
Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent


Abstract
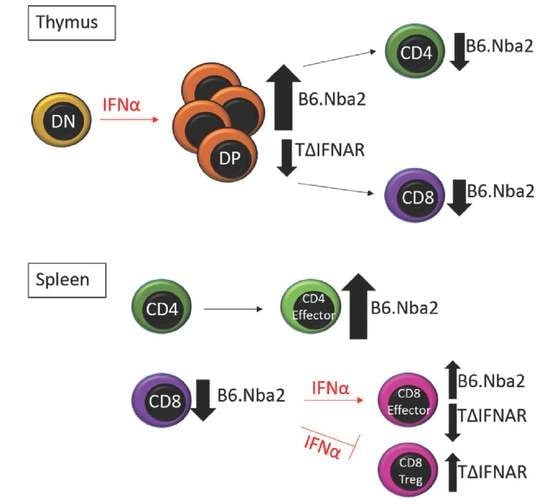
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
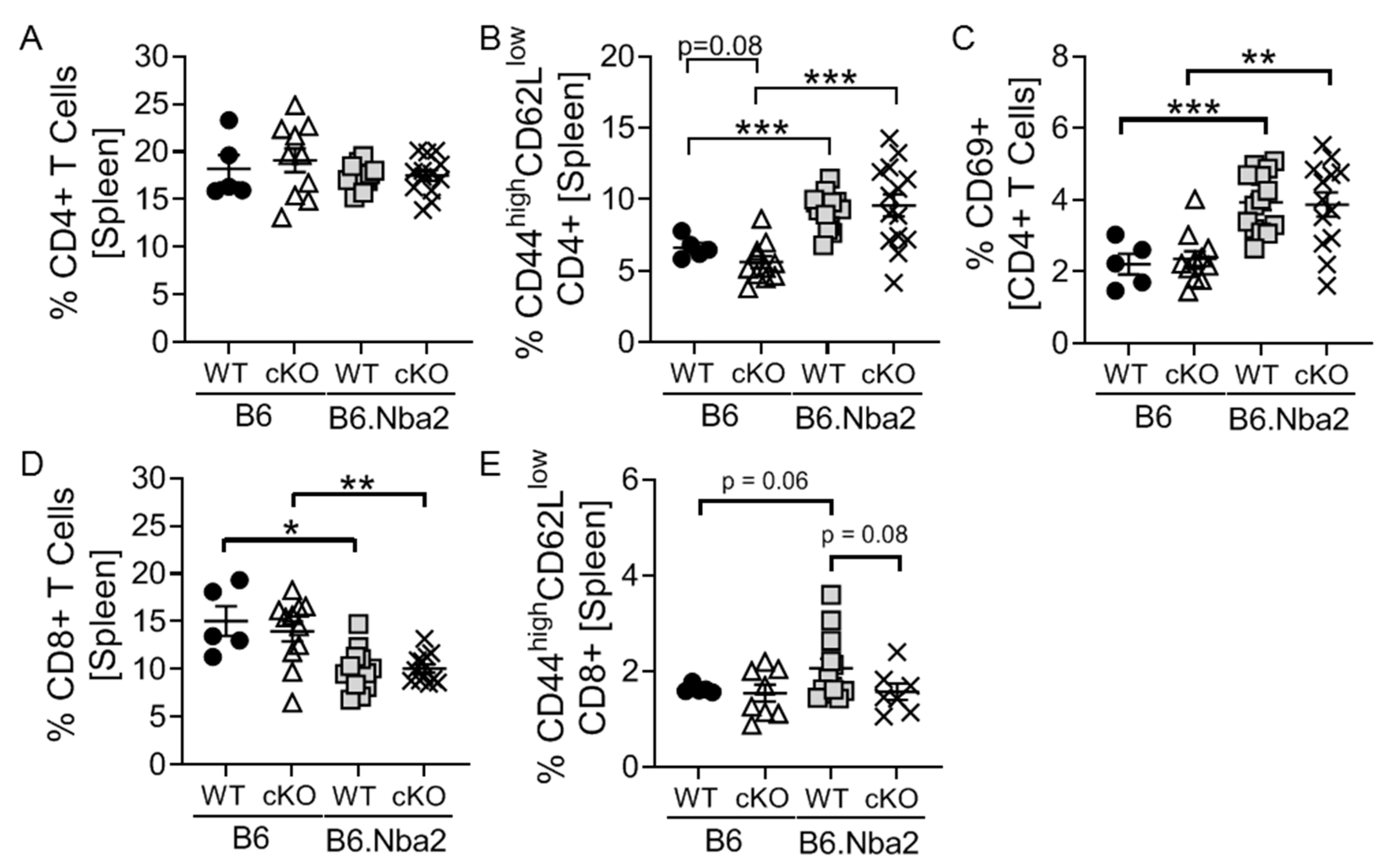
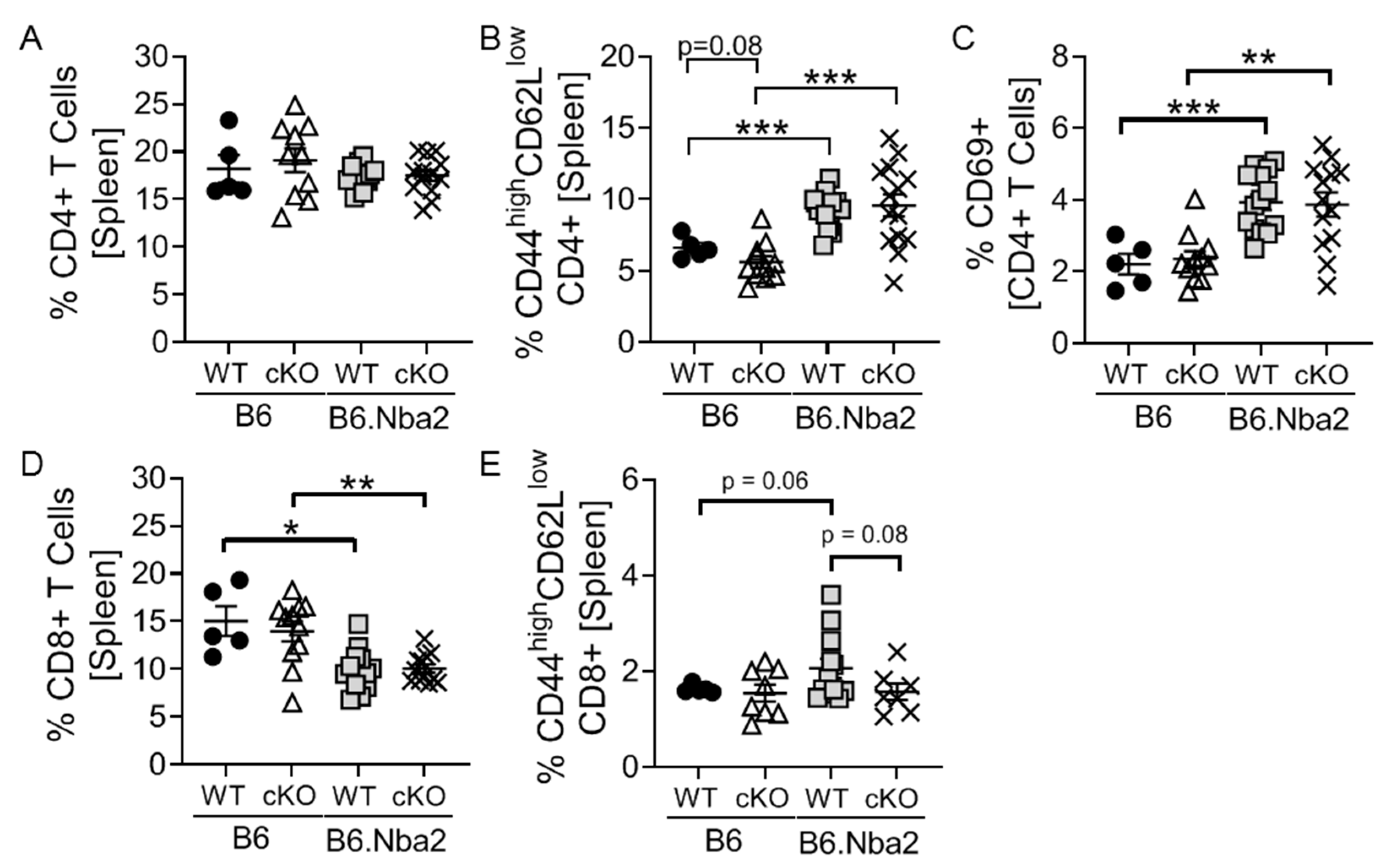
2.1. T-Cell Specific IFNAR Deficiency Does Not Affect Splenic CD4 and CD8 T Cell Dysregulation in B6.Nba2 Mice
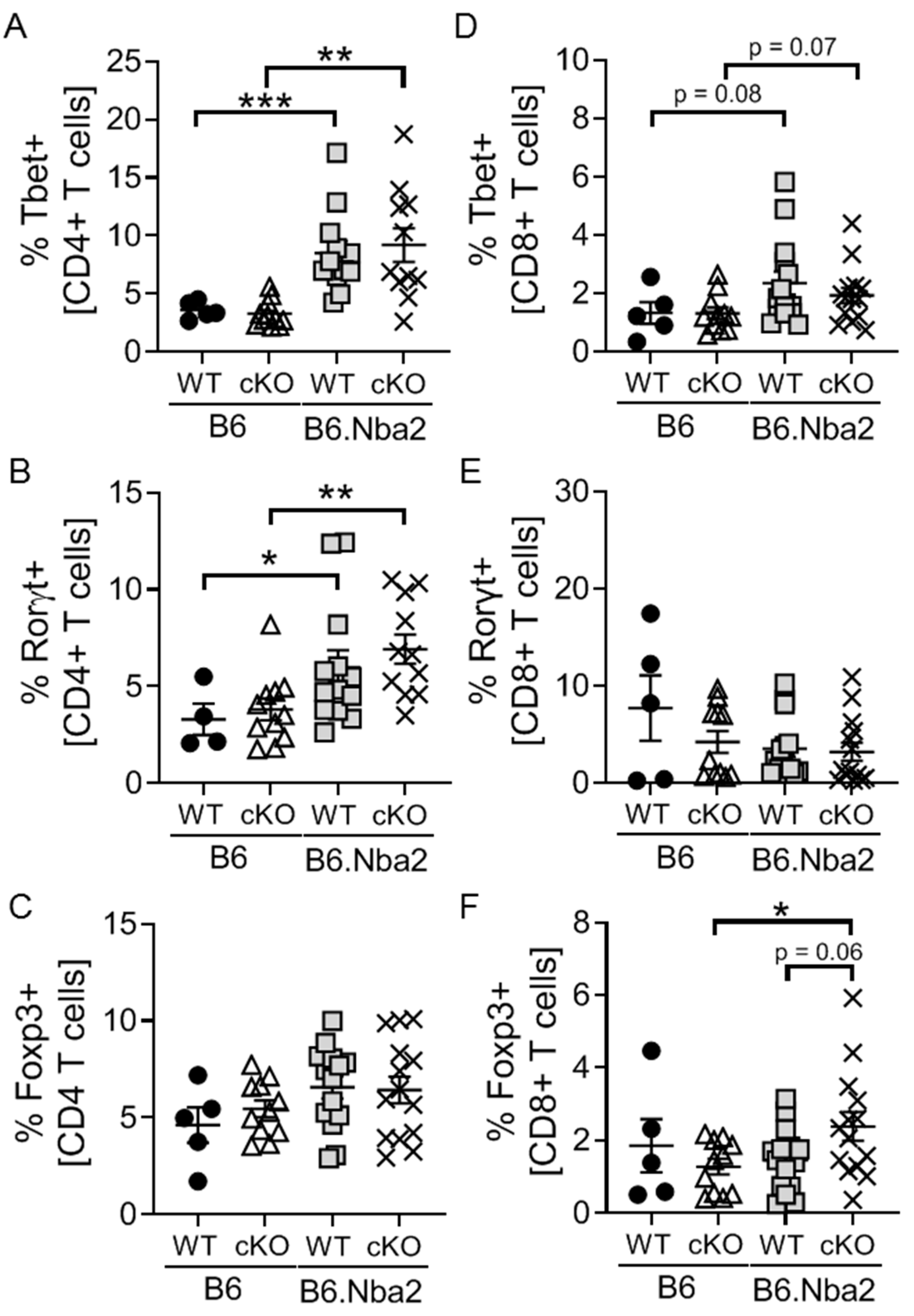
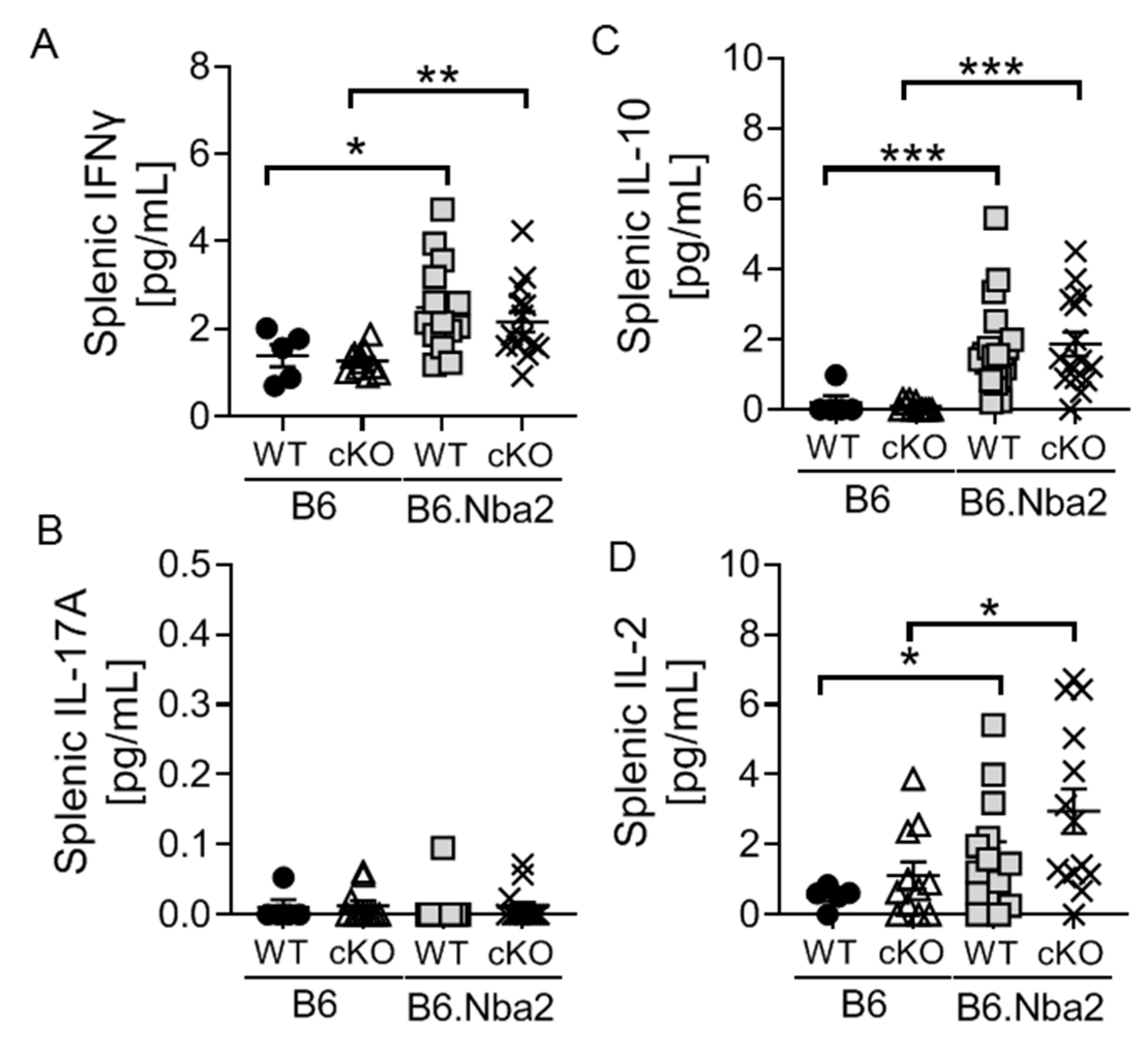
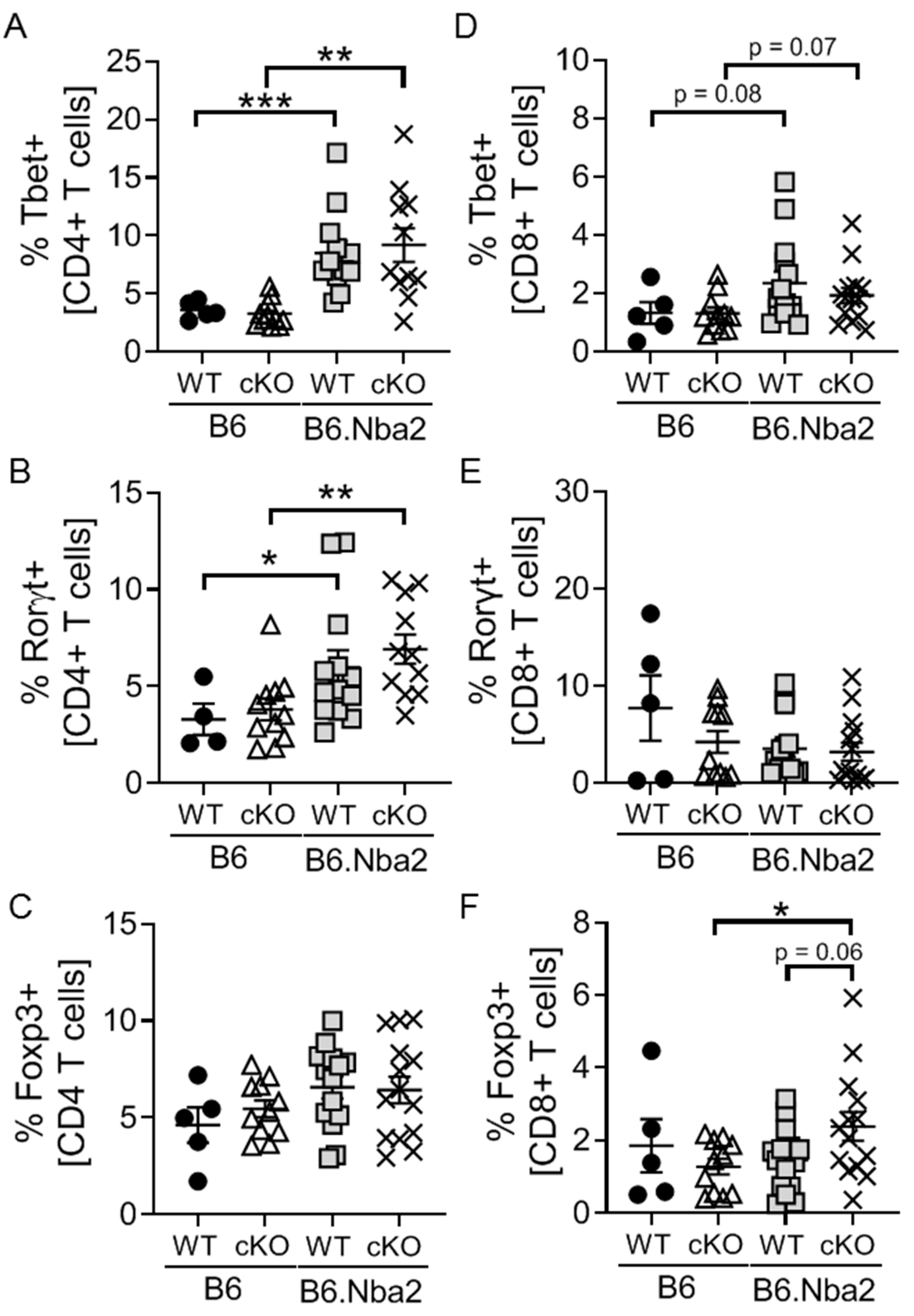
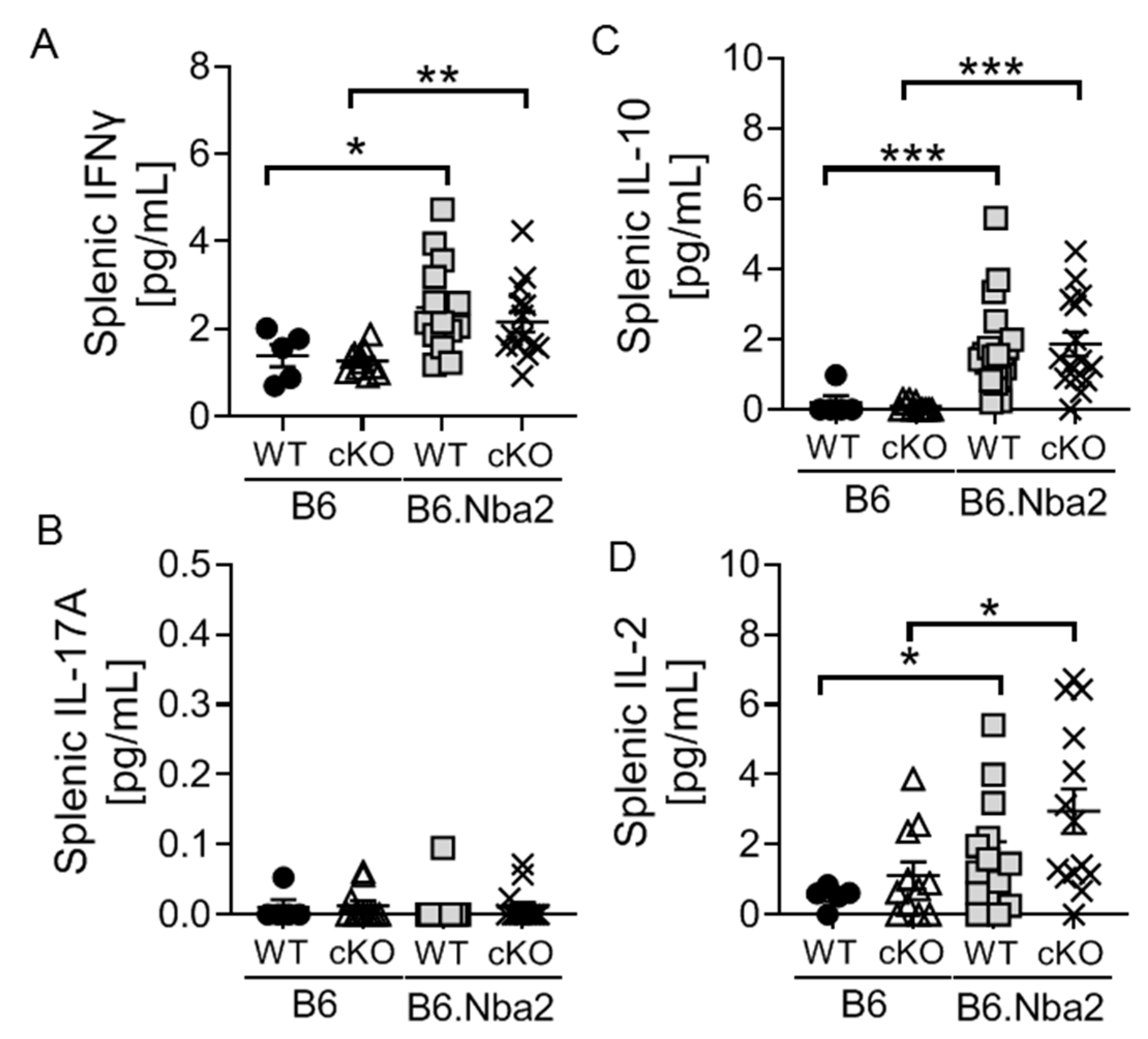
2.2. B6.Nba2 Mice Accumulate Th1 and Th17 Cells in an IFNAR-Independent Manner
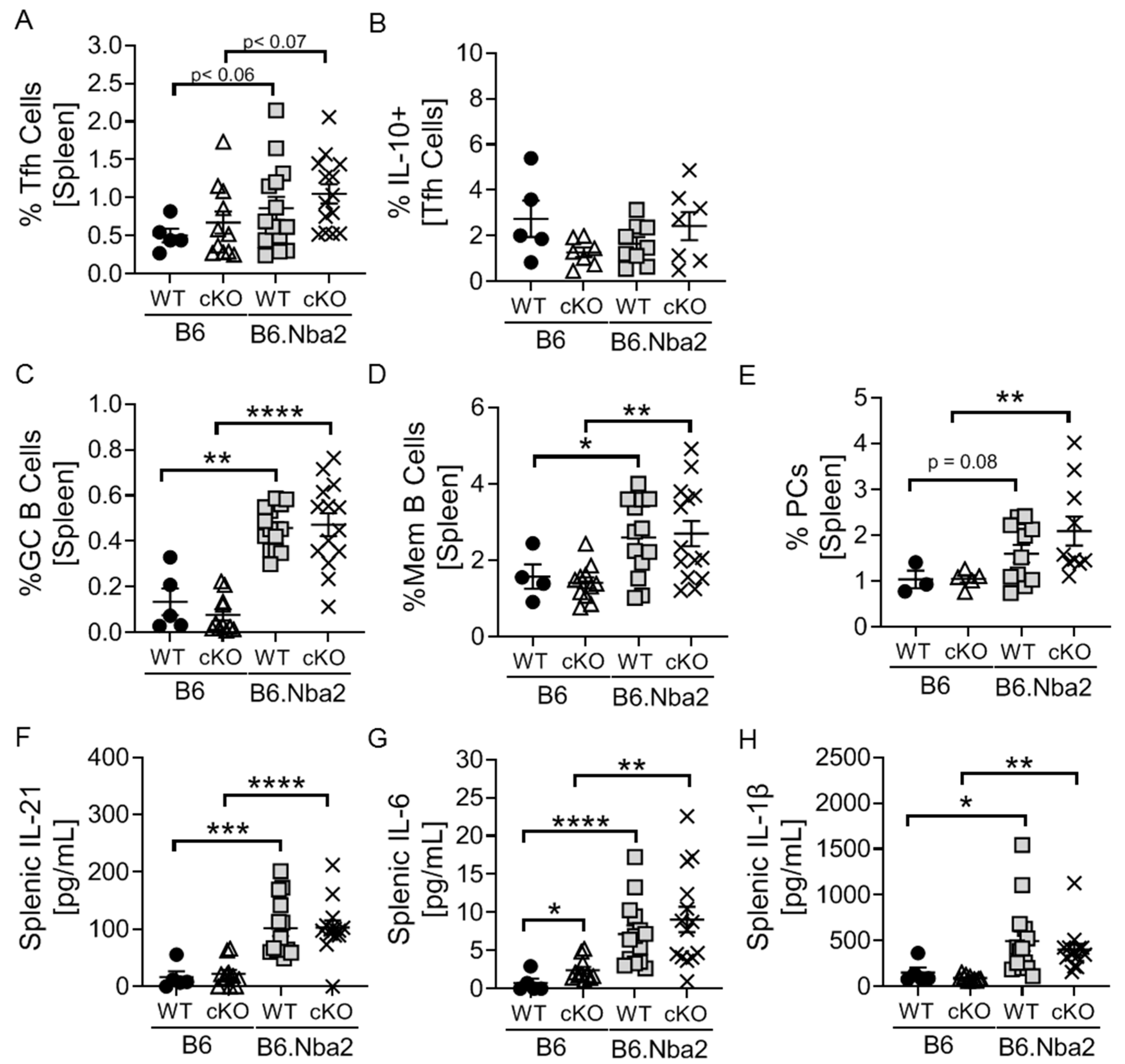
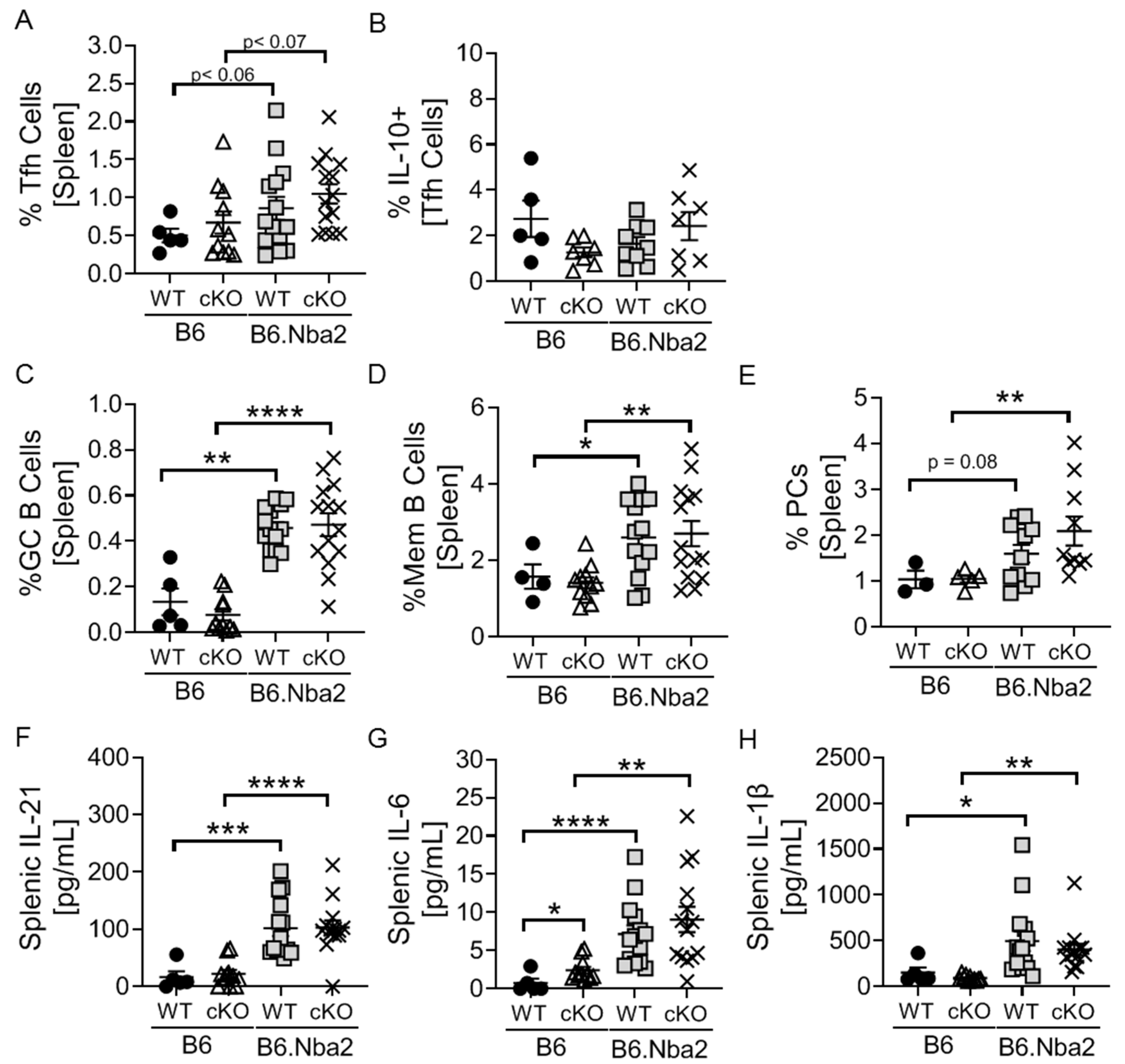
2.3. Nba2-Driven Dysregulation of T Follicular Helper (Tfh) Cells, Germinal Center Associated B Cells and Associated Cytokines Is Independent IFNAR Expression
2.4. Nba2-Driven Splenomegaly and ANA Production Is Intact in B6.Nba2.cKO Mice
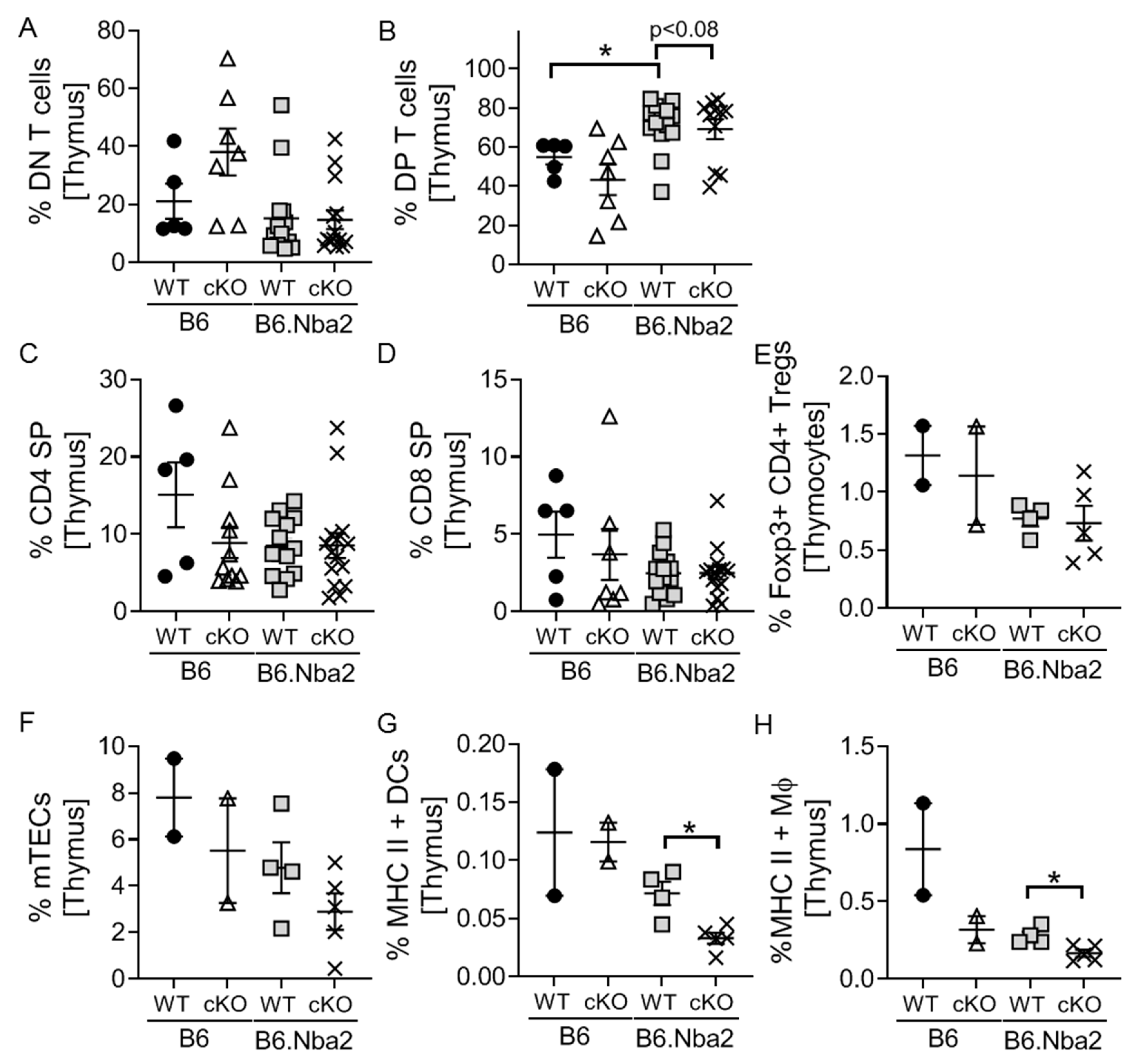
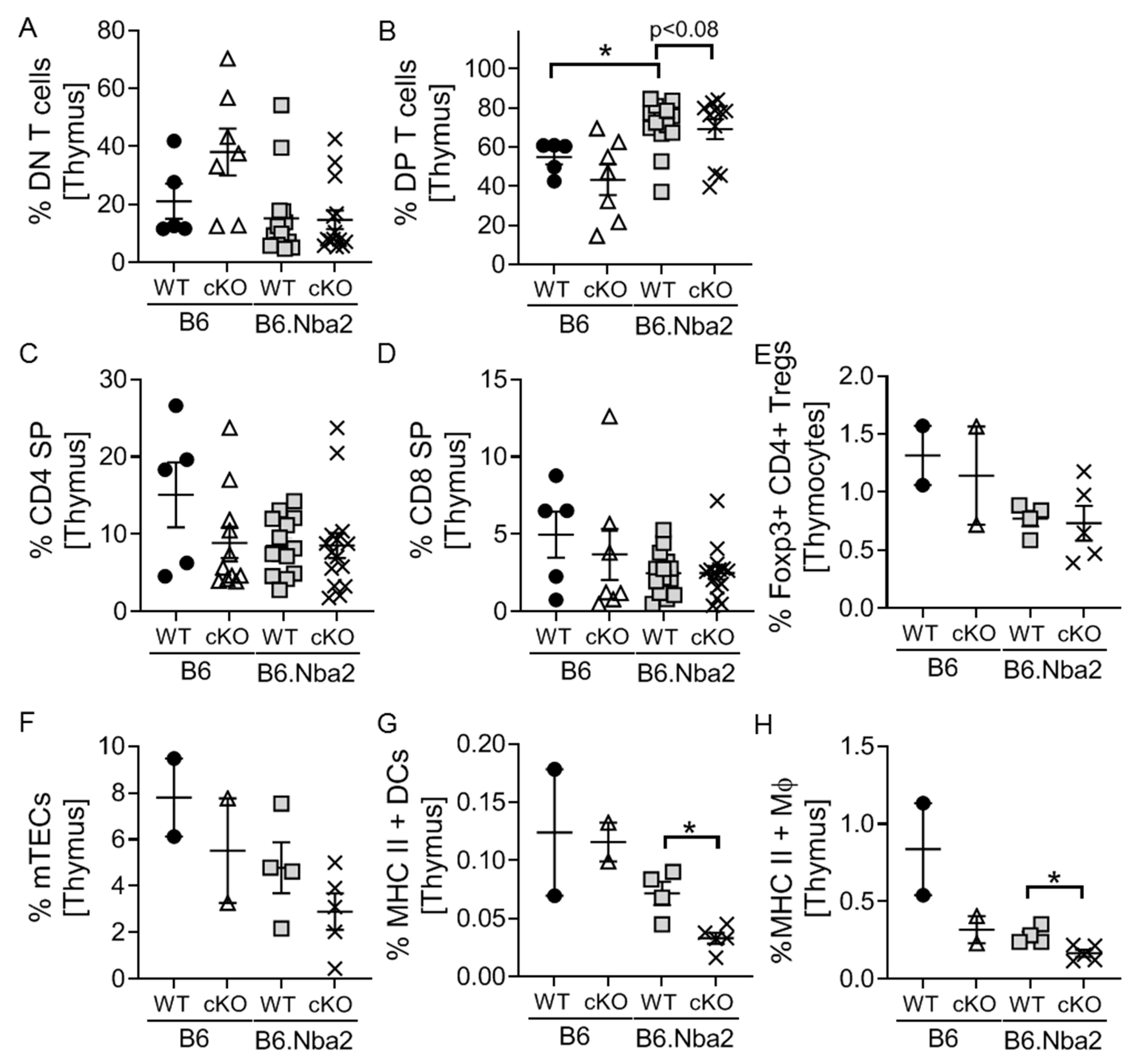
2.5. DP Thymocytes Accumulate in B6.Nba2 Mice in a Partially IFNAR-Dependent Manner
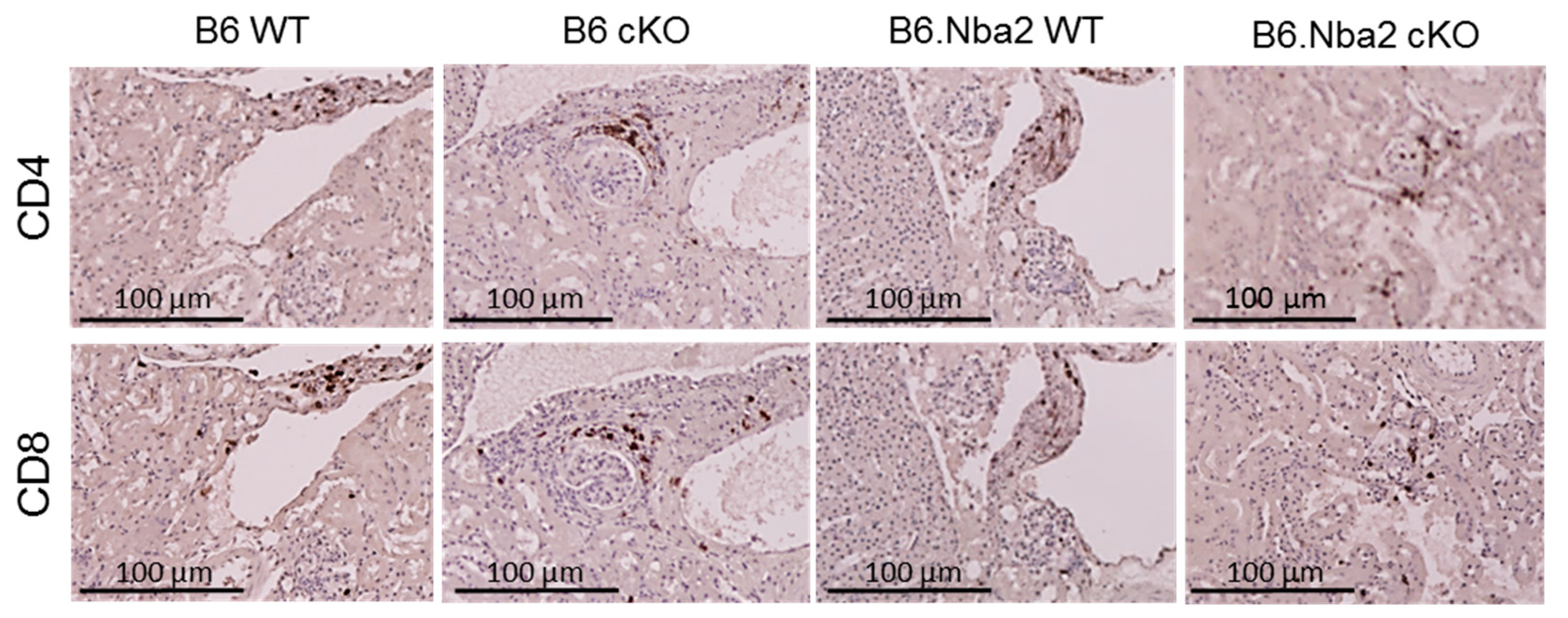
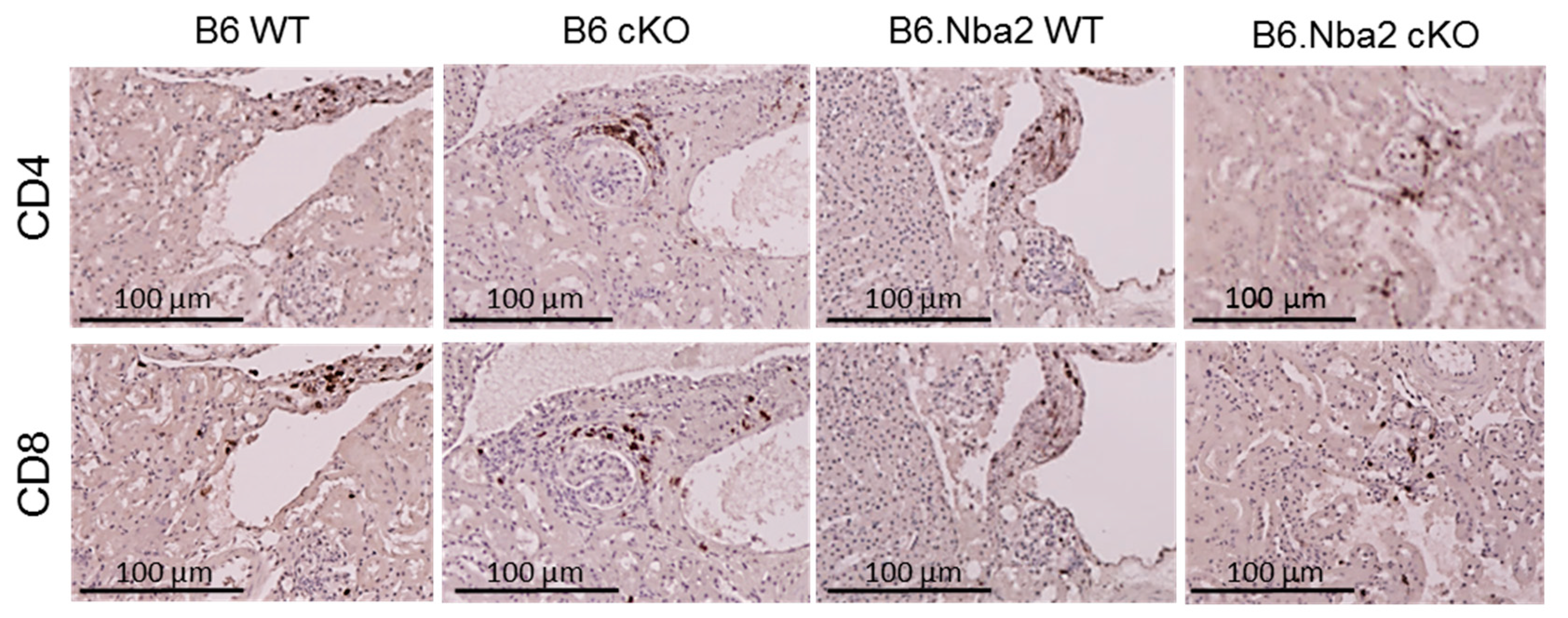
2.6. Kidney-Infiltrating T Cells Are Unaffected by Intrinsic IFNAR Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Flow Cytometry
4.3. Intra-Splenic Cytokine Levels Using Electrochemiluminescence
4.4. Anti-dsDNA Autoantibody ELISA
4.5. Immunohistochemistry
4.6. Microscopy
4.7. Statistic
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Talaat, R.M.; Mohamed, S.F.; Bassyouni, I.H.; Raouf, A.A. Th1/Th2/Th17/Treg cytokine imbalance in systemic lupus erythematosus (SLE) patients: Correlation with disease activity. Cytokine 2015, 72, 146–153. [Google Scholar] [CrossRef]
- Shah, K.; Lee, W.-W.; Lee, S.-H.; Kim, S.H.; Kang, S.W.; Craft, J.; Kang, I. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R53. [Google Scholar] [CrossRef] [Green Version]
- Shivakumar, S.; Tsokos, G.C.; Datta, S.K. T cell receptor alpha/beta expressing double-negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J. Immunol. 1989, 143, 103–112. [Google Scholar]
- Li, H.; Adamopoulos, I.E.; Moulton, V.R.; Stillman, I.E.; Herbert, Z.; Moon, J.J.; Sharabi, A.; Krishfield, S.; Tsokos, M.G.; Tsokos, G.C. Systemic lupus erythematosus favors the generation of IL-17 producing double negative T cells. Nat. Commun. 2020, 11, 2859. [Google Scholar] [CrossRef] [PubMed]
- Tulunay, A.; Yavuz, S.; Direskeneli, H.; Eksioglu-Demiralp, E. CD8+CD28-, suppressive T cells in systemic lupus erythematosus. Lupus 2008, 17, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lit, L.C.W.; Tam, L.S.; Li, E.K.M.; Wong, P.T.Y.; Lam, C.W.K. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Lombardi, L.; Richards, H.B.; Dammacco, F.; Silvestris, F. Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clin. Exp. Immunol. 2008, 154, 247–254. [Google Scholar] [CrossRef]
- Zhang, X.; Lindwall, E.; Gauthier, C.; Lyman, J.; Spencer, N.; Alarakhia, A.; Fraser, A.; Ing, S.; Chen, M.; Webb-Detiege, T.; et al. Circulating CXCR5+CD4+helper T cells in systemic lupus erythematosus patients share phenotypic properties with germinal center follicular helper T cells and promote antibody production. Lupus 2015, 24, 909–917. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Ho, J.H.-e.; Pasoto, S.G.; Bunin, V.; Kim, S.T.; Carrasco, S.; Borba, E.F.; Gonçalves, C.R.; Costa, P.R.; Kallas, E.G.; et al. Circulating Follicular Helper–Like T Cells in Systemic Lupus Erythematosus: Association With Disease Activity. Arthritis Rheumatol. 2015, 67, 988–999. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, P.; Ma, L.; Shan, Y.; Jiang, Z.; Wang, J.; Jiang, Y. Increased Interleukin 21 and Follicular Helper T-like Cells and Reduced Interleukin 10+ B cells in Patients with New-onset Systemic Lupus Erythematosus. J. Rheumatol. 2014, 41, 1781–1792. [Google Scholar] [CrossRef]
- Sobel, E.S.; Satoh, M.; Chen, Y.; Wakeland, E.K.; Morel, L. The Major Murine Systemic Lupus Erythematosus Susceptibility Locus Sle1 Results in Abnormal Functions of Both B and T Cells. J. Immunol. 2002, 169, 2694–2700. [Google Scholar] [CrossRef] [Green Version]
- Davison, L.M.; Jorgensen, T.N. Sialic acid-binding immunoglobulin-type lectin H-positive plasmacytoid dendritic cells drive spontaneous lupus-like disease development in B6.Nba2 mice. Arthritis Rheumatol. 2015, 67, 1012–1022. [Google Scholar] [CrossRef]
- Keller, E.J.; Patel, N.B.; Patt, M.; Nguyen, J.K.; Jørgensen, T.N. Partial Protection From Lupus-Like Disease by B-Cell Specific Type I Interferon Receptor Deficiency. Front. Immunol. 2021, 11, 3371. [Google Scholar] [CrossRef]
- Humrich, J.Y.; Morbach, H.; Undeutsch, R.; Enghard, P.; Rosenberger, S.; Weigert, O.; Kloke, L.; Heimann, J.; Gaber, T.; Brandenburg, S.; et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc. Natl. Acad. Sci. USA 2010, 107, 204–209. [Google Scholar] [CrossRef] [Green Version]
- Sadlack, B.; Löhler, J.; Schorle, H.; Klebb, G.; Haber, H.; Sickel, E.; Noelle, R.J.; Horak, I. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur. J. Immunol. 1995, 25, 3053–3059. [Google Scholar] [CrossRef]
- Stohl, W.; Xu, D.; Metzger, T.E.; Kim, K.S.; Morel, L.; Kotzin, B.L. Dichotomous effects of complete versus partial class II major histocompatibility complex deficiency on circulating autoantibody levels in autoimmune-prone mice. Arthritis Rheum. 2004, 50, 2227–2239. [Google Scholar] [CrossRef]
- Schmidt, T.; Paust, H.J.; Krebs, C.F.; Turner, J.E.; Kaffke, A.; Bennstein, S.B.; Koyro, T.; Peters, A.; Velden, J.; Hünemörder, S.; et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol. 2015, 67, 475–487. [Google Scholar] [CrossRef]
- Mizui, M.; Koga, T.; Lieberman, L.A.; Beltran, J.; Yoshida, N.; Johnson, M.C.; Tisch, R.; Tsokos, G.C. IL-2 Protects Lupus-Prone Mice from Multiple End-Organ Damage by Limiting CD4−CD8− IL-17–Producing T Cells. J. Immunol. 2014, 193, 2168–2177. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, S.H.; Seo, H.B.; Ryu, J.G.; Jung, K.; Choi, J.W.; Jhun, J.; Park, J.S.; Kwon, J.Y.; Kwok, S.K.; et al. Inhibition of IL-17 ameliorates systemic lupus erythematosus in Roquin(san/san) mice through regulating the balance of TFH cells, GC B cells, Treg and Breg. Sci. Rep. 2019, 9, 5227. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.; Theofilopoulos, A.N.; Weiner, R.; Katz, D.H.; Dixon, F.J. Analysis of T cell function in autoimmune murine strains. Defects in production and responsiveness to interleukin 2. J. Exp. Med. 1981, 154, 791–808. [Google Scholar] [CrossRef] [Green Version]
- Chodisetti, S.B.; Fike, A.J.; Domeier, P.P.; Singh, H.; Choi, N.M.; Corradetti, C.; Kawasawa, Y.I.; Cooper, T.K.; Caricchio, R.; Rahman, Z.S.M. Type II but Not Type I IFN Signaling Is Indispensable for TLR7-Promoted Development of Autoreactive B Cells and Systemic Autoimmunity. J. Immunol. 2020, 204, 796–809. [Google Scholar] [CrossRef]
- Linterman, M.A.; Rigby, R.J.; Wong, R.K.; Yu, D.; Brink, R.; Cannons, J.L.; Schwartzberg, P.L.; Cook, M.C.; Walters, G.D.; Vinuesa, C.G. Follicular helper T cells are required for systemic autoimmunity. J. Exp. Med. 2009, 206, 561–576. [Google Scholar] [CrossRef]
- Iwai, H.; Abe, M.; Hirose, S.; Tsushima, F.; Tezuka, K.; Akiba, H.; Yagita, H.; Okumura, K.; Kohsaka, H.; Miyasaka, N.; et al. Involvement of Inducible Costimulator-B7 Homologous Protein Costimulatory Pathway in Murine Lupus Nephritis. J. Immunol. 2003, 171, 2848–2854. [Google Scholar] [CrossRef] [Green Version]
- Kalled, S.L.; Cutler, A.H.; Datta, S.K.; Thomas, D.W. Anti-CD40 ligand antibody treatment of SNF1 mice with established nephritis: Preservation of kidney function. J. Immunol. 1998, 160, 2158–2165. [Google Scholar]
- Jorgensen, T.N.; Roper, E.; Thurman, J.M.; Marrack, P.; Kotzin, B.L. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2× NZW)F(1) mice. Genes Immun. 2007, 8, 653–662. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, H.; Jacob, N.; Carreras, E.; Bajana, S.; Putterman, C.; Turner, S.; Neas, B.; Mathian, A.; Koss, M.N.; Stohl, W.; et al. Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J. Immunol. 2009, 183, 6021–6029. [Google Scholar] [CrossRef] [Green Version]
- Fairhurst, A.M.; Mathian, A.; Connolly, J.E.; Wang, A.; Gray, H.F.; George, T.A.; Boudreaux, C.D.; Zhou, X.J.; Li, Q.Z.; Koutouzov, S.; et al. Systemic IFN-alpha drives kidney nephritis in B6.Sle123 mice. Eur. J. Immunol. 2008, 38, 1948–1960. [Google Scholar] [CrossRef] [Green Version]
- Mathian, A.; Weinberg, A.; Gallegos, M.; Banchereau, J.; Koutouzov, S. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J. Immunol. 2005, 174, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Raber, M.L.; Baccala, R.; Haraldsson, K.M.; Choubey, D.; Stewart, T.A.; Kono, D.H.; Theofilopoulos, A.N. Type-I Interferon Receptor Deficiency Reduces Lupus-like Disease in NZB Mice. J. Exp. Med. 2003, 197, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Kono, D.H.; Baccala, R.; Theofilopoulos, A.N. Inhibition of lupus by genetic alteration of the interferon-alpha/beta receptor. Autoimmunity 2003, 36, 503–510. [Google Scholar] [CrossRef]
- Bennett, L.; Palucka, A.K.; Arce, E.; Cantrell, V.; Borvak, J.; Banchereau, J.; Pascual, V. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med. 2003, 197, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [Green Version]
- Marrack, P.; Kappler, J.; Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 1999, 189, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Havenar-Daughton, C.; Kolumam, G.A.; Murali-Krishna, K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 2006, 176, 3315–3319. [Google Scholar] [CrossRef] [Green Version]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Le Bon, A.; Durand, V.; Kamphuis, E.; Thompson, C.; Bulfone-Paus, S.; Rossmann, C.; Kalinke, U.; Tough, D.F. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J. Immunol. 2006, 176, 4682–4689. [Google Scholar] [CrossRef] [Green Version]
- Nakayamada, S.; Poholek, A.C.; Lu, K.T.; Takahashi, H.; Kato, M.; Iwata, S.; Hirahara, K.; Cannons, J.L.; Schwartzberg, P.L.; Vahedi, G.; et al. Type I IFN induces binding of STAT1 to Bcl6: Divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J. Immunol. 2014, 192, 2156–2166. [Google Scholar] [CrossRef]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.W.; Lyons, P.A.; Smith, K.G.C. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Skaggs, B.J.; Singh, R.P.; Hahn, B.H. Induction of immune tolerance by activation of CD8+ T suppressor/regulatory cells in lupus-prone mice. Hum. Immunol. 2008, 69, 790–796. [Google Scholar] [CrossRef] [Green Version]
- Dinesh, R.K.; Skaggs, B.J.; La Cava, A.; Hahn, B.H.; Singh, R.P. CD8+ Tregs in lupus, autoimmunity, and beyond. Autoimmun. Rev. 2010, 9, 560–568. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; La Cava, A.; Hahn, B.H. pConsensus peptide induces tolerogenic CD8+ T cells in lupus-prone (NZB x NZW)F1 mice by differentially regulating Foxp3 and PD1 molecules. J. Immunol. 2008, 180, 2069–2080. [Google Scholar] [CrossRef] [Green Version]
- Tilstra, J.S.; Avery, L.; Menk, A.V.; Gordon, R.A.; Smita, S.; Kane, L.P.; Chikina, M.; Delgoffe, G.M.; Shlomchik, M.J. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Investig. 2018, 128, 4884–4897. [Google Scholar] [CrossRef]
- Jorgensen, T.N.; Alfaro, J.; Enriquez, H.L.; Jiang, C.; Loo, W.M.; Atencio, S.; Bupp, M.R.; Mailloux, C.M.; Metzger, T.; Flannery, S.; et al. Development of murine lupus involves the combined genetic contribution of the SLAM and FcgammaR intervals within the Nba2 autoimmune susceptibility locus. J. Immunol. 2010, 184, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Dinesh, R.; Hahn, B.H.; La Cava, A.; Singh, R.P. Interferon-inducible gene 202b controls CD8(+) T cell-mediated suppression in anti-DNA Ig peptide-treated (NZB × NZW) F1 lupus mice. Genes Immun. 2011, 12, 360–369. [Google Scholar] [CrossRef] [Green Version]
- Couzi, L.; Merville, P.; Deminière, C.; Moreau, J.F.; Combe, C.; Pellegrin, J.L.; Viallard, J.F.; Blanco, P. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. 2007, 56, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Durelli, L.; Conti, L.; Clerico, M.; Boselli, D.; Contessa, G.; Ripellino, P.; Ferrero, B.; Eid, P.; Novelli, F. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann. Neurol. 2009, 65, 499–509. [Google Scholar] [CrossRef]
- Filaci, G.; Fravega, M.; Negrini, S.; Procopio, F.; Fenoglio, D.; Rizzi, M.; Brenci, S.; Contini, P.; Olive, D.; Ghio, M.; et al. Nonantigen specific CD8+ T suppressor lymphocytes originate from CD8+CD28- T cells and inhibit both T-cell proliferation and CTL function. Hum. Immunol. 2004, 65, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Inaba, M.; Sugihara, A.; Koshiji, M.; Sugiura, K.; Amoh, Y.; Mori, S.; Kamiya, T.; Genba, H.; Ikehara, S. Effects of administration of monoclonal antibodies (anti-CD4 or anti-CD8) on the development of autoimmune diseases in (NZW x BXSB)F1 mice. Immunobiology 1998, 198, 451–464. [Google Scholar] [CrossRef]
- Hahn, B.H.; Singh, R.P.; La Cava, A.; Ebling, F.M. Tolerogenic treatment of lupus mice with consensus peptide induces Foxp3-expressing, apoptosis-resistant, TGFbeta-secreting CD8+ T cell suppressors. J. Immunol. 2005, 175, 7728–7737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.P.; Fitzpatrick, D.R.; Beard, C.; Jessup, H.K.; Lehar, S.; Makar, K.W.; Pérez-Melgosa, M.; Sweetser, M.T.; Schlissel, M.S.; Nguyen, S.; et al. A Critical Role for Dnmt1 and DNA Methylation in T Cell Development, Function, and Survival. Immunity 2001, 15, 763–774. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keller, E.J.; Dvorina, N.; Jørgensen, T.N. Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent. Int. J. Mol. Sci. 2022, 23, 874. https://doi.org/10.3390/ijms23020874
Keller EJ, Dvorina N, Jørgensen TN. Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent. International Journal of Molecular Sciences. 2022; 23(2):874. https://doi.org/10.3390/ijms23020874
Chicago/Turabian StyleKeller, Emma J., Nina Dvorina, and Trine N. Jørgensen. 2022. "Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent" International Journal of Molecular Sciences 23, no. 2: 874. https://doi.org/10.3390/ijms23020874
APA StyleKeller, E. J., Dvorina, N., & Jørgensen, T. N. (2022). Spontaneous CD4+ T Cell Activation and Differentiation in Lupus-Prone B6.Nba2 Mice Is IFNAR-Independent. International Journal of Molecular Sciences, 23(2), 874. https://doi.org/10.3390/ijms23020874