
Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic


 ,
, 
Abstract
1. Introduction
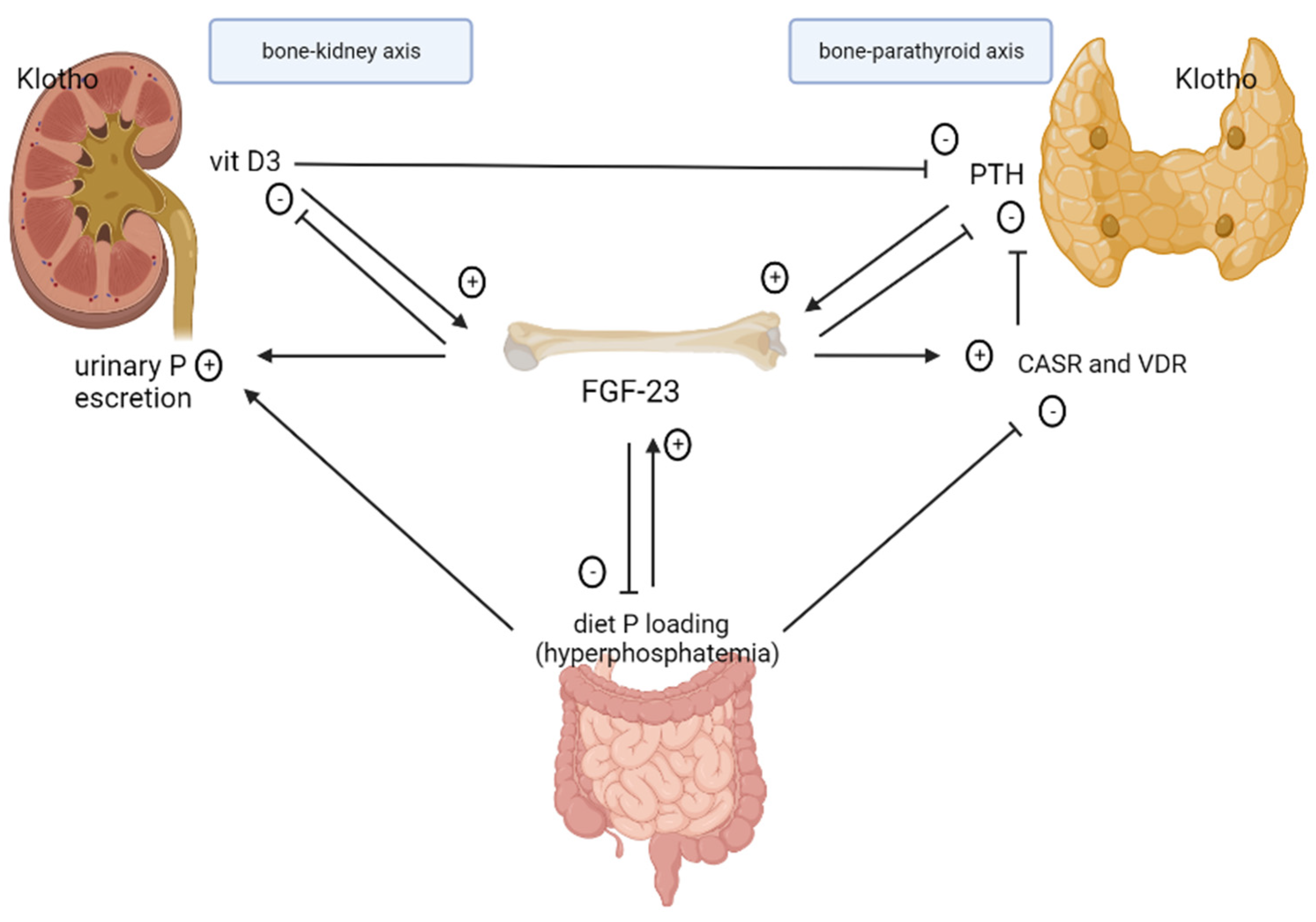
2. Pathophysiology of CKD–MBD
2.1. Role of FGF-23 in CKD
2.2. Role of Klotho in CKD
2.3. Role and Regulation of Phosphate
2.4. Role of Calcitriol
2.5. Secondary Hyperparathyroidism
2.6. Renal Osteodystrophy
3. Association between CKD–MBD and Prognosis
4. Risk Reduction Induced by Specific Treatments Acting on CKD–MBD
4.1. Management of Hyperphosphatemia
4.1.1. Dietary Restriction of Phosphate
4.1.2. Intestinal Phosphate Binders
4.1.3. Phosphate Removal through Dialysis for Patients with CKD Stage G5D
4.2. Treatment of Secondary Hyperparathyroidism
4.2.1. Vitamin D and Analogues
4.2.2. Calcimimetic Agents
4.2.3. Parathyroidectomy
4.3. Treatment of CKD–MBD/Osteoporosis

{kind=link}
{kind=link}
| Study (Year) | Type | Drugs | Sample Size and Population | Outcome | Results |
|---|---|---|---|---|---|
| CALMAG De Francisco (2010) [82] | Phase 4 | Calcium acetate/magnesium carbonate vs. sevelamer hydrochloride | 326 patients HD | Efficacy of CaMg compared with sevelamer-HCl as an active control of serum phosphorus at week 25. | CaMg was noninferior to the comparator at controlling serum phosphorus levels at week 25. |
| D’Haese (2003) [92] | Phase 3 | Lanthanum carbonate vs. calcium carbonate | 98 Patients HD | Tolerability, phosphate binder efficacy, incidence of hypercalcemia, and evolution to low bone turnover | LC-treated patients show almost no evolution toward low bone turnover over one year. |
| LANDMARK Ogata (2021) [95] | Phase 3 | Lanthanum carbonate vs. Calcium carbonate | 2374 patients HD | Reduction in cardiovascular events. Overall survival, secondary hyperparathyroidism-free survival, hip fracture-free survival, and adverse events. | Treatment of hyperphosphatemia with LC compared with CC did not result in a significant difference in composite CV events. |
| PRIMO Thadhani (2012) [104] | Phase 3 | Paricalcitol vs. placebo | 227 CKD stages 3–4 patients with LV hypertrophy, preserved left ventricular ejection fraction | Change in LV mass index over 48 weeks by cardiac magnetic resonance imaging. Echocardiographic changes in left ventricular diastolic function. | Paricalcitol did not alter left ventricular mass index or improve diastolic dysfunction. |
| OPERA Wang (2014) [105] | Not applicable | Paricalcitol vs. placebo | 60 CKD stages 3–5 patients with LV hypertrophy | Change in LV mass index over 52 weeks by cardiac magnetic resonance imaging. Changes in LV volume, echocardiographic measures of systolic and diastolic function, biochemical parameters of MBD, and measures of renal function. | 52 weeks of treatment with oral paricalcitol significantly improved secondary hyperparathyroidism but did not alter measures of LV structure and function. |
| IMPACT-SHPT Ketteler (2012) [107] | Phase 4 | Paricalcitol vs. cinacalcet | 272 patients HD | PTH 150–300 pg/mL | Overall superiority of paricalcitol (56.0%) over cinacalcet (38.2%; p = 0.010) in achieving PTH 150–300 pg/mL during Weeks 21–28. |
| PARADIGM Wetmore (2015) [110] | Phase 4 | Cinacalcet vs. vitamin D analogs | 312 patients HD | Mean percentage change in plasma PTH levels. Proportion of participants achieving plasma PTH <300 pg/mL or a ≥30% decrease in PTH. | Modest reductions in PTH with either cinacalcet or vitamin D analog monotherapy over 52 weeks of treatment. |
| EVOLVE (2012) [117] | Phase 3 | Cinacalcet vs. placebo | 3883 patients HD | All-cause mortality, major cardiovascular events, development of severe unremitting HPT. | Cinacalcet did not significantly reduce the risk of death or major cardiovascular events |
| Block (2017) [122] | Phase 3 | Etelcalcetide vs. cinacalcet | 683 patients HD | Noninferiority of etelcalcetide at achieving more than a 30% reduction from baseline in PHT compared to cinacalcet. Superiority in achieving >50% and >30% reduction in PTH. Self-reported nausea and vomiting. | Non-inferiority of etelcalcetide in reduction in PTH concentrations compared to cinacalcet. |
| FREEDOM Cummings (2009) [132] | Phase 3 | Denosumab vs. placebo | 7868 postmenopausal women with osteoporosis | New vertebral fractures. Nonvertebral and hip fractures. | Denosumab given subcutaneously twice yearly for 36 months was associated with a reduction in the risk of vertebral, nonvertebral, and hip fractures in women with osteoporosis. |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jager, K.J.; Kovesdy, C.; Langham, R.; Rosenberg, M.; Jha, V.; Zoccali, C. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Nephrol. Dial. Transplant. 2019, 34, 1803–1805. [Google Scholar] [CrossRef]
- Kidney Disease Improving Global Outcomes Work Group. Chapter 4: Other complications of CKD: CVD, medication dosage, patient safety, infections, hospitalizations, and caveats for investigating complications of CKD. Kidney Int. Suppl. 2013, 3, 91–111. [Google Scholar] [CrossRef]
- Lau, W.L.; Obi, Y.; Kalantar-Zadeh, K. Parathyroidectomy in the Management of Secondary Hyperparathyroidism. Clin. J. Am. Soc. Nephrol. 2018, 13, 952–961. [Google Scholar] [CrossRef]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G.; et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef]
- Fang, Y.; Ginsberg, C.; Sugatani, T.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. Early chronic kidney disease-mineral bone disorder stimulates vascular calcification. Kidney Int. 2014, 85, 142–150. [Google Scholar] [CrossRef]
- Albright, F.; Bauer, W.; Cockrill, J.R.; Ellsworth, R. Studies on The Physiology of The Parathyroid Glands: II. The Relation of the Serum Calcium to the Serum Phosphorus at Different Levels of Parathyroid Activity. J. Clin. Investig. 1931, 9, 659–677. [Google Scholar] [CrossRef]
- Bricker, N.S.; Morrin, P.A.; Kime, S.W., Jr. The pathologic physiology of chronic Bright’s disease. An exposition of the “intact nephron hypothesis”. Am. J. Med. 1960, 28, 77–98. [Google Scholar] [CrossRef]
- Bricker, N.S. On the pathogenesis of the uremic state. An exposition of the “trade-off hypothesis”. N. Engl. J. Med. 1972, 286, 1093–1099. [Google Scholar] [CrossRef]
- Slatopolsky, E. The intact nephron hypothesis: The concept and its implications for phosphate management in CKD-related mineral and bone disorder. Kidney Int. 2011, 79, S3–S8. [Google Scholar] [CrossRef]
- Moranne, O.; Froissart, M.; Rossert, J.; Gauci, C.; Boffa, J.-J.; Haymann, J.P.; Ben M’Rad, M.; Jacquot, C.; Houillier, P.; Stengel, B.; et al. Timing of onset of CKD-related metabolic complications. J. Am. Soc. Nephrol. 2009, 20, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Coppolino, G.; Nicotera, R.; Cernaro, V.; Calimeri, S.; Leonardi, G.; Cosentino, S.; Comi, A.; Donato, C.; Lucia, C.M.; Provenzano, M.; et al. Iron Infusion and Induced Hypophosphatemia: The Role of Fibroblast Growth Factor-23. Ther. Apher. Dial. 2020, 24, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Kuro-O, M. Overview of the FGF23-Klotho axis. Pediatr. Nephrol. 2010, 25, 583–590. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Moe, O.W. Role of alphaKlotho and FGF23 in regulation of type II Na-dependent phosphate co-transporters. Pflügers Arch.-Eur. J. Physiol. 2019, 471, 99–108. [Google Scholar] [CrossRef]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef]
- Fliser, D.; Kollerits, B.; Neyer, U.; Ankerst, D.P.; Lhotta, K.; Lingenhel, A.; Ritz, E.; Kronenberg, F.; Group, M.S.; Kuen, E.; et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: The Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 2007, 18, 2600–2608. [Google Scholar] [CrossRef]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef]
- Liu, S.; Tang, W.; Zhou, J.; Stubbs, J.R.; Luo, Q.; Pi, M.; Quarles, L.D. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J. Am. Soc. Nephrol. 2006, 17, 1305–1315. [Google Scholar] [CrossRef]
- Dai, B.; David, V.; Martin, A.; Huang, J.; Li, H.; Jiao, Y.; Gu, W.; Quarles, L.D. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS ONE 2012, 7, e44161. [Google Scholar] [CrossRef]
- Vaidya, A.; Williams, J.S. The relationship between vitamin D and the renin-angiotensin system in the pathophysiology of hypertension, kidney disease, and diabetes. Metabolism 2012, 61, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Ito, M.; Kuwahata, M.; Kato, S.; Segawa, H. Inhibition of intestinal sodium-dependent inorganic phosphate transport by fibroblast growth factor 23. Ther. Apher. Dial. 2005, 9, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.; Jaiswal, A.; Agarwal, V.; Kumar, S.; Chaturvedi, S.; Yadav, S.; Gupta, A.; Sharma, R.K.; Bhadauria, D.; Kaul, A. FGF23 is associated with early post-transplant hypophosphataemia and normalizes faster than iPTH in living donor renal transplant recipients: A longitudinal follow-up study. Clin. Kidney J. 2016, 9, 669–676. [Google Scholar] [CrossRef]
- Tsujikawa, H.; Kurotaki, Y.; Fujimori, T.; Fukuda, K.; Nabeshima, Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol. Endocrinol. 2003, 17, 2393–2403. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Quinones, H.; Griffith, C.; Kuro-o, M.; Moe, O.W. Klotho deficiency causes vascular calcification in chronic kidney disease. J. Am. Soc. Nephrol. 2011, 22, 124–136. [Google Scholar] [CrossRef]
- Galitzer, H.; Ben-Dov, I.Z.; Silver, J.; Naveh-Many, T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010, 77, 211–218. [Google Scholar] [CrossRef]
- Thongprayoon, C.; Neyra, J.A.; Hansrivijit, P.; Medaura, J.; Leeaphorn, N.; Davis, P.W.; Kaewput, W.; Bathini, T.; Salim, S.A.; Chewcharat, A.; et al. Serum Klotho in Living Kidney Donors and Kidney Transplant Recipients: A Meta-Analysis. J. Clin. Med. 2020, 9, 1834. [Google Scholar] [CrossRef]
- Juppner, H. Phosphate and FGF-23. Kidney Int. Suppl. 2011, 79, S24–S27. [Google Scholar] [CrossRef]
- Jacquillet, G.; Unwin, R.J. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflügers Arch.-Eur. J. Physiol. 2019, 471, 83–98. [Google Scholar] [CrossRef]
- Bergwitz, C.; Juppner, H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef]
- Centeno, P.P.; Herberger, A.; Mun, H.C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 4693. [Google Scholar] [CrossRef]
- Moallem, E.; Kilav, R.; Silver, J.; Naveh-Many, T. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J. Biol. Chem. 1998, 273, 5253–5259. [Google Scholar] [CrossRef]
- Vervloet, M.G.; Massy, Z.A.; Brandenburg, V.M.; Mazzaferro, S.; Cozzolino, M.; Ureña-Torres, P.; Bover, J.; Goldsmith, D. Bone: A new endocrine organ at the heart of chronic kidney disease and mineral and bone disorders. Lancet Diabetes Endocrinol. 2014, 2, 427–436. [Google Scholar] [CrossRef]
- Mehta, R.; Cai, X.; Lee, J.; Xie, D.; Wang, X.; Scialla, J.; Anderson, A.H.; Taliercio, J.; Dobre, M.; Chen, J.; et al. Serial Fibroblast Growth Factor 23 Measurements and Risk of Requirement for Kidney Replacement Therapy: The CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2020, 75, 908–918. [Google Scholar] [CrossRef]
- De Nicola, L.; Provenzano, M.; Chiodini, P.; Borrelli, S.; Russo, L.; Bellasi, A.; Santoro, D.; Conte, G.; Minutolo, R. Epidemiology of low-proteinuric chronic kidney disease in renal clinics. PLoS ONE 2017, 12, e0172241. [Google Scholar] [CrossRef]
- Dusso, A.S.; Brown, A.J.; Slatopolsky, E. Vitamin D. Am. J. Physiol. Renal. Physiol. 2005, 289, 8–28. [Google Scholar] [CrossRef]
- Hyder, R.; Sprague, S.M. Secondary Hyperparathyroidism in a Patient with CKD. Clin. J. Am. Soc. Nephrol. 2020, 15, 1041–1043. [Google Scholar] [CrossRef]
- Goltzman, D.; Mannstadt, M.; Marcocci, C. Physiology of the Calcium-Parathyroid Hormone-Vitamin D Axis. Front. Horm. Res. 2018, 50, 1–13. [Google Scholar] [CrossRef]
- Cunningham, J.; Locatelli, F.; Rodriguez, M. Secondary hyperparathyroidism: Pathogenesis, disease progression, and therapeutic options. Clin. J. Am. Soc. Nephrol. 2011, 6, 913–921. [Google Scholar] [CrossRef]
- Brown, E.M. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, M.; Kazama, J.J. The making of a bone in blood vessels: From the soft shell to the hard bone. Kidney Int. 2007, 72, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef]
- Houben, E.; Neradova, A.; Schurgers, L.J.; Vervloet, M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G. Ital. Nefrol. 2016, 33, 1724–5590. [Google Scholar]
- Alem, A.M.; Sherrard, D.J.; Gillen, D.L.; Weiss, N.S.; Beresford, S.A.; Heckbert, S.R.; Wong, C.; Stehman-Breen, C. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int. 2000, 58, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Malluche, H.H.; Porter, D.S.; Monier-Faugere, M.C.; Mawad, H.; Pienkowski, D. Differences in bone quality in low- and high-turnover renal osteodystrophy. J. Am. Soc. Nephrol. 2012, 23, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Moorthi, R.N.; Moe, S.M. CKD-mineral and bone disorder: Core curriculum 2011. Am. J. Kidney Dis. 2011, 58, 1022–1036. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Bellorin-Font, E.; Jorgetti, V.; Carvalho, A.B.; Malluche, H.H.; Ferreira, A.; D’Haese, P.C.; Drueke, T.B.; Du, H.; Manley, T.; et al. Diagnostic Accuracy of Bone Turnover Markers and Bone Histology in Patients With CKD Treated by Dialysis. Am. J. Kidney Dis. 2016, 67, 559–566. [Google Scholar] [CrossRef]
- Seibel, M.J. Biochemical markers of bone turnover: Part I: Biochemistry and variability. Clin. Biochem. Rev. 2005, 26, 97–122. [Google Scholar]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Bohm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, A.K.; Chhabra, Y.K.; Mahajan, S. Cardiovascular disease in patients with chronic kidney disease: A neglected subgroup. Heart Asia 2016, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Nitta, K. Vascular calcification in patients with chronic kidney disease. Ther. Apher. Dial. 2011, 15, 513–521. [Google Scholar] [CrossRef]
- Paloian, N.J.; Giachelli, C.M. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal. Physiol. 2014, 307, F891–F900. [Google Scholar] [CrossRef]
- Lopes, M.B.; Karaboyas, A.; Bieber, B.; Pisoni, R.L.; Walpen, S.; Fukagawa, M.; Christensson, A.; Evenepoel, P.; Pegoraro, M.; Robinson, B.M.; et al. Impact of longer term phosphorus control on cardiovascular mortality in hemodialysis patients using an area under the curve approach: Results from the DOPPS. Nephrol. Dial. Transplant. 2020, 35, 1794–1801. [Google Scholar] [CrossRef]
- Hou, Y.; Li, X.; Sun, L.; Qu, Z.; Jiang, L.; Du, Y. Phosphorus and mortality risk in end-stage renal disease: A meta-analysis. Clin. Chim. Acta 2017, 474, 108–113. [Google Scholar] [CrossRef]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B., Sr.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Bellasi, A.; Mandreoli, M.; Baldrati, L.; Corradini, M.; Di Nicolo, P.; Malmusi, G.; Santoro, A. Chronic kidney disease progression and outcome according to serum phosphorus in mild-to-moderate kidney dysfunction. Clin. J. Am. Soc. Nephrol. 2011, 6, 883–891. [Google Scholar] [CrossRef]
- Sim, J.J.; Bhandari, S.K.; Smith, N.; Chung, J.; Liu, I.L.; Jacobsen, S.J.; Kalantar-Zadeh, K. Phosphorus and risk of renal failure in subjects with normal renal function. Am. J. Med. 2013, 126, 311–318. [Google Scholar] [CrossRef]
- Sekiguchi, S.; Suzuki, A.; Asano, S.; Nishiwaki-Yasuda, K.; Shibata, M.; Nagao, S.; Yamamoto, N.; Matsuyama, M.; Sato, Y.; Yan, K.; et al. Phosphate overload induces podocyte injury via type III Na-dependent phosphate transporter. Am. J. Physiol. Renal. Physiol. 2011, 300, F848–F856. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues-With and Without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef]
- Lee, T.W.; Chung, C.C.; Lee, T.I.; Lin, Y.K.; Kao, Y.H.; Chen, Y.J. Fibroblast Growth Factor 23 Stimulates Cardiac Fibroblast Activity through Phospholipase C-Mediated Calcium Signaling. Int. J. Mol. Sci. 2021, 23, 166. [Google Scholar] [CrossRef]
- Yang, H.; Luo, H.; Tang, X.; Zeng, X.; Yu, Y.; Ma, L.; Fu, P. Prognostic value of FGF23 among patients with end-stage renal disease: A systematic review and meta-analysis. Biomark Med. 2016, 10, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Grams, M.E.; Coresh, J.; Selvin, E.; Inker, L.A.; Levey, A.S.; Kimmel, P.L.; Vasan, R.S.; Eckfeldt, J.H.; Feldman, H.I.; et al. Serum fibroblast growth factor-23 is associated with incident kidney disease. J. Am. Soc. Nephrol. 2015, 26, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Chiodini, P.; De Nicola, L.; Minutolo, R.; Provenzano, M.; Garofalo, C.; Remuzzi, G.; Ronco, C.; Cozzolino, M.G.; Manno, C.; et al. Prognosis and determinants of serum PTH changes over time in 1-5 CKD stage patients followed in tertiary care. PLoS ONE 2018, 13, e0202417. [Google Scholar] [CrossRef]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Executive summary of the 2017 KDIGO Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD) Guideline Update: What’s changed and why it matters. Kidney Int. 2017, 92, 26–36. [Google Scholar] [CrossRef]
- Floege, J.; Kim, J.; Ireland, E.; Chazot, C.; Drueke, T.; de Francisco, A.; Kronenberg, F.; Marcelli, D.; Passlick-Deetjen, J.; Schernthaner, G.; et al. Serum iPTH, calcium and phosphate, and the risk of mortality in a European haemodialysis population. Nephrol. Dial. Transplant. 2011, 26, 1948–1955. [Google Scholar] [CrossRef]
- Wheeler, D.C.; Winkelmayer, W.C. KDIGO 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int. Suppl. 2017, 7, 1–59. [Google Scholar] [CrossRef]
- Massry, S.G.; Coburn, J.W.; Chertow, G.M.; Hruska, K.; Langman, C.; Malluche, H.; Martin, K.; McCann, L.M.; McCarthy, J.T.; Moe, S.; et al. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am. J. Kidney Dis. 2003, 42, S1–S201. [Google Scholar]
- Cupisti, A.; Gallieni, M.; Rizzo, M.A.; Caria, S.; Meola, M.; Bolasco, P. Phosphate control in dialysis. Int. J. Nephrol. Renov. Dis. 2013, 6, 193–205. [Google Scholar] [CrossRef]
- Kalantar-Zadeh, K.; Gutekunst, L.; Mehrotra, R.; Kovesdy, C.P.; Bross, R.; Shinaberger, C.S.; Noori, N.; Hirschberg, R.; Benner, D.; Nissenson, A.R.; et al. Understanding sources of dietary phosphorus in the treatment of patients with chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 519–530. [Google Scholar] [CrossRef]
- Cupisti, A.; Kalantar-Zadeh, K. Management of natural and added dietary phosphorus burden in kidney disease. Semin. Nephrol. 2013, 33, 180–190. [Google Scholar] [CrossRef]
- Noori, N.; Kalantar-Zadeh, K.; Kovesdy, C.P.; Bross, R.; Benner, D.; Kopple, J.D. Association of dietary phosphorus intake and phosphorus to protein ratio with mortality in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2010, 5, 683–692. [Google Scholar] [CrossRef]
- Alfrey, A.C. Aluminum toxicity in patients with chronic renal failure. Ther. Drug Monit. 1993, 15, 593–597. [Google Scholar] [CrossRef]
- Levin, N.W.; Hoenich, N.A. Consequences of hyperphosphatemia and elevated levels of the calcium-phosphorus product in dialysis patients. Curr. Opin. Nephrol. Hypertens. 2001, 10, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Raggi, P.; Bellasi, A.; Kooienga, L.; Spiegel, D.M. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007, 71, 438–441. [Google Scholar] [CrossRef]
- Malluche, H.H.; Mawad, H.; Monier-Faugere, M.C. Effects of treatment of renal osteodystrophy on bone histology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S157–S163. [Google Scholar] [CrossRef]
- Kanbay, M.; Goldsmith, D.; Uyar, M.E.; Turgut, F.; Covic, A. Magnesium in chronic kidney disease: Challenges and opportunities. Blood Purif. 2010, 29, 280–292. [Google Scholar] [CrossRef] [PubMed]
- de Francisco, A.L.; Leidig, M.; Covic, A.C.; Ketteler, M.; Benedyk-Lorens, E.; Mircescu, G.M.; Scholz, C.; Ponce, P.; Passlick-Deetjen, J. Evaluation of calcium acetate/magnesium carbonate as a phosphate binder compared with sevelamer hydrochloride in haemodialysis patients: A controlled randomized study (CALMAG study) assessing efficacy and tolerability. Nephrol. Dial. Transplant. 2010, 25, 3707–3717. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.A.; Vandermeer, B.; Raggi, P.; Mendelssohn, D.C.; Chatterley, T.; Dorgan, M.; Lok, C.E.; Fitchett, D.; Tsuyuki, R.T. Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: An updated systematic review and meta-analysis. Lancet 2013, 382, 1268–1277. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Chen, H.; Zhu, X.; Zhou, L.; Yang, Y. The effects of non-calcium-based phosphate binders versus calcium-based phosphate binders on cardiovascular calcification and bone remodeling among dialysis patients: A meta-analysis of randomized trials. Ren. Fail. 2014, 36, 1244–1252. [Google Scholar] [CrossRef]
- Delmez, J.; Block, G.; Robertson, J.; Chasan-Taber, S.; Blair, A.; Dillon, M.; Bleyer, A.J. A randomized, double-blind, crossover design study of sevelamer hydrochloride and sevelamer carbonate in patients on hemodialysis. Clin. Nephrol. 2007, 68, 386–391. [Google Scholar] [CrossRef]
- Garg, J.P.; Chasan-Taber, S.; Blair, A.; Plone, M.; Bommer, J.; Raggi, P.; Chertow, G.M. Effects of sevelamer and calcium-based phosphate binders on uric acid concentrations in patients undergoing hemodialysis: A randomized clinical trial. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2005, 52, 290–295. [Google Scholar] [CrossRef]
- Spaia, S. Phosphate binders: Sevelamer in the prevention and treatment of hyperphosphataemia in chronic renal failure. Hippokratia 2011, 15, 22–26. [Google Scholar]
- Ferreira, A.; Frazao, J.M.; Monier-Faugere, M.C.; Gil, C.; Galvao, J.; Oliveira, C.; Baldaia, J.; Rodrigues, I.; Santos, C.; Ribeiro, S.; et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J. Am. Soc. Nephrol. 2008, 19, 405–412. [Google Scholar] [CrossRef]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef]
- Swainston Harrison, T.; Scott, L.J. Lanthanum carbonate. Drugs 2004, 64, 985–996; discussion 997–988. [Google Scholar] [CrossRef]
- Damment, S.J. Pharmacology of the phosphate binder, lanthanum carbonate. Ren. Fail. 2011, 33, 217–224. [Google Scholar] [CrossRef]
- D’Haese, P.C.; Spasovski, G.B.; Sikole, A.; Hutchison, A.; Freemont, T.J.; Sulkova, S.; Swanepoel, C.; Pejanovic, S.; Djukanovic, L.; Balducci, A.; et al. A multicenter study on the effects of lanthanum carbonate (Fosrenol) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int. Suppl. 2003, 63, S73–S78. [Google Scholar] [CrossRef] [PubMed]
- Finn, W.F. Lanthanum carbonate versus standard therapy for the treatment of hyperphosphatemia: Safety and efficacy in chronic maintenance hemodialysis patients. Clin. Nephrol. 2006, 65, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wen, J.; Li, Z.; Fan, J. Efficacy and safety of lanthanum carbonate on chronic kidney disease-mineral and bone disorder in dialysis patients: A systematic review. BMC Nephrol. 2013, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Fukagawa, M.; Hirakata, H.; Kagimura, T.; Fukushima, M.; Akizawa, T.; Investigators, L.; Committees. Effect of Treating Hyperphosphatemia With Lanthanum Carbonate vs Calcium Carbonate on Cardiovascular Events in Patients With Chronic Kidney Disease Undergoing Hemodialysis: The LANDMARK Randomized Clinical Trial. JAMA 2021, 325, 1946–1954. [Google Scholar] [CrossRef] [PubMed]
- Sinsakul, M.; Sika, M.; Koury, M.; Shapiro, W.; Greene, T.; Dwyer, J.; Smith, M.; Korbet, S.; Lewis, J. The safety and tolerability of ferric citrate as a phosphate binder in dialysis patients. Nephron Clin. Pract. 2012, 121, c25–c29. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.B.; Sika, M.; Koury, M.J.; Chuang, P.; Schulman, G.; Smith, M.T.; Whittier, F.C.; Linfert, D.R.; Galphin, C.M.; Athreya, B.P.; et al. Ferric citrate controls phosphorus and delivers iron in patients on dialysis. J. Am. Soc. Nephrol. 2015, 26, 493–503. [Google Scholar] [CrossRef]
- Geisser, P.; Philipp, E. PA21: A novel phosphate binder for the treatment of hyperphosphatemia in chronic kidney disease. Clin. Nephrol. 2010, 74, 4–11. [Google Scholar] [CrossRef]
- Floege, J.; Covic, A.C.; Ketteler, M.; Rastogi, A.; Chong, E.M.; Gaillard, S.; Lisk, L.J.; Sprague, S.M.; Group, P.A.S. A phase III study of the efficacy and safety of a novel iron-based phosphate binder in dialysis patients. Kidney Int. 2014, 86, 638–647. [Google Scholar] [CrossRef]
- Hwang, E.; Choi, B.S.; Oh, K.H.; Kwon, Y.J.; Kim, G.H. Management of chronic kidney disease-mineral and bone disorder: Korean working group recommendations. Kidney Res. Clin. Pr. 2015, 34, 4–12. [Google Scholar] [CrossRef][Green Version]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Coburn, J.W.; Maung, H.M.; Elangovan, L.; Germain, M.J.; Lindberg, J.S.; Sprague, S.M.; Williams, M.E.; Bishop, C.W. Doxercalciferol safely suppresses PTH levels in patients with secondary hyperparathyroidism associated with chronic kidney disease stages 3 and 4. Am. J. Kidney Dis. 2004, 43, 877–890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hamdy, N.A.; Kanis, J.A.; Beneton, M.N.; Brown, C.B.; Juttmann, J.R.; Jordans, J.G.; Josse, S.; Meyrier, A.; Lins, R.L.; Fairey, I.T. Effect of alfacalcidol on natural course of renal bone disease in mild to moderate renal failure. BMJ 1995, 310, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Appelbaum, E.; Pritchett, Y.; Chang, Y.; Wenger, J.; Tamez, H.; Bhan, I.; Agarwal, R.; Zoccali, C.; Wanner, C.; et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: The PRIMO randomized controlled trial. JAMA 2012, 307, 674–684. [Google Scholar] [CrossRef]
- Wang, A.Y.; Fang, F.; Chan, J.; Wen, Y.Y.; Qing, S.; Chan, I.H.; Lo, G.; Lai, K.N.; Lo, W.K.; Lam, C.W.; et al. Effect of paricalcitol on left ventricular mass and function in CKD--the OPERA trial. J. Am. Soc. Nephrol. 2014, 25, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.; Cianciolo, G.; Tripepi, G.; Plebani, M.; Aghi, A.; Politi, C.; Zaninotto, M.; Nickolas, T.L.; Ferrari, S.; Ketteler, M.; et al. Oral Calcitriol Use, Vertebral Fractures, and Vitamin K in Hemodialysis Patients: A Cross-Sectional Study. J. Bone Miner. Res. 2021, 36, 2361–2370. [Google Scholar] [CrossRef]
- Ketteler, M.; Martin, K.J.; Wolf, M.; Amdahl, M.; Cozzolino, M.; Goldsmith, D.; Sharma, A.; Marx, S.; Khan, S. Paricalcitol versus cinacalcet plus low-dose vitamin D therapy for the treatment of secondary hyperparathyroidism in patients receiving haemodialysis: Results of the IMPACT SHPT study. Nephrol. Dial. Transplant. 2012, 27, 3270–3278. [Google Scholar] [CrossRef]
- Cozzolino, M.; Ketteler, M.; Martin, K.J.; Sharma, A.; Goldsmith, D.; Khan, S. Paricalcitol- or cinacalcet-centred therapy affects markers of bone mineral disease in patients with secondary hyperparathyroidism receiving haemodialysis: Results of the IMPACT-SHPT study. Nephrol. Dial. Transplant. 2014, 29, 899–905. [Google Scholar] [CrossRef]
- Sprague, S.M.; Wetmore, J.B.; Gurevich, K.; Da Roza, G.; Buerkert, J.; Reiner, M.; Goodman, W.; Cooper, K. Effect of Cinacalcet and Vitamin D Analogs on Fibroblast Growth Factor-23 during the Treatment of Secondary Hyperparathyroidism. Clin. J. Am. Soc. Nephrol. 2015, 10, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Gurevich, K.; Sprague, S.; Da Roza, G.; Buerkert, J.; Reiner, M.; Goodman, W.; Cooper, K. A Randomized Trial of Cinacalcet versus Vitamin D Analogs as Monotherapy in Secondary Hyperparathyroidism (PARADIGM). Clin. J. Am. Soc. Nephrol. 2015, 10, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Mascia, S.; Garofalo, C.; Donnarumma, G.; Di Pietro, R.; Liberti, M.E.; Pacilio, M.; Sagliocca, A.; Zamboli, P.; Minutolo, R.; Conte, G.; et al. Role of paracalcitol in the management of non-dialysis CKD: State of art and … Unmet needs. G. Ital. Nefrol. 2010, 27, 616–628. [Google Scholar]
- Garofalo, C.; Secondulfo, C.; Apicella, L.; Bilancio, G.; De Nicola, L.; Minutolo, R.; Borrelli, S.; Provenzano, M.; Luciani, R.; Bellizzi, V. Antiproteinuric effect of paricalcitol in kidney transplant recipients with severe proteinuria: A prospective cohort study. J. Nephrol. 2022, 35, 1943–1945. [Google Scholar] [CrossRef]
- Barman Balfour, J.A.; Scott, L.J. Cinacalcet hydrochloride. Drugs 2005, 65, 271–281. [Google Scholar] [CrossRef]
- Goodman, W.G.; Hladik, G.A.; Turner, S.A.; Blaisdell, P.W.; Goodkin, D.A.; Liu, W.; Barri, Y.M.; Cohen, R.M.; Coburn, J.W. The Calcimimetic agent AMG 073 lowers plasma parathyroid hormone levels in hemodialysis patients with secondary hyperparathyroidism. J. Am. Soc. Nephrol. 2002, 13, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Martin, K.J.; de Francisco, A.L.; Turner, S.A.; Avram, M.M.; Suranyi, M.G.; Hercz, G.; Cunningham, J.; Abu-Alfa, A.K.; Messa, P.; et al. Cinacalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N. Engl. J. Med. 2004, 350, 1516–1525. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, J.S.; Moe, S.M.; Goodman, W.G.; Coburn, J.W.; Sprague, S.M.; Liu, W.; Blaisdell, P.W.; Brenner, R.M.; Turner, S.A.; Martin, K.J. The calcimimetic AMG 073 reduces parathyroid hormone and calcium x phosphorus in secondary hyperparathyroidism. Kidney Int. 2003, 63, 248–254. [Google Scholar] [CrossRef]
- Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; Mahaffey, K.W.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, P.S.; Drueke, T.B.; Block, G.A.; Correa-Rotter, R.; Floege, J.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; Moe, S.M.; Wheeler, D.C.; et al. The Effects of Cinacalcet in Older and Younger Patients on Hemodialysis: The Evaluation of Cinacalcet HCl Therapy to Lower Cardiovascular Events (EVOLVE) Trial. Clin. J. Am. Soc. Nephrol. 2015, 10, 791–799. [Google Scholar] [CrossRef]
- Parfrey, P.S.; Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Floege, J.; Herzog, C.A.; London, G.M.; Mahaffey, K.W.; Moe, S.M.; et al. The clinical course of treated hyperparathyroidism among patients receiving hemodialysis and the effect of cinacalcet: The EVOLVE trial. J. Clin. Endocrinol. Metab. 2013, 98, 4834–4844. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Kubo, Y.; Floege, A.; Chertow, G.M.; Parfrey, P.S. The Effect of Cinacalcet on Calcific Uremic Arteriolopathy Events in Patients Receiving Hemodialysis: The EVOLVE Trial. Clin. J. Am. Soc. Nephrol. 2015, 10, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Walter, S.; Baruch, A.; Dong, J.; Tomlinson, J.E.; Alexander, S.T.; Janes, J.; Hunter, T.; Yin, Q.; Maclean, D.; Bell, G.; et al. Pharmacology of AMG 416 (Velcalcetide), a novel peptide agonist of the calcium-sensing receptor, for the treatment of secondary hyperparathyroidism in hemodialysis patients. J. Pharmacol. Exp. Ther. 2013, 346, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Bushinsky, D.A.; Cheng, S.; Cunningham, J.; Dehmel, B.; Drueke, T.B.; Ketteler, M.; Kewalramani, R.; Martin, K.J.; Moe, S.M.; et al. Effect of Etelcalcetide vs Cinacalcet on Serum Parathyroid Hormone in Patients Receiving Hemodialysis With Secondary Hyperparathyroidism: A Randomized Clinical Trial. JAMA 2017, 317, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Friedl, C.; Zitt, E. Role of etelcalcetide in the management of secondary hyperparathyroidism in hemodialysis patients: A review on current data and place in therapy. Drug Des. Dev. Ther. 2018, 12, 1589–1598. [Google Scholar] [CrossRef]
- Chen, J.; Jia, X.; Kong, X.; Wang, Z.; Cui, M.; Xu, D. Total parathyroidectomy with autotransplantation versus subtotal parathyroidectomy for renal hyperparathyroidism: A systematic review and meta-analysis. Nephrology 2017, 22, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of action and role in clinical practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef]
- Damasiewicz, M.J.; Nickolas, T.L. Bisphosphonate therapy in CKD: The current state of affairs. Curr. Opin. Nephrol. Hypertens. 2020, 29, 221–226. [Google Scholar] [CrossRef]
- Khosla, S.; Burr, D.; Cauley, J.; Dempster, D.W.; Ebeling, P.R.; Felsenberg, D.; Gagel, R.F.; Gilsanz, V.; Guise, T.; Koka, S.; et al. Bisphosphonate-associated osteonecrosis of the jaw: Report of a task force of the American Society for Bone and Mineral Research. J. Bone Miner. Res. 2007, 22, 1479–1491. [Google Scholar] [CrossRef]
- Schilcher, J.; Koeppen, V.; Aspenberg, P.; Michaëlsson, K. Risk of atypical femoral fracture during and after bisphosphonate use. N. Engl. J. Med. 2014, 371, 974–976. [Google Scholar] [CrossRef]
- Rosen, C.J.; Brown, S. Severe hypocalcemia after intravenous bisphosphonate therapy in occult vitamin D deficiency. N. Engl. J. Med. 2003, 348, 1503–1504. [Google Scholar] [CrossRef]
- Dempster, D.W.; Lambing, C.L.; Kostenuik, P.J.; Grauer, A. Role of RANK ligand and denosumab, a targeted RANK ligand inhibitor, in bone health and osteoporosis: A review of preclinical and clinical data. Clin. Ther. 2012, 34, 521–536. [Google Scholar] [CrossRef]
- Bone, H.G.; Bolognese, M.A.; Yuen, C.K.; Kendler, D.L.; Wang, H.; Liu, Y.; San Martin, J. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J. Clin. Endocrinol. Metab. 2008, 93, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.P.; Reid, I.R.; Wagman, R.B.; Kendler, D.; Miller, P.D.; Jensen, J.E.; Bolognese, M.A.; Daizadeh, N.; Valter, I.; Zerbini, C.A.; et al. Effects of up to 5 years of denosumab treatment on bone histology and histomorphometry: The FREEDOM study extension. J. Bone Miner. Res. 2014, 29, 2051–2056. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.A.; Ljunggren, O.; Stehman-Breen, C.; Cummings, S.R.; McClung, M.R.; Goemaere, S.; Ebeling, P.R.; Franek, E.; Yang, Y.C.; Egbuna, O.I.; et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J. Bone Miner. Res. 2011, 26, 1829–1835. [Google Scholar] [CrossRef]
- Block, G.A.; Bone, H.G.; Fang, L.; Lee, E.; Padhi, D. A single-dose study of denosumab in patients with various degrees of renal impairment. J. Bone Miner. Res. 2012, 27, 1471–1479. [Google Scholar] [CrossRef]
- Gopaul, A.; Kanagalingam, T.; Thain, J.; Khan, T.; Cowan, A.; Sultan, N.; Clemens, K.K. Denosumab in chronic kidney disease: A narrative review of treatment efficacy and safety. Arch. Osteoporos. 2021, 16, 116. [Google Scholar] [CrossRef]
- Nitta, K.; Yajima, A.; Tsuchiya, K. Management of Osteoporosis in Chronic Kidney Disease. Intern. Med. 2017, 56, 3271–3276. [Google Scholar] [CrossRef]


| Study (Year) | Type | Population | Sample Size | Outcome | Results |
|---|---|---|---|---|---|
| Lopes et al. (2020) [56] | Prospective cohort study | HD | 17,414 patients | CV mortality | Patients with poor control of serum phosphorus levels (expressed as AUC) during a 6-month period have a higher risk of CV mortality (for AUC > 2 = HR 2.03, 95% CI 1.53–2.69). |
| Hou Y. et al. (2017) [57] | Meta-analysis of 9 cohort studies | HD or PD | 1,992,869 patients | All-cause mortality | Compared to reference phosphorus category, both very high (HR 1.39; 95% CI 1.31–1.47) and very low (HR 1.16, 95% CI 1.06–1.28) phosphorus levels are associated with a greater risk for all-cause mortality. |
| Dhingra et al. (2007) [58] | Observational prospective study | Not CKD | 3368 patients | Incident of Cardiovascular disease | Patients with elevated phosphorus serum levels have a greater risk of CVD (HR 1.55, 95% CI 1.16–2.07%; p = 0.004). |
| Bellasi et al. (2011) [59] | Observational retrospective study | CKD 3–5, not requiring dialysis | 1716 patients | Composite end point of progression to ESKD or death | A worse control of phosphorus levels (≥4.3 mg/dL) exposes to an increased risk of progression to ESKD or death (HR ratio 2.04; 95% CI 1.44–2.90). |
| Johhn J. Sim et al. (2013) [60] | Observational retrospective longitudinal study | Not CKD | 94,989 patients | Incident of ESKD | Iperphosphatemia is associated with greater risk for ESKD. Risk increased by 40% for each increase of 0.5 mg/dL of serum phosphate. |
| Yang et al. (2016) [65] | Meta-analysis of 7 studies | HD or PD | 1406 patients | All-cause mortality | Higher serum FGF23 levels are associated with increased risk of death (HR 1.53; 95% CI: 1.05–2.25). |
| Rebholz et al. (2015) [67] | Observational prospective study | Not CKD | 13,488 patients | Incident of ESKD | Elevated fibroblast growth factor serum concentration is associated with greater risk of ESKD (HR 2.10; 95% CI 1.31 to 3.36; trend p < 0.001). |
| Isakova et al. (2011) [12] | Observational prospective study | CKD 2–4 | 3879 patients | All-cause mortality and ESKD | Patients with CKD 2–4 with higher FGF23 levels have an augmented risk of death (quartile 1, reference; quartile 2, HR 1.3; 95%CI 0.8–2.2; quartile 3, HR 2.0; 95%CI 1.2–3.3; quartile 4, and HR 3.0; 95%CI 1.8–5.1). Elevated FGF23 values are associated with higher risk of ESKD (for patients with eGFR 30–44 mL/min = HR 1.3 per SD of lnFGF23; 95%CI 1.04–1.6 and for patients with eGFR ≥ 45 mL/min = HR 1.7; 95% CI 1.1–2.4). This association has not been demonstrated for patients with eGFR< 30 mL/min. |
| Borrelli et al. (2018) [68] | Observational prospective study | CKD 1–5, not requiring dialysis | 543 patients | Renal death (composite of ESKD or all-causes death before ESKD) | In the same subject, higher PTH variation over time (∆PTH) with respect to baseline value is associated with an augmented risk of renal death (for the highest ΔPTH quartile = HR 1.91; 95%CI:1.08–3.38; p = 0.026). |
| Floege J. et al. (2011) [70] | Observational prospective study | HD | 7970 patients | All-cause mortality | Compared to reference PTH range (150–300 pg/mL), both elevated (>600 pg/mL) and low (<75 pg/mL) PTH levels are, respectively, associated with a 2-fold (HR 2.10; 95%CI 1.62–2.73, p < 0.001) and 1,5-fold (HR 1.46; 95% CI 1.17–1.83, p = 0.001) risk of death. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, L.; Napoletano, A.; Provenzano, M.; Garofalo, C.; Bini, C.; Comai, G.; La Manna, G. Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic. Int. J. Mol. Sci. 2022, 23, 12223. https://doi.org/10.3390/ijms232012223
Hu L, Napoletano A, Provenzano M, Garofalo C, Bini C, Comai G, La Manna G. Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic. International Journal of Molecular Sciences. 2022; 23(20):12223. https://doi.org/10.3390/ijms232012223
Chicago/Turabian StyleHu, Lilio, Angelodaniele Napoletano, Michele Provenzano, Carlo Garofalo, Claudia Bini, Giorgia Comai, and Gaetano La Manna. 2022. "Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic" International Journal of Molecular Sciences 23, no. 20: 12223. https://doi.org/10.3390/ijms232012223
APA StyleHu, L., Napoletano, A., Provenzano, M., Garofalo, C., Bini, C., Comai, G., & La Manna, G. (2022). Mineral Bone Disorders in Kidney Disease Patients: The Ever-Current Topic. International Journal of Molecular Sciences, 23(20), 12223. https://doi.org/10.3390/ijms232012223