez, S.; Obermoser, G.; Schmitt, N.; Mueller, C.; Harrod, C.; Flano, E.; Mejias, A.; Albrecht, R.A.; Blankenship, D.; et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci. Transl. Med. 2013, 5, 176ra32. [Google Scholar] [CrossRef]
- Fonseca, V.R.; Agua-Doce, A.; Maceiras, A.R.; Pierson, W.; Ribeiro, F.; Romao, V.C.; Pires, A.R.; da Silva, S.L.; Fonseca, J.E.; Sousa, A.E.; et al. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci. Immunol. 2017, 2, 1487. [Google Scholar] [CrossRef]
- Wing, J.B.; Kitagawa, Y.; Locci, M.; Hume, H.; Tay, C.; Morita, T.; Kidani, Y.; Matsuda, K.; Inoue, T.; Kurosaki, T.; et al. A distinct subpopulation of CD25(−) T-follicular regulatory cells localizes in the germinal centers. Proc. Natl. Acad. Sci. USA 2017, 114, E6400–E6409. [Google Scholar] [CrossRef]
- Simpson, N.; Gatenby, P.A.; Wilson, A.; Malik, S.; Fulcher, D.A.; Tangye, S.G.; Manku, H.; Vyse, T.J.; Roncador, G.; Huttley, G.A.; et al. Expansion of circulating T cells resembling follicular helper T cells is a fixed phenotype that identifies a subset of severe systemic lupus erythematosus. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2010, 62, 234–244. [Google Scholar] [CrossRef]
- Choi, J.Y.; Ho, J.H.; Pasoto, S.G.; Bunin, V.; Kim, S.T.; Carrasco, S.; Borba, E.F.; Goncalves, C.R.; Costa, P.R.; Kallas, E.G.; et al. Circulating follicular helper-like T cells in systemic lupus erythematosus: Association with disease activity. Arthritis Rheumatol. 2015, 67, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Le Coz, C.; Joublin, A.; Pasquali, J.L.; Korganow, A.S.; Dumortier, H.; Monneaux, F. Circulating TFH subset distribution is strongly affected in lupus patients with an active disease. PLoS ONE 2013, 8, e75319. [Google Scholar] [CrossRef]
- Xu, B.; Wang, S.; Zhou, M.; Huang, Y.; Fu, R.; Guo, C.; Chen, J.; Zhao, J.; Gaskin, F.; Fu, S.M.; et al. The ratio of circulating follicular T helper cell to follicular T regulatory cell is correlated with disease activity in systemic lupus erythematosus. Clin. Immunol. 2017, 183, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, D.; Song, Y.; Lu, S.; Zhao, J.; Wang, H. Increased circulating CD4(+)CXCR5(+)FoxP3(+) follicular regulatory T cells correlated with severity of systemic lupus erythematosus patients. Int. Immunopharmacol. 2018, 56, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.P.; Fuhrmann, F.; Hutloff, A. T-follicular helper cells survive as long-term memory cells. Eur. J. Immunol. 2012, 42, 1981–1988. [Google Scholar] [CrossRef]
- Sage, P.T.; Alvarez, D.; Godec, J.; von Andrian, U.H.; Sharpe, A.H. Circulating T follicular regulatory and helper cells have memory-like properties. J. Clin. Investig. 2014, 124, 5191–5204. [Google Scholar] [CrossRef]
- Zhang, X.; Lindwall, E.; Gauthier, C.; Lyman, J.; Spencer, N.; Alarakhia, A.; Fraser, A.; Ing, S.; Chen, M.; Webb-Detiege, T.; et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus 2015, 24, 909–917. [Google Scholar] [CrossRef]
- Yang, X.; Yang, J.; Chu, Y.; Xue, Y.; Xuan, D.; Zheng, S.; Zou, H. T follicular helper cells and regulatory B cells dynamics in systemic lupus erythematosus. PLoS ONE 2014, 9, e88441. [Google Scholar] [CrossRef]
- Feng, X.; Wang, D.; Chen, J.; Lu, L.; Hua, B.; Li, X.; Tsao, B.P.; Sun, L. Inhibition of aberrant circulating Tfh cell proportions by corticosteroids in patients with systemic lupus erythematosus. PLoS ONE 2012, 7, e51982. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.; Cui, X.; Zuo, Y.; Zhang, Z.; Li, Y.; Tao, R.; Li, Y.; Pang, J. Increased frequency of circulating follicular helper T cells in lupus patients is associated with autoantibody production in a CD40L-dependent manner. Cell Immunol. 2015, 295, 46–51. [Google Scholar] [CrossRef]
- Zhou, H.; Hu, B.; Huang, N.; Mo, X.; Li, W.; Zhang, B.; Wei, B.; Gao, M.; Wang, Y.; Liu, X.; et al. Aberrant T cell subsets and cytokines expression profile in systemic lupus erythematosus. Clin. Rheumatol. 2018, 37, 2405–2413. [Google Scholar] [CrossRef]
- Szabo, K.; Papp, G.; Szanto, A.; Tarr, T.; Zeher, M. A comprehensive investigation on the distribution of circulating follicular T helper cells and B cell subsets in primary Sjogren’s syndrome and systemic lupus erythematosus. Clin. Exp. Immunol. 2016, 183, 76–89. [Google Scholar] [CrossRef]
- Sasaki, T.; Bracero, S.; Keegan, J.; Chen, L.; Cao, Y.; Stevens, E.; Qu, Y.; Wang, G.; Nguyen, J.; Sparks, J.A.; et al. Longitudinal immune cell profiling in early systemic lupus erythematosus. Arthritis Rheumatol. 2022, 42248. [Google Scholar] [CrossRef]
- Turner, J.E.; Paust, H.J.; Steinmetz, O.M.; Peters, A.; Riedel, J.H.; Erhardt, A.; Wegscheid, C.; Velden, J.; Fehr, S.; Mittrucker, H.W.; et al. CCR6 recruits regulatory T cells and Th17 cells to the kidney in glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 974–985. [Google Scholar] [CrossRef]
- Hao, H.; Nakayamada, S.; Yamagata, K.; Ohkubo, N.; Iwata, S.; Inoue, Y.; Zhang, M.; Zhang, T.; Kanda Satoh, Y.; Shan, Y.; et al. Conversion of T Follicular Helper Cells to T Follicular Regulatory Cells by Interleukin-2 Through Transcriptional Regulation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2021, 73, 132–142. [Google Scholar] [CrossRef]
- Kurata, I.; Mikami, N.; Ohyama, A.; Osada, A.; Kondo, Y.; Tsuboi, H.; Sumida, T.; Matsumoto, I. Impaired function of PD-1(+) follicular regulatory T cells in systemic lupus erythematosus. Clin. Exp. Immunol. 2021, 206, 28–35. [Google Scholar] [CrossRef]
- Liu, W.; Putnam, A.L.; Xu-Yu, Z.; Szot, G.L.; Lee, M.R.; Zhu, S.; Gottlieb, P.A.; Kapranov, P.; Gingeras, T.R.; Fazekas de St Groth, B.; et al. CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J. Exp. Med. 2006, 203, 1701–1711. [Google Scholar] [CrossRef]
- Liang, K.; He, J.; Wei, Y.; Zeng, Q.; Gong, D.; Qin, J.; Ding, H.; Chen, Z.; Zhou, P.; Niu, P.; et al. Sustained low-dose interleukin-2 therapy alleviates pathogenic humoral immunity via elevating the Tfr/Tfh ratio in lupus. Clin. Transl. Immunol. 2021, 10, e1293. [Google Scholar] [CrossRef]
- Miao, M.; Xiao, X.; Tian, J.; Zhufeng, Y.; Feng, R.; Zhang, R.; Chen, J.; Zhang, X.; Huang, B.; Jin, Y.; et al. Therapeutic potential of targeting Tfr/Tfh cell balance by low-dose-IL-2 in active SLE: A post hoc analysis from a double-blind RCT study. Arthritis Res. Ther. 2021, 23, 167. [Google Scholar] [CrossRef]
- Mao, M.; Xu, S.; Lin, L.; Dong, D.; Xue, M.; He, S.; Cai, G. Impact of Corticosteroids on the Proportions of Circulating Tfh Cell Subsets in Patients With Systemic Lupus Erythematous. Front. Med. 2022, 9, 949334. [Google Scholar] [CrossRef]
- Yang, M.; Cao, P.; Zhao, Z.; Wang, Z.; Jia, C.; Guo, Y.; Yin, H.; Zhao, M.; Ding, Y.; Wu, H.; et al. An Enhanced Expression Level of CXCR3 on Tfh-like Cells from Lupus Skin Lesions Rather Than Lupus Peripheral Blood. Clin. Immunol. 2021, 226, 108717. [Google Scholar] [CrossRef]
- Paquissi, F.C.; Abensur, H. The Th17/IL-17 Axis and Kidney Diseases, With Focus on Lupus Nephritis. Front. Med. 2021, 8, 654912. [Google Scholar] [CrossRef]
- Riedel, J.H.; Turner, J.E.; Panzer, U. T helper cell trafficking in autoimmune kidney diseases. Cell Tissue Res. 2021, 385, 281–292. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Ding, R.; Lv, B.; Bi, L. CXCL13 blockade attenuates lupus nephritis of MRL/lpr mice. Acta Histochem. 2015, 117, 732–737. [Google Scholar] [CrossRef]
- Abraham, R.; Durkee, M.S.; Ai, J.; Veselits, M.; Casella, G.; Asano, Y.; Chang, A.; Ko, K.; Oshinsky, C.; Peninger, E.; et al. Specific in situ inflammatory states associate with progression to renal failure in lupus nephritis. J. Clin. Investig. 2022, 132, 5350. [Google Scholar] [CrossRef]
- Liarski, V.M.; Kaverina, N.; Chang, A.; Brandt, D.; Yanez, D.; Talasnik, L.; Carlesso, G.; Herbst, R.; Utset, T.O.; Labno, C.; et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci. Transl. Med. 2014, 6, 230ra46. [Google Scholar] [CrossRef]
- Dunlap, G.S.; Billi, A.C.; Xing, X.; Ma, F.; Maz, M.P.; Tsoi, L.C.; Wasikowski, R.; Hodgin, J.B.; Gudjonsson, J.E.; Kahlenberg, J.M.; et al. Single-cell transcriptomics reveals distinct effector profiles of infiltrating T cells in lupus skin and kidney. JCI Insight 2022, 7, e156341. [Google Scholar] [CrossRef]
- Koga, T.; Otomo, K.; Mizui, M.; Yoshida, N.; Umeda, M.; Ichinose, K.; Kawakami, A.; Tsokos, G.C. Calcium/Calmodulin-Dependent Kinase IV Facilitates the Recruitment of Interleukin-17-Producing Cells to Target Organs Through the CCR6/CCL20 Axis in Th17 Cell-Driven Inflammatory Diseases. Arthritis Rheumatol. 2016, 68, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bayona, B.; Ramos-Amaya, A.; Perez-Venegas, J.J.; Rodriguez, C.; Brieva, J.A. Decreased frequency and activated phenotype of blood CD27 IgD IgM B lymphocytes is a permanent abnormality in systemic lupus erythematosus patients. Arthritis Res. Ther. 2010, 12, R108. [Google Scholar] [CrossRef] [PubMed]
- Dorner, T.; Lipsky, P.E. Correlation of circulating CD27high plasma cells and disease activity in systemic lupus erythematosus. Lupus 2004, 13, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef]
- Horisberger, A.; Humbel, M.; Fluder, N.; Bellanger, F.; Fenwick, C.; Ribi, C.; Comte, D. Measurement of circulating CD21(−)CD27(−) B lymphocytes in SLE patients is associated with disease activity independently of conventional serological biomarkers. Sci. Rep. 2022, 12, 9189. [Google Scholar] [CrossRef]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.H.; Milner, E.C.; Sanz, I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J. Immunol. 2007, 178, 6624–6633. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11c(hi)T-bet(+) B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef]
- Ding, Y.; Li, J.; Yang, P.; Luo, B.; Wu, Q.; Zajac, A.J.; Wildner, O.; Hsu, H.C.; Mountz, J.D. Interleukin-21 promotes germinal center reaction by skewing the follicular regulatory T cell to follicular helper T cell balance in autoimmune BXD2 mice. Arthritis Rheumatol. 2014, 66, 2601–2612. [Google Scholar] [CrossRef]
- Jandl, C.; Liu, S.M.; Canete, P.F.; Warren, J.; Hughes, W.E.; Vogelzang, A.; Webster, K.; Craig, M.E.; Uzel, G.; Dent, A.; et al. IL-21 restricts T follicular regulatory T cell proliferation through Bcl-6 mediated inhibition of responsiveness to IL-2. Nat. Commun. 2017, 8, 14647. [Google Scholar] [CrossRef]
- Kim, V.; Lee, K.; Tian, H.; Jang, S.H.; Diamond, B.; Kim, S.J. IL-17-producing follicular Th cells enhance plasma cell differentiation in lupus-prone mice. JCI Insight 2022, 7, 157332. [Google Scholar] [CrossRef]
- Herber, D.; Brown, T.P.; Liang, S.; Young, D.A.; Collins, M.; Dunussi-Joannopoulos, K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J. Immunol. 2007, 178, 3822–3830. [Google Scholar] [CrossRef]
- Wu, H.; Deng, Y.; Long, D.; Yang, M.; Li, Q.; Feng, Y.; Chen, Y.; Qiu, H.; Huang, X.; He, Z.; et al. The IL-21-TET2-AIM2-c-MAF pathway drives the T follicular helper cell response in lupus-like disease. Clin. Transl. Med. 2022, 12, e781. [Google Scholar] [CrossRef]
- Choi, J.Y.; Seth, A.; Kashgarian, M.; Terrillon, S.; Fung, E.; Huang, L.; Wang, L.C.; Craft, J. Disruption of Pathogenic Cellular Networks by IL-21 Blockade Leads to Disease Amelioration in Murine Lupus. J. Immunol. 2017, 198, 2578–2588. [Google Scholar] [CrossRef]
- Terrier, B.; Costedoat-Chalumeau, N.; Garrido, M.; Geri, G.; Rosenzwajg, M.; Musset, L.; Klatzmann, D.; Saadoun, D.; Cacoub, P. Interleukin 21 correlates with T cell and B cell subset alterations in systemic lupus erythematosus. J. Rheumatol. 2012, 39, 1819–1828. [Google Scholar] [CrossRef]
- Ignatenko, S.; Skrumsager, B.K.; Mouritzen, U. Safety, PK, and PD of recombinant anti-interleukin-21 monoclonal antibody in a first-in-human trial. Int. J. Clin. Pharmacol. Ther. 2016, 54, 243–252. [Google Scholar] [CrossRef]
- Petri, M.; Orbai, A.M.; Alarcon, G.S.; Gordon, C.; Merrill, J.T.; Fortin, P.R.; Bruce, I.N.; Isenberg, D.; Wallace, D.J.; Nived, O.; et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2677–2686. [Google Scholar] [CrossRef]
- Linderman, G.C.; Rachh, M.; Hoskins, J.G.; Steinerberger, S.; Kluger, Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 2019, 16, 243–245. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. Available online: http://www.rstudio.com/ (accessed on 8 June 2022).
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 8 June 2022).
- Harrell, F.E.; Dupont, C. R Package, Version 4.7-0; R Package ‘Hmisc’: Hmisc: Harrell Miscellaneous; 2022. Available online: https://cran.r-project.org/web/packages/Hmisc/ (accessed on 8 June 2022).
- Wei, T.; Simko, V. R Package, Version 0.92; R Package ‘corrplot’: Visualization of a Correlation Matrix; 2021. Available online: https://cran.r-project.org/web/packages/corrplot/index.html (accessed on 8 June 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | SLE (n = 48) | HC (n = 36) | p Value |
|---|---|---|---|
| Demographics | |||
| Age, median years | 40 (32–45) | 43 (34–47) | 0.3121 |
| Gender, M/F | 1/47 | 1/36 | - |
| SLEDAI, mean ± SD | 3.83 ± 3.14 | NA | - |
| SLEDAI, median | 2 (2–6) | NA | - |
| Laboratory variables | |||
| Lymphocytes, 109/L | 1.17 (0.75–1.71) | 1.71 (1.39–2.20) | 0.0004 |
| Anti-dsDNA titer, IU/mL | 28.7 (21.3–68.1) | NA | - |
| Anti-dsDNA, n (%) | 36 (75.00) | NA | - |
| IC, titer, extinction | 72 (61–109) | NA | - |
| C3, titer, g/L | 0.96 (0.75–1.12) | NA | - |
| C3, n (%) | 19 (39.58) | NA | - |
| C4, titer, g/L | 0.13 (0.08–0.16) | NA | - |
| C4, n (%) | 15 (31.25) | NA | - |
| Organ involvement | |||
| Renal, n (%) | 15 (31.25) | NA | - |
| Cutaneous, n (%) | 13 (27.08) | NA | - |
| Medications at time of study | |||
| No therapy, n (%) | 6 (12.50) | NA | - |
| Methylprednisolone, n (%) | 27 (56.25) | NA | - |
| Methylprednisolone, median, mg/day | 4 (4–4) | NA | - |
| Chloroquine, n (%) | 27 (56.25) | NA | - |
| Hydroxychloroquine, n (%) | 3 (6.25) | NA | - |
| Azathioprine, n (%) | 8 (16.67) | NA | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, K.; Jámbor, I.; Pázmándi, K.; Nagy, N.; Papp, G.; Tarr, T. Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R. Int. J. Mol. Sci. 2022, 23, 12209. https://doi.org/10.3390/ijms232012209
Szabó K, Jámbor I, Pázmándi K, Nagy N, Papp G, Tarr T. Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R. International Journal of Molecular Sciences. 2022; 23(20):12209. https://doi.org/10.3390/ijms232012209
Chicago/Turabian StyleSzabó, Krisztina, Ilona Jámbor, Kitti Pázmándi, Nikolett Nagy, Gábor Papp, and Tünde Tarr. 2022. "Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R" International Journal of Molecular Sciences 23, no. 20: 12209. https://doi.org/10.3390/ijms232012209
APA StyleSzabó, K., Jámbor, I., Pázmándi, K., Nagy, N., Papp, G., & Tarr, T. (2022). Altered Circulating Follicular T Helper Cell Subsets and Follicular T Regulatory Cells Are Indicators of a Derailed B Cell Response in Lupus, Which Could Be Modified by Targeting IL-21R. International Journal of Molecular Sciences, 23(20), 12209. https://doi.org/10.3390/ijms232012209