
The Epithelial–Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression

 ,
, 
 and
and 
Abstract
1. Introduction
2. The Epithelial–Mesenchymal Transition in Cancer
2.1. Cellular and Molecular Changes
2.2. EMT Transcription Factors
2.3. Pathways Regulating EMT
2.4. EMT, Chemoresistance, and Cancer Stem Cells
3. Interplay between EMT and Metabolism
3.1. From EMT to Metabolic Changes
3.2. From Metabolic Changes to EMT
4. Role of EMT and Metabolism in CSCs
5. The Role of LncRNAs in EMT and Metabolism
6. EMT and Metabolism in Selected Cancers
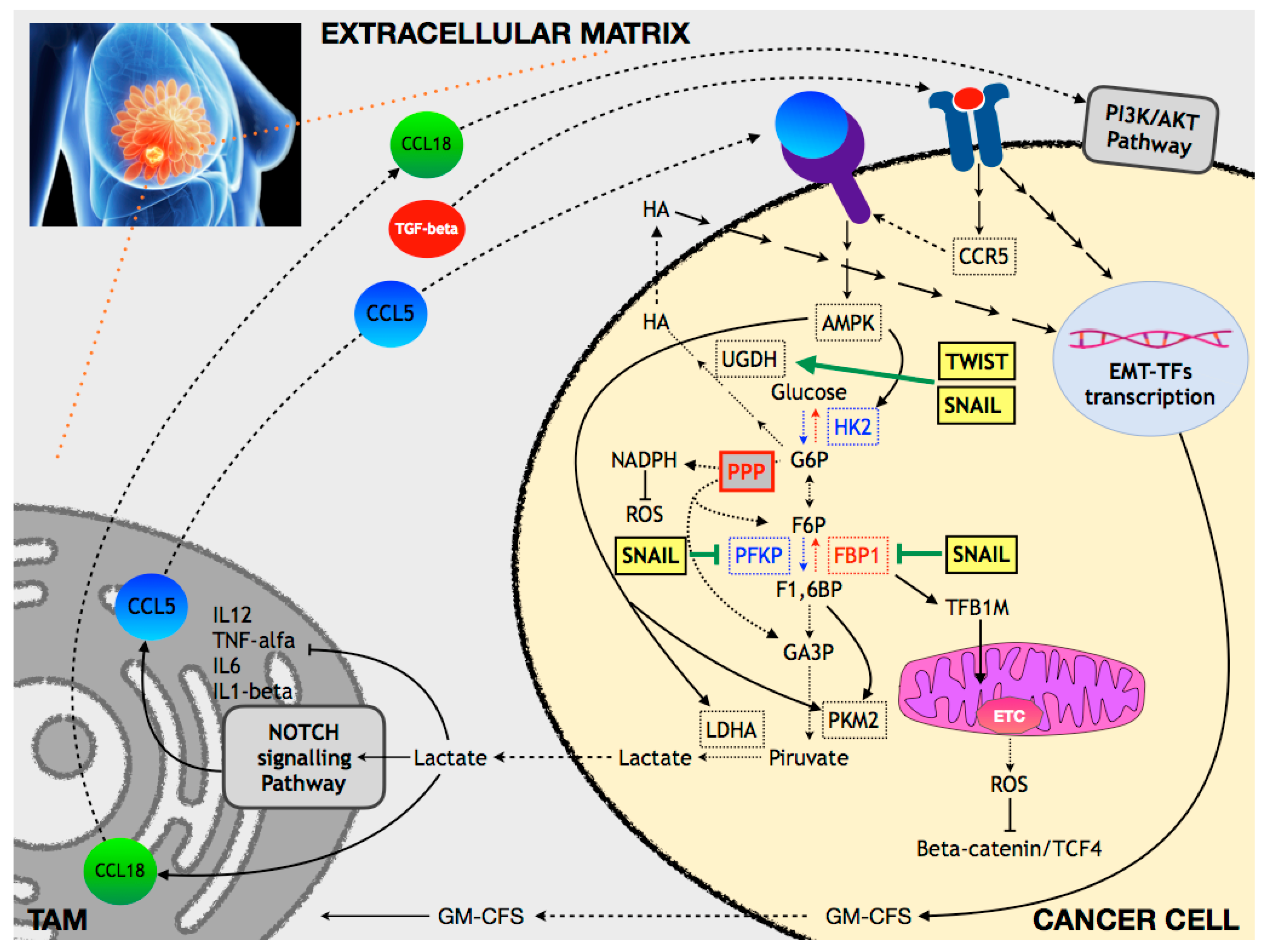
6.1. Breast Cancer
6.1.1. Glucose and OXPHOS Metabolism
6.1.2. Lipid Metabolism
6.1.3. Amino Acid Metabolism
6.1.4. ROS Metabolism
6.2. Lung Cancer
6.2.1. Glucose and OXPHOS Metabolism
6.2.2. Amino Acid Metabolism
6.2.3. ROS Metabolism
6.2.4. Others
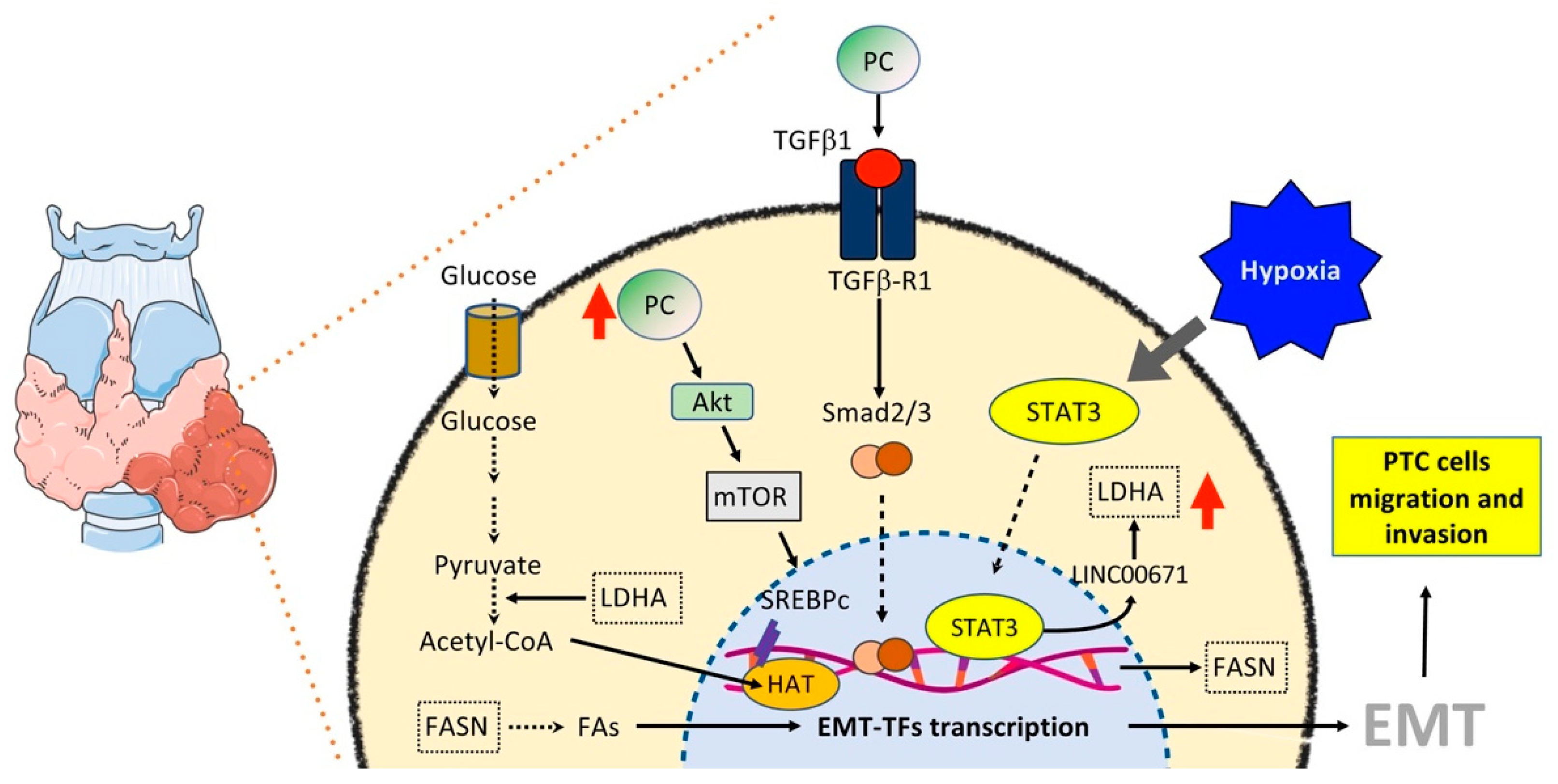
6.3. Thyroid Cancer
6.3.1. Glucose and OXPHOS Metabolism
6.3.2. Lipid Metabolism
7. Therapeutic Perspectives
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta. Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Li, C.F.; Chen, J.Y.; Ho, Y.H.; Hsu, W.H.; Wu, L.C.; Lan, H.Y.; Hsu, D.S.S.; Tai, S.K.; Chang, Y.C.; Yang, M.H. Snail-induced Claudin-11 prompts collective migration for tumour progression. Nat. Cell Biol. 2019, 21, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Rovcanin, B.R.; Gopcevic, K.R.; Kekic, D.L.; Zivaljevic, V.R.; Diklic, A.D.; Paunovic, I.R. Papillary thyroid carcinoma: A malignant tumor with increased antioxidant defense capacity. Tohoku J. Exp. Med. 2016, 240, 101–111. [Google Scholar] [CrossRef]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Robert, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Sun, L.; Li, Q.; Han, X.; Lei, L.; Zhang, H.; Shang, Y. SET8 promotes epithelial-mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 2012, 31, 110–123. [Google Scholar] [CrossRef]
- Sánchez-Tilló, E.; Lázaro, A.; Torrent, R.; Cuatrecasas, M.; Vaquero, E.C.; Castells, A.; Engel, P.; Postigo, A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene 2010, 29, 3490–3500. [Google Scholar] [CrossRef]
- Postigo, A.A.; Depp, J.J.; Taylor, J.J.; Kroll, K.L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 2003, 22, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, S.; Ros, G.; Piazza, S.; Sommaggio, R.; Ciani, Y.; Rosato, A.; Sgarra, R.; Del Sal, G.; Manfioletti, G. HMGA1 promotes metastatic processes in basal-like breast cancer regulating EMT and stemness. Oncotarget 2013, 4, 1293–1308. [Google Scholar] [CrossRef]
- Zhong, J.; Liu, C.; Zhang, Q.H.; Chen, L.; Shen, Y.Y.; Chen, Y.J.; Zeng, X.; Zu, X.Y.; Cao, R.X. TGF-beta1 induces HMGA1 expression: The role of HMGA1 in thyroid cancer proliferation and invasion. Int. J. Oncol. 2017, 50, 1567–1578. [Google Scholar] [CrossRef]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.H.; Moustakas, A.J. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 17, 175–183. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Z.; Shao, C.; Gong, Y.; Hernando, E.; Lee, P.; Narita, M.; Muller, W.; Liu, J.; Wei, J.J. HMGA2 overexpression-induced ovarian surface epithelial transformation is mediated through regulation of EMT genes. Cancer Res. 2011, 71, 349–359. [Google Scholar] [CrossRef]
- Jin, G.H.; Shi, Y.; Tian, Y.; Cao, T.T.; Mao, Y.; Tang, T.Y. HMGA1 accelerates the malignant progression of gastric cancer through stimulating EMT. Eur. Rev. Med. Pharmacol. Sci. 2020, 2, 3642–3647. [Google Scholar]
- Li, Z.; Liu, J.; Chen, T.; Sun, R.; Liu, Z.; Qiu, B.; Xu, Y.; Zhang, Z. HMGA1-TRIP13 axis promotes stemness and epithelial mesenchymal transition of perihilar cholangiocarcinoma in a positive feedback loop dependent on c-Myc. J. Exp. Clin. Cancer Res. 2021, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lu, C.; Wang, W.; Gao, W.; Yu, C. circBANP promotes colorectal cancer growth and metastasis via sponging let-7d-5p to modulate HMGA1/Wnt/beta-catenin signaling. Mol. Ther. Oncolytics 2021, 21, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Cao, G.; Fu, C. HMGA1 interacts with β-catenin to positively regulate Wnt/β-catenin signaling in colorectal cancer cells. Pathol. Oncol. Res. 2014, 2, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Raman, B.K.; Van Slyck, E.J.; Riddle, J.; Sawdyk, M.A.; Abraham, J.P.; Saeed, S.M. Platelet function and structure in myeloproliferative disease, myelodysplastic syndrome, and secondary thrombocytosis. Am. J. Clin. Pathol. 1989, 91, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, M.; Giordano, A. Signal transduction growth factors: The effective governance of transcription and cellular adhesion in cancer invasion. Oncotarget 2017, 8, 36869–36884. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Horiuchi, A.; Wang, C.; Oka, K.; Ohira, S.; Nikaido, T.; Konishi, I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am. J. Pathol. 2003, 163, 1437–1447. [Google Scholar] [CrossRef]
- Shirakihara, T.; Horiguchi, K.; Miyazawa, K.; Ehata, S.; Shibata, T.; Morita, I.; Miyazono, K.; Saitoh, M. TGF-β regulates isoform switching of FGF receptors and epithelial-mesenchymal transition. EMBO J. 2011, 30, 783–795. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Scheel, C.; Eaton, E.N.; Li, S.H.J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Anderle, P.; Cameron, D.; Wirapati, P.; Becette, V.; André, S.; Piccart, M.; Campone, M.; Brain, E.; et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nat. Med. 2009, 15, 68–74. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.K.; Her, N.G.; Lee, M.G.; Ryu, B.K.; Lee, J.H.; Han, J.; Jeong, S.I.; Kang, M.J.; Kim, N.H.; Kim, H.J.; et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Res. 2012, 72, 4097–4109. [Google Scholar] [CrossRef]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.J.; Lin, T.Y.; Lin, X.L.; Liu, Y.; Zhao, W.T.; Li, X.Y.; Lian, M.; Chen, H.W.; Li, Y.L.; Zhang, X.L.; et al. Loss of PDK4 expression promotes proliferation, tumorigenicity, motility and invasion of hepatocellular carcinoma cells. J. Cancer 2020, 11, 4397–4405. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Daemen, A.; Hatzivassiliou, G.; Arnott, D.; Wilson, C.; Zhuang, G.; Gao, M.; Liu, P.; Boudreau, A.; Johnson, L.; et al. Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells. Cancer Metab. 2014, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, N.; Jaumot, J.; Tauler, R.; Bedia, C. Epithelial-to-mesenchymal transition involves triacylglycerol accumulation in DU145 prostate cancer cells. Mol. Biosyst. 2015, 11, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Cha, Y.H.; Lee, J.; Lee, S.H.; Yang, J.H.; Yun, J.S.; Cho, E.S.; Zhang, X.; Nam, M.; Kim, N.; et al. Snail reprograms glucose metabolism by repressing phosphofructokinase PFKP allowing cancer cell survival under metabolic stress. Nat. Commun. 2017, 8, 14374. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Kim, C.H.; Yoon, G.; Han, S.I.; Park, H.G.; Kang, H.S. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012, 72, 3607–3617. [Google Scholar] [CrossRef] [PubMed]
- Hamabe, A.; Konno, M.; Tanuma, N.; Shima, H.; Tsunekuni, K.; Kawamoto, K.; Nishida, N.; Koseki, J.; Mimori, K.; Gotoh, N.; et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2014, 111, 15526–15531. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, K.; Liu, T.; Chen, J.; Lv, W.; Yang, W.; Xu, S.; Wang, X.; Li, L. Decreased glucose bioavailability and elevated aspartate metabolism in prostate cancer cells undergoing epithelial-mesenchymal transition. J. Cell Physiol. 2020, 235, 5602–5612. [Google Scholar] [CrossRef]
- Funasaka, T.; Hogan, V.; Raz, A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial and mesenchymal phenotype conversions in breast cancer. Cancer Res. 2009, 69, 5349–5356. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Aboukameel, A.; Kong, D.; Wang, Z.; Sethi, S.; Chen, W.; Sarkar, F.H.; Raz, A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial-mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. 2011, 7, 3400–3409. [Google Scholar] [CrossRef] [PubMed]
- Funasaka, T.; Hu, H.; Yanagawa, T.; Hogan, V.; Raz, A. Down-regulation of phosphoglucose isomerase/autocrine motility factor results in mesenchymal-to-epithelial transition of human lung fibrosarcoma cells. Cancer Res. 2007, 67, 4236–4243. [Google Scholar] [CrossRef]
- Aspuria, P.P.; Lunt, S.Y.; Väremo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Yan, B.; Liu, S.; Jia, J.; Lai, W.; Xin, X.; Tang, C.E.; Luo, D.; Tan, T.; Jiang, Y.; et al. Chromatin Remodeling Factor LSH Drives Cancer Progression by Suppressing the Activity of Fumarate Hydratase. Cancer Res. 2016, 76, 5743–5755. [Google Scholar] [CrossRef]
- Liu, X.; Qiao, Y.; Ting, X.; Si, W. Isocitrate dehydrogenase 3A, a rate-limiting enzyme of the TCA cycle, promotes hepatocellular carcinoma migration and invasion through regulation of MTA1, a core component of the NuRD complex. Am. J. Cancer Res. 2020, 10, 3212–3229. [Google Scholar]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.; et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Lin, F.; Barrett, R.; Liu, Y.; Jiang, W.; Korpal, M.; Astley, H.; Gitterman, D.; Henley, T.; Howes, R.; et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J. Biol. Chem. 2012, 287, 42180–42194. [Google Scholar] [CrossRef]
- Lin, C.C.; Cheng, T.L.; Tsai, W.H.; Tsai, H.J.; Hu, K.H.; Chang, H.C.; Yeh, C.W.; Chen, Y.C.; Liao, C.C.; Chang, W.T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep. 2012, 2, 785. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef]
- Lee, S.Y.; Jeon, H.M.; Ju, M.K.; Jeong, E.K.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget 2016, 7, 7925–7939. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.C.; Chen, C.K.; Hua, K.T.; Yu, P.; Lee, W.J.; Chen, M.W.; Jeng, Y.M.; Chien, M.H.; Kuo, K.T.; Hsiao, M.; et al. Glutaminase 2 stabilizes Dicer to repress Snail and metastasis in hepatocellular carcinoma cells. Cancer Lett. 2016, 383, 282–294. [Google Scholar] [CrossRef]
- Ramirez-Peña, E.; Arnold, J.; Shivakumar, V.; Joseph, R.; Vidhya Vijay, G.; den Hollander, P.; Bhangre, N.; Allegakoen, P.; Prasad, R.; Conley, Z.; et al. The Epithelial to Mesenchymal Transition Promotes Glutamine Independence by Suppressing GLS2 Expression. Cancers 2019, 11, 1610. [Google Scholar] [CrossRef]
- Recouvreux, M.V.; Moldenhauer, M.R.; Galenkamp, K.M.O.; Jung, M.; James, B.; Zhang, Y.; Lowy, A.; Bagchi, A.; Commisso, C. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J. Exp. Med. 2020, 217, e20200388. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martínez, R.; Cruz-Gil, S.; de Cedrón, M.G.; Álvarez-Fernández, M.; Vargas, T.; Molina, S.; García, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Gil, S.; Sanchez-Martinez, R.; de Cedron, M.G.; Martin-Hernandez, R.; Vargas, T.; Molina, S.; Herranz, J.; Davalos, A.; Reglero, G.; de Molina, A.R. Targeting the lipid metabolic axis ACSL/SCD in colorectal cancer progression by therapeutic miRNAs: miR-19b-1 role. J. Lipid. Res. 2018, 59, 14–24. [Google Scholar] [CrossRef]
- Nath, A.; Li, I.; Roberts, L.R.; Chan, C. Elevated free fatty acid uptake via CD36 promotes epithelial-mesenchymal transition in hepatocellular carcinoma. Sci. Rep. 2015, 5, 14752. [Google Scholar] [CrossRef]
- Martinez-Orozco, R.; Navarro-Tito, N.; Soto-Guzman, A.; Castro-Sanchez, L.; Perez Salazar, E. Arachidonic acid promotes epithelial-to-mesenchymal-like transition in mammary epithelial cells MCF10A. Eur. J. Cell Biol. 2010, 89, 476–488. [Google Scholar] [CrossRef]
- Espinosa-Neira, R.; Mejia-Rangel, J.; Cortes-Reynosa, P.; Salazar, E.P. Linoleic acid induces an EMT-like process in mammary epithelial cells MCF10A. Int. J. Biochem. Cell Biol. 2011, 43, 1782–1791. [Google Scholar] [CrossRef]
- Morandi, A.; Taddei, M.L.; Chiarugi, P.; Giannoni, E. Targeting the Metabolic Reprogramming That Controls Epithelial-to-Mesenchymal Transition in Aggressive Tumors. Front. Oncol. 2017, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Daniel, Y.; Lelou, E.; Aninat, C.; Corlu, A.; Cabillic, F. Interplay between Metabolism Reprogramming and Epithelial-to-Mesenchymal Transition in Cancer Stem Cells. Cancers 2021, 13, 1973. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.M.; Yuan, H.; Cheng, J.; Hunt, A.J. Polarity in stem cell division: Asymmetric stem cell division in tissue homeostasis. Cold Spring Harb. Perspect. Biol. 2010, 2, a001313. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Biagioni, A.; Papucci, L.; Magnelli, L.; Mazzanti, B.; Stecca, B.; Calorini, L. The acidic tumor microenvironment drives a stem-like phenotype in melanoma cells. J. Mol. Med. 2020, 98, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Puca, F.; Colamaio, M.; Federico, A.; Gemei, M.; Tosti, N.; Bastos, A.U.; Del Vecchio, L.; Pece, S.; Battista, S.; Fusco, A. HMGA1 silencing restores normal stem cell characteristics in colon cancer stem cells by increasing p53 levels. Oncotarget 2014, 5, 3234–3245. [Google Scholar] [CrossRef]
- Puca, F.; Tosti, N.; Federico, A.; Kuzay, Y.; Pepe, A.; Morlando, S.; Savarese, T.; D’Alessio, F.; Colamaio, M.; Sarnataro, D.; et al. HMGA1 negatively regulates NUMB expression at transcriptional and post transcriptional level in glioblastoma stem cells. Cell Cycle 2019, 18, 1446–1457. [Google Scholar] [CrossRef]
- Battista, S.; National Research Council (CNR), Institute of Experimental Endocrinology and Oncology “G. Salvatore” (IEOS), Naples, Italy. Personal communication, 2021.
- Schmidt, J.M.; Panzilius, E.; Bartsch, H.S.; Irmler, M.; Beckers, J.; Kari, V.; Linnemann, J.R.; Dragoi, D.; Hirschi, B.; Kloos, U.J.; et al. Stem-cell-like properties and epithelial plasticity arise as stable traits after transient Twist1 activation. Cell Rep. 2015, 10, 131–139. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Fekir, K.; Dubois-Pot-Schneider, H.; Désert, R.; Daniel, Y.; Glaise, D.; Rauch, C.; Morel, F.; Fromenty, B.; Musso, O.; Cabillic, F.; et al. Retrodifferentiation of Human Tumor Hepatocytes to Stem Cells Leads to Metabolic Reprogramming and Chemoresistance. Cancer Res. 2019, 79, 1869–1883. [Google Scholar] [CrossRef]
- Chiefari, E.; Foti, D.P.; Sgarra, R.; Pegoraro, S.; Arcidiacono, B.; Brunetti, F.S.; Greco, M.; Manfioletti, G.; Brunetti, A. Transcriptional Regulation of Glucose Metabolism: The Emerging Role of the HMGA1 Chromatin Factor. Front. Endocrinol. 2018, 9, 357. [Google Scholar] [CrossRef]
- Cao, X.P.; Cao, Y.; Zhao, H.; Yin, J.; Hou, P. HMGA1 promoting gastric cancer oncogenic and glycolytic phenotypes by regulating c-myc expression. Biochem. Biophys. Res. Commun. 2019, 516, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Chiefari, E.; Tanyolac, S.; Paonessa, F.; Pullinger, C.R.; Capula, C.; Iiritano, S.; Mazza, T.; Forlin, M.; Fusco, A.; Durlach, V.; et al. Functional variants of the HMGA1 gene and type 2 diabetes mellitus. JAMA 2011, 305, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shen, W.; Ma, L.; Zhao, M.; Zheng, J.; Bu, S.; Hino, S.; Nakao, M. HMGA2 promotes adipogenesis by activating C/EBPβ-mediated expression of PPARγ. Biochem. Biophys. Res. Commun. 2016, 472, 617–623. [Google Scholar] [CrossRef]
- Morlando, S.; Savarese, T.; D’Alessio, F.; Romano, M.; Fedele, M.; Fusco, A.; Battista, S. Diet affects stem cell fate by modifying NUMB phosphorylation: Role of HMGA1. In Proceedings of the SIBBM 2021—Frontiers in Metabolic research, Online. 7–10 June 2021. [Google Scholar]
- Rafalski, V.A.; Mancini, E.; Brunet, A. Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 2012, 125, 5597–5608. [Google Scholar] [CrossRef] [PubMed]
- Cliff, T.S.; Dalton, S. Metabolic switching and cell fate decisions: Implications for pluripotency, reprogramming and development. Curr. Opin. Genet. Dev. 2017, 46, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.M.; Mitschke, J.; Losada, M.L.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.O.; Berenguer, A.; Prats, N.; Hueto, J.A.; Bescós, C.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Otvos, B.; Sinyuk, M.; Alvarado, A.G.; Hitomi, M.; Stoltz, K.; Wu, Q.; Flavahan, W.; Levison, B.; Johansen, M.L.; et al. Cancer Stem Cell-Specific Scavenger Receptor CD36 Drives Glioblastoma Progression. Stem. Cells 2014, 32, 1746–1758. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.N.; Belli, A.; Di Pietro, V. Small Non-coding RNAs: New Class of Biomarkers and Potential Therapeutic Targets in Neurodegenerative Disease. Front. Genet. 2019, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef]
- Heery, R.; Finn, S.P.; Cuffe, S.; Gray, S.G. Long Non-Coding RNAs: Key regulators of epithelial-mesenchymal transition, tumour drug resistance and cancer stem cells. Cancers 2017, 9, 38. [Google Scholar] [CrossRef]
- Xu, Q.; Deng, F.; Qin, Y.; Zhao, Z.; Wu, Z.; Xing, Z.; Ji, A.; Wang, Q.J. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016, 7, e2254. [Google Scholar] [CrossRef]
- Li, S.P.; Xu, H.X.; Yu, Y.; He, J.D.; Wang, Z.; Xu, Y.J.; Wang, C.Y.; Zhang, H.M.; Zhang, R.X.; Zhang, J.J.; et al. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget 2016, 7, 42431–42446. [Google Scholar] [CrossRef]
- Hua, Q.; Mi, B.; Huang, G. The emerging co-regulatory role of long noncoding RNAs in epithelial mesenchymal transition and the Warburg effect in aggressive tumors. Crit. Rev. Oncol. Hematol. 2018, 126, 112–120. [Google Scholar] [CrossRef]
- Liu, X.; Gan, B. lncRNA NBR2 modulates cancer cell sensitivity to phenformin through GLUT1. Cell Cycle 2016, 15, 3471–3481. [Google Scholar] [CrossRef]
- Liu, Y.W.; Sun, M.; Xia, R.; Zhang, E.B.; Liu, X.H.; Zhang, Z.H.; Xu, T.P.; De, W.; Liu, B.R.; Wang, Z.X. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015, 6, e1802. [Google Scholar] [CrossRef]
- Ma, J.; Fan, Y.; Feng, T.; Chen, F.; Xu, Z.; Li, S.; Lin, Q.; He, X.; Shi, W.; Liu, Y.; et al. HOTAIR regulates HK2expression by binding endogenous miR-125 and miR-143 in oesophageal squamous cell carcinoma progression. Oncotarget 2017, 8, 86410–86422. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Fan, Q.; Yang, L.; Zhang, X.; Ma, Y.; Zong, Z.; Hua, X.; Su, D.; Sun, H.; Li, H.; et al. Promotion of glycolysis by HOTAIR through GLUT1 upregulation via mTOR signaling. Oncol. Rep. 2017, 38, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; de Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, J. Long non-coding RNA HOTAIR functions as miRNA sponge to promote the epithelial to mesenchymal transition in esophageal cancer. Biomed. Pharm. 2017, 90, 888–896. [Google Scholar] [CrossRef]
- Li, J.; Wang, J.; Chen, Y.; Li, S.; Jin, M.; Wang, H.; Chen, Z.; Yu, W. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am. J. Cancer Res. 2016, 6, 1099–1107. [Google Scholar]
- Shi, B.; Wang, Y.; Yin, F. MALAT1/miR-124/Capn4 axis regulates proliferation, invasion and EMT in nasopharyngeal carcinoma cells. Cancer Biol. Ther. 2017, 18, 792–800. [Google Scholar] [CrossRef]
- Luo, F.; Liu, X.; Ling, M.; Lu, L.; Shi, L.; Lu, X.; Li, J.; Zhang, A.; Liu, Q. The lncRNA MALAT1, acting through HIF-1α stabilization, enhances arsenite-induced glycolysis in human hepatic L-02 cells. Biochim. Biophys. Acta. 2016, 1862, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, X.; Wang, H.; Wang, L.; Liu, T.; Du, L.; Yang, Y.; Wang, C. MALAT1 Is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol. Cancer Ther. 2017, 16, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Jia, S.; Wu, M.; An, J.; Zheng, Q.; Zhang, W.; Lu, D. miR675 upregulates long noncoding RNA H19 through activating EGR1 in human liver cancer. Oncotarget 2015, 6, 31958–31984. [Google Scholar] [CrossRef]
- Liang, W.C.; Fu, W.M.; Wong, C.W.; Wang, Y.; Wang, W.M.; Hu, G.X.; Zhang, L.; Xiao, L.J.; Wan, D.C.C.; Zhang, J.F.; et al. The lncRNA H19 promotes epithelial to mesenchymal transition by functioning as miRNA sponges in colorectal cancer. Oncotarget 2015, 6, 22513–22525. [Google Scholar] [CrossRef]
- Ma, C.; Nong, K.; Zhu, H.; Wang, W.; Huang, X.; Yuan, Z.; Ai, K. H19 promotes pancreatic cancer metastasis by derepressing let-7’s suppression on its target HMGA2-mediated EMT. Tumor Biol. 2014, 35, 9163–9169. [Google Scholar] [CrossRef]
- Peng, F.; Wang, J.H.; Fan, W.J.; Meng, Y.T.; Li, M.M.; Li, T.T.; Cui, B.; Wang, H.F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2018, 37, 1119. [Google Scholar] [CrossRef]
- Zhang, D.M.; Lin, Z.Y.; Yang, Z.H.; Wang, Y.Y.; Wan, D.; Zhong, J.L.; Zhuang, P.L.; Huang, Z.Q.; Zhou, B.; Chen, W.L. IncRNA H19 promotes tongue squamous cell carcinoma progression through β-catenin/GSK3β/EMT signaling via association with EZH2. Am. J. Transl. Res. 2017, 9, 3474–3486. [Google Scholar]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef]
- Xue, M.; Pang, H.; Li, X.; Li, H.; Pan, J.; Chen, W. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer Sci. 2016, 107, 18–27. [Google Scholar] [CrossRef]
- Xiao, J.N.; Yan, T.H.; Yu, R.M.; Gao, Y.; Zeng, W.L.; Lu, S.W.; Que, H.X.; Liu, Z.P.; Jiang, J.H. Long non-coding RNA UCA1 regulates the expression of Snail2 by miR-203 to promote hepatocellular carcinoma progression. J. Cancer Res. Clin. Oncol. 2017, 143, 981–990. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Q.; Wang, S.; Zhang, J. miR-485-5p inhibits bladder cancer metastasis by targeting HMGA2. Int. J. Mol. Med. 2015, 36, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Wu, C.H.; Hu, H.Z. LncRNA UCA1 promotes epithelial-mesenchymal transition (EMT) of breast cancer cells via enhancing Wnt/beta-catenin signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2819–2824. [Google Scholar]
- Lei, H.; Gao, Y.; Xu, X. LncRNA TUG1 influences papillary thyroid cancer cell proliferation, migration and EMT formation through targeting miR-145. Acta. Biochim. Biophys. Sin. 2017, 49, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Qiu, K.; Li, M.; Liang, Y. Double-negative feedback loop between long noncoding RNA TUG1 and miR-145 promotes epithelial to mesenchymal transition and radioresistance in human bladder cancer cells. FEBS Lett. 2015, 589, 3175–3181. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Wu, M.H.; Huang, Y.H.; Yeh, C.T.; Cheng, M.L.; Chi, H.C.; Tsai, C.Y.; Chung, I.H.; Chen, C.Y.; Lin, K.H. Taurine up-regulated gene 1 functions as a master regulator to coordinate glycolysis and metastasis in hepatocellular carcinoma. Hepatology 2018, 67, 188–203. [Google Scholar] [CrossRef]
- Zhao, L.; Sun, H.; Kong, H.; Chen, Z.; Chen, B.; Zhou, M. The Lncrna-TUG1/EZH2 axis promotes pancreatic cancer cell proliferation, migration and EMT phenotype formation through sponging Mir-382. Cell Physiol. Biochem. 2017, 42, 2145–2158. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Han, X.; Qi, X.; Jin, X.; Li, X. TUG1 promotes osteosarcoma tumorigenesis by upregulating EZH2 expression via miR-144-3p. Int. J. Oncol. 2017, 51, 1115–1123. [Google Scholar] [CrossRef]
- Song, J.; Wu, X.; Liu, F.; Li, M.; Sun, Y.; Wang, Y.; Wang, C.; Zhu, K.; Jia, X.; Wang, B.; et al. Long non-coding RNA PVT1 promotes glycolysis and tumor progression by regulating miR-497/HK2 axis in osteosarcoma. Biochem. Biophys. Res. Commun. 2017, 490, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Yu, F.; Dong, P.; Wu, L.; Zhang, Y.; Hu, Y.; Zheng, L. Long non-coding RNA PVT1 activates hepatic stellate cells through competitively binding microRNA-152. Oncotarget 2016, 7, 62886–62897. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, J.Q.; Chen, J.Z.; Chen, H.X.; Qiu, F.N.; Yan, M.L.; Chen, Y.L.; Peng, C.H.; Tian, Y.F.; Wang, Y.D. The over expression of long non-coding RNA AnRIL promotes epithelial-mesenchymal transition by activating the ATM-E2F1 signaling pathway in pancreatic cancer: An in vivo and in vitro study. Int. J. Biol. Macromol. 2017, 102, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.W.; Ma, C.; Medoro, L.; Chen, L.; Wang, B.; Gupta, R.; Liu, T.; Yang, X.Z.; Chen, T.T.; Wang, R.Z.; et al. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing sidepopulation stem-like cancer cells. Oncotarget 2016, 7, 61741–61754. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Tang, Y.; Xing, W.; Dong, W.; Wang, Z. LncRNA, CRNDE promotes osteosarcoma cell proliferation, invasion and migration by regulating Notch1 signaling and epithelial-mesenchymal transition. Exp. Mol. Pathol. 2018, 104, 19–25. [Google Scholar] [CrossRef]
- Ellis, B.C.; Graham, L.D.; Molloy, P.L. CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim. Biophys. Acta 2014, 1843, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yan, I.K.; Haga, H.; Patel, T. Modulation of hypoxia-signaling pathways by extracellular linc-RoR. J. Cell Sci. 2014, 127, 1585–1594. [Google Scholar] [PubMed]
- Hou, P.; Zhao, Y.; Li, Z.; Yao, R.; Ma, M.; Gao, Y.; Zhao, L.; Zhang, Y.; Huang, B.; Lu, J. LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis. 2014, 5, e1287. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wang, P.; Hua, Y.; Xi, H.; Meng, Z.; Liu, T.; Chen, Z.; Liu, L. ROR functions as a ceRNA to regulate Nanog expression by sponging miR-145 and predicts poor prognosis in pancreatic cancer. Oncotarget 2016, 7, 1608–1618. [Google Scholar] [CrossRef]
- Zhan, H.X.; Wang, Y.; Li, C.; Xu, J.W.; Zhou, B.; Zhu, J.K.; Han, H.F.; Wang, L.; Wang, Y.S.; Hu, S.Y. LincRNA-ROR promotes invasion, metastasis and tumor growth in pancreatic cancer through activating ZEB1 pathway. Cancer Lett. 2016, 374, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xie, H.; Duan, L.; Zhao, D.; Ding, J.; Jiang, G. Long Non-Coding RNA CASC9 and HIF-1alpha Form A Positive Feedback Loop To Facilitate Cell Proliferation And Metastasis In Lung Cancer. Onco. Targets Ther. 2019, 12, 9017–9027. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, F.; Zhan, H.; Chen, L.; Deng, Q.; Xiong, T.; Li, Y.; Ye, J. lncRNA CASC9 sponges miR-758-3p to promote proliferation and EMT in bladder cancer by upregulating TGF-beta2. Oncol. Rep. 2021, 45, 265–277. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Zhou, Y.; Li, B. Long non-coding RNA CASC9 promotes the progression and development of gastric cancer via regulating miR-370/EGFR axis. Dig. Liver Dis. 2021, 53, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Jiang, L.; Wang, Y.D.; Huang, J.Z.; Yu, M.; Xue, H.Z. lincRNA-p21 inhibits invasion and metastasis of hepatocellular carcinoma through Notch signaling-induced epithelial-mesenchymal transition. Hepatol. Res. 2016, 46, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wei, G.; Xia, H.; Yu, H.; Tang, Q.; Bi, F. Down regulation of lincRNA-p21 contributes to gastric cancer development through Hippo-independent activation of YAP. Oncotarget 2017, 8, 63813–63824. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal regulation of HIF-1α and lincRNAp21 modulates the Warburg effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Y.; Wang, X.; Jiang, C.; Han, S.; Dong, K.; Shen, M.; Xu, D. LincRNA-p21suppresses development of human prostate cancer through inhibition of PKM2. Cell Prolif. 2017, 50, e12395. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Wang, S.; Zhang, H.; Han, H.; Ma, B.; Nan, W. Long noncoding RNA GAS5 suppresses cell growth and epithelial-mesenchymal transition in osteosarcoma by regulating the miR-221/ARHI pathway. J. Cell Biochem. 2017, 118, 4772–4781. [Google Scholar] [CrossRef] [PubMed]
- Barthel, A.; Schmoll, D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E685–E692. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Hussain, F. Higher Glucose Enhances Breast Cancer Cell Aggressiveness. Nutr. Cancer 2020, 72, 734–746. [Google Scholar] [CrossRef]
- Nilchian, A.; Giotopoulou, N.; Sun, W.; Fuxe, J. Different Regulation of Glut1 Expression and Glucose Uptake during the Induction and Chronic Stages of TGFβ1-Induced EMT in Breast Cancer Cells. Biomolecules 2020, 10, 1621. [Google Scholar] [CrossRef]
- Kondaveeti, Y.; Guttilla Reed, I.K.; White, B.A. Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. 2015, 364, 44–58. [Google Scholar] [CrossRef]
- Lunetti, P.; Di Giacomo, M.; Vergara, D.; De Domenico, S.; Maffia, M.; Zara, V.; Capobianco, L.; Ferramosca, A. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J. 2019, 286, 688–709. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.M.; Gu, F.; Ambati, C.R.; Rasaily, U.; Ramirez-Pena, E.; Joseph, R.; Manikkam, M.; San Martin, R.; Charles, C.; Pan, Y.; et al. UDP-glucose 6-dehydrogenase regulates hyaluronic acid production and promotes breast cancer progression. Oncogene 2020, 39, 3089–3101. [Google Scholar] [CrossRef]
- Ran, F.; Zhang, Y.; Shi, Y.; Liu, J.; Li, H.; Ding, L.; Ye, Q. miR-1224-3p Promotes Breast Cancer Cell Proliferation and Migration through PGM5-Mediated Aerobic Glycolysis. J. Oncol. 2021, 2021, 5529770. [Google Scholar] [CrossRef]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.E.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.S.; Kim, N.H.; Yun, J.S.; Cho, S.B.; Kim, H.S.; Yook, J.I. Breast Cancer Subtypes Underlying EMT-Mediated Catabolic Metabolism. Cells 2020, 9, 2064. [Google Scholar] [CrossRef]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Liu, Q.Q.; Huo, H.Y.; Ao, S.; Liu, T.; Yang, L.; Fei, Z.Y.; Zhang, Z.Q.; Ding, L.; Cui, Q.H.; Lin, J.; et al. TGF-β1-induced epithelial-mesenchymal transition increases fatty acid oxidation and OXPHOS activity via the p-AMPK pathway in breast cancer cells. Oncol. Rep. 2020, 44, 1206–1215. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, L.; Wei, P.; Zhou, R.; Hua, C.; Xiao, M.; Tu, Y.; Gu, Z.; Wei, T. PEG-GO@XN nanocomposite suppresses breast cancer metastasis via inhibition of mitochondrial oxidative phosphorylation and blockade of epithelial-to-mesenchymal transition. Eur. J. Pharmacol. 2021, 895, 173866. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Xu, W.; Zeng, H.; He, Z.; Lu, X.; Zuo, D.; Qin, G.; Chen, W. OXPHOS-dependent metabolic reprogramming prompts metastatic potential of breast cancer cells under osteogenic differentiation. Br. J. Cancer 2020, 123, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Fite, K.; Gomez-Cambronero, J. Down-regulation of MicroRNAs (MiRs) 203, 887, 3619 and 182 Prevents Vimentin-triggered, Phospholipase D (PLD)-mediated Cancer Cell Invasion. J. Biol. Chem. 2016, 291, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.; Qiao, X.; Gu, X.; Xue, J.; Han, Y.; Sun, L.; Cui, M.; Liu, C. LIPH promotes metastasis by enriching stem-like cells in triple-negative breast cancer. J. Cell Mol. Med. 2020, 24, 9125–9134. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Kim, N.H.; Yun, J.S.; Cho, E.S.; Cha, Y.H.; Cho, S.B.; Lee, S.H.; Cha, S.Y.; Kim, S.Y.; Choi, J.; et al. Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci. Alliance 2020, 3, e202000683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, H.S.; Sun, H.L.; Liu, H.Y.; Liu, M.Y.; Zhou, Z. KDM5B promotes breast cancer cell proliferation and migration via AMPK-mediated lipid metabolism reprogramming. Exp. Cell Res. 2019, 379, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.C.; Chou, S.K.; Kan, J.Y.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. Solute Carrier Family 27 Member 4 (SLC27A4) Enhances Cell Growth, Migration, and Invasion in Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 3434. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Yoon, J.H.; Long, N.P.; Koo, G.B.; Noh, H.J.; Oh, S.J.; Lee, S.B.; Kim, H.M.; Hong, J.Y.; Lee, W.J.; et al. Spheroid-Induced Epithelial-Mesenchymal Transition Provokes Global Alterations of Breast Cancer Lipidome: A Multi-Layered Omics Analysis. Front. Oncol. 2019, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, C.; Maulucci, G.; Colabianchi, A.; Iacopino, F.; D’Alessio, A.; Maiorana, A.; Palmieri, V.; Papi, M.; De Spirito, M.; Di Leone, A.; et al. Stearoyl-CoA desaturase 1 and paracrine diffusible signals have a major role in the promotion of breast cancer cell migration induced by cancer-associated fibroblasts. Br. J. Cancer 2015, 112, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, C.; D’Alessio, A.; Iacopino, F.; Proietti, G.; Di Leone, A.; Masetti, R.; Sica, G. Pivotal role of human stearoyl-CoA desaturases (SCD1 and 5) in breast cancer progression: Oleic acid-based effect of SCD1 on cell migration and a novel pro-cell survival role for SCD5. Oncotarget 2018, 9, 24364–24380. [Google Scholar] [CrossRef]
- Giudetti, A.M.; De Domenico, S.; Ragusa, A.; Lunetti, P.; Gaballo, A.; Franck, J.; Simeone, P.; Nicolardi, G.; De Nuccio, F.; Santino, A.; et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. BBA Mol. Cell Biol. Lipids 2019, 1864, 344–357. [Google Scholar] [CrossRef]
- Yu, R.; Longo, J.; van Leeuwen, J.E.; Zhang, C.; Branchard, E.; Elbaz, M.; Cescon, D.W.; Drake, R.R.; Dennis, J.W.; Penn, L.Z. Mevalonate Pathway Inhibition Slows Breast Cancer Metastasis via Reduced N-glycosylation Abundance and Branching. Cancer Res. 2021, 81, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Jariyal, H.; Gupta, C.; Andhale, S.; Gadge, S.; Srivastava, A. Comparative stemness and differentiation of luminal and basal breast cancer stem cell type under glutamine-deprivation. J. Cell Commun. Signal. 2021, 15, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Ding, C.K.; Wu, J.; Sjol, J.; Wardell, S.; Spasojevic, I.; George, D.; McDonnell, D.P.; Hsu, D.S.; Chang, J.T.; et al. Cystine addiction of triple-negative breast cancer associated with EMT augmented death signaling. Oncogene 2017, 36, 4235–4242. [Google Scholar] [CrossRef]
- Khatib, A.; Solaimuthu, B.; Ben Yosef, M.; Abu Rmaileh, A.; Tanna, M.; Oren, G.; Schlesinger Frisch, M.; Axelrod, J.H.; Lichtenstein, M.; Shaul, Y.D. The glutathione peroxidase 8 (GPX8)/IL-6/STAT3 axis is essential in maintaining an aggressive breast cancer phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 21420–21431. [Google Scholar] [CrossRef]
- Feng, S.; Zhang, L.; Liu, X.; Li, G.; Zhang, B.; Wang, Z.; Zhang, H.; Ma, H. Low levels of AMPK promote epithelial-mesenchymal transition in lung cancer primarily through HDAC4- and HDAC5-mediated metabolic reprogramming. J. Cell Mol. Med. 2020, 24, 7789–7801. [Google Scholar] [CrossRef]
- Masin, M.; Vazquez, J.; Rossi, S.; Groeneveld, S.; Samson, N.; Schwalie, P.C.; Deplancke, B.; Frawley, L.E.; Gouttenoire, J.; Moradpour, D.; et al. GLUT3 is induced during epithelial-mesenchymal transition and promotes tumor cell proliferation in non-small cell lung cancer. Cancer Metab. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Osumi, H.; Horiguchi, H.; Kadomatsu, T.; Tashiro, K.; Morinaga, J.; Takahashi, T.; Ikeda, K.; Ito, T.; Suzuki, M.; Endo, M.; et al. Tumor cell-derived angiopoietin-like protein 2 establishes a preference for glycolytic metabolism in lung cancer cells. Cancer Sci. 2020, 111, 1241–1253. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, R.; Zhu, W.; Chu, H.; Yu, H.; Wei, P.; Wu, X.; Zhu, H.; Gao, H.; Liang, J.; et al. UDP-glucose accelerates SNAI1 mRNA decay and impairs lung cancer metastasis. Nature 2019, 571, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Szymura, S.J.; Zaemes, J.P.; Allison, D.F.; Clift, S.H.; D’Innocenzi, J.M.; Gray, L.G.; McKenna, B.D.; Morris, B.B.; Bekiranov, S.; LeGallo, R.D.; et al. NF-κB upregulates glutamine-fructose-6-phosphate transaminase 2 to promote migration in non-small cell lung cancer. Cell Commun. Signal. 2019, 17, 24. [Google Scholar] [CrossRef]
- Fu, Q.F.; Liu, Y.; Fan, Y.; Hua, S.N.; Qu, H.Y.; Dong, S.W.; Li, R.L.; Zhao, M.Y.; Zhen, Y.; Yu, X.L.; et al. Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway. J. Hematol. Oncol. 2015, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Kim, J.; Hwang, Y.; Im, S.; Moon, Y.; Kang, D.M. TGF-β suppresses the expression of genes related to mitochondrial function in lung A549 cells. Cell Mol. Biol. 2012, 58, 1763–1767. [Google Scholar] [PubMed]
- Zhang, L.; Yang, Y.; Chai, L.; Bu, H.; Yang, Y.; Huang, H.; Ran, J.; Zhu, Y.; Li, L.; Chen, F.; et al. FRK plays an oncogenic role in non-small cell lung cancer by enhancing the stemness phenotype via induction of metabolic reprogramming. Int. J. Cancer 2020, 146, 208–222. [Google Scholar] [CrossRef]
- Nakasuka, F.; Tabata, S.; Sakamoto, T.; Hirayama, A.; Ebi, H.; Yamada, T.; Umetsu, K.; Ohishi, M.; Ueno, A.; Goto, H.; et al. TGF-β-dependent reprogramming of amino acid metabolism induces epithelial-mesenchymal transition in non-small cell lung cancers. Commun. Biol. 2021, 4, 782. [Google Scholar] [CrossRef]
- Chen, G.; Wu, J.; Li, J.; Wang, J. Identification and Characterization of Glycine Decarboxylase as a Direct Target of Snail in the Epithelial-Mesenchymal Transition of Cancer Cells. Tumor Microenviron. 2021, 1, 55–62. [Google Scholar] [PubMed]
- Wang, Y.; Fu, L.; Cui, M.; Wang, Y.; Xu, Y.; Li, M.; Mi, J. Amino acid transporter SLC38A3 promotes metastasis of non-small cell lung cancer cells by activating PDK1. Cancer Lett. 2017, 393, 8–15. [Google Scholar] [CrossRef]
- Lei, H.M.; Zhang, K.R.; Wang, C.H.; Wang, Y.; Zhuang, G.L.; Lu, L.M.; Zhang, J.; Shen, Y.; Chen, H.Z.; Zhu, L. Aldehyde dehydrogenase 1A1 confers erlotinib resistance via facilitating the reactive oxygen species-reactive carbonyl species metabolic pathway in lung adenocarcinomas. Theranostics 2019, 9, 7122–7139. [Google Scholar] [CrossRef]
- Wang, W.; Hu, Y.; Yang, C.; Zhu, S.; Wang, X.; Zhang, Z.; Deng, H. Decreased NAD Activates STAT3 and Integrin Pathways to Drive Epithelial-Mesenchymal Transition. Mol. Cell Proteom. 2018, 17, 2005–2017. [Google Scholar] [CrossRef]
- Shi, Y.; Xu, Y.; Yao, J.; Yan, C.; Su, H.; Zhang, X.; Chen, E.; Ying, K. MTHFD2 promotes tumorigenesis and metastasis in lung adenocarcinoma by regulating AKT/GSK-3β/β-catenin signalling. J. Cell Mol. Med. 2021, 25, 7013–7027. [Google Scholar] [CrossRef]
- Koufaris, C.; Nilsson, R. Protein interaction and functional data indicate MTHFD2 involvement in RNA processing and translation. Cancer Metab. 2018, 6, 12. [Google Scholar] [CrossRef]
- Green, N.H.; Galvan, D.L.; Badal, S.S.; Chang, B.H.; LeBleu, V.S.; Long, J.; Jonasch, E.; Danesh, F.R. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene 2019, 38, 6211–6225. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, J.; Kong, D.; Wang, X.; Wang, Z.; Liu, J.; Yu, W.; Wu, H.; Long, Z.; Zhang, W.; et al. Silent information regulator 1 suppresses epithelial-to-mesenchymal transition in lung cancer cells via its regulation of mitochondria status. Life Sci. 2021, 280, 119716. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef]
- Haley, J.A.; Ruiz, C.F.; Montal, E.D.; Wang, D.; Haley, J.D.; Girnun, G.D. Decoupling of Nrf2 Expression Promotes Mesenchymal State Maintenance in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1488. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Battista, S.; Cerchia, L. Metabolic Reprogramming in Thyroid Cancer: Role of the Epithelial-Mesenchymal Transition. Endocrines 2021, 2, 427–438. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, Y.; Zhao, Q.; Pan, Y.; Zhang, Y. Value of Pyruvate Carboxylase in Thyroid Fine-Needle Aspiration Wash-Out Fluid for Predicting Papillary Thyroid Cancer Lymph Node Metastasis. Front. Oncol. 2021, 11, 643416. [Google Scholar] [CrossRef] [PubMed]
- Huo, N.; Cong, R.; Sun, Z.J.; Li, W.C.; Zhu, X.; Xue, C.Y.; Chen, Z.; Ma, L.Y.; Chu, Z.; Han, Y.C.; et al. STAT3/LINC00671 axis regulates papillary thyroid tumor growth and metastasis via LDHA-mediated glycolysis. Cell Death Dis. 2021, 12, 799. [Google Scholar] [CrossRef]
- Hou, X.; Shi, X.; Zhang, W.; Li, D.; Hu, L.; Yang, J.; Zhao, J.; Wei, S.; Wei, X.; Ruan, X.; et al. LDHA induces EMT gene transcription and regulates autophagy to promote the metastasis and tumorigenesis of papillary thyroid carcinoma. Cell Death Dis. 2021, 12, 347. [Google Scholar] [CrossRef]
- Ren, H.; Song, Z.; Chao, C.; Mao, W. circCCDC66 promotes thyroid cancer cell proliferation, migratory an invasive abilities and glycolysis through the miR-211-5p/PDK4 axis. Oncol. Lett. 2021, 21, 416. [Google Scholar] [CrossRef]
- Qu, N.; Hu, J.Q.; Liu, L.; Zhang, T.T.; Sun, G.H.; Shi, R.L.; Ji, Q.H. SIRT6 is upregulated and associated with cancer aggressiveness in papillary thyroid cancer via BRAF/ERK/Mcl1 pathway. Int. J. Oncol. 2017, 50, 1683–1692. [Google Scholar] [CrossRef]
- Yang, Z.; Yu, W.; Huang, R.; Ye, M.; Min, Z. SIRT6/HIF-1a axis promotes papillary thyroid cancer progression by inducing epithelial-mesenchymal transition. Cancer Cell Int. 2019, 19, 17. [Google Scholar] [CrossRef]
- Yu, W.; Yang, Z.; Huang, R.; Min, Z.; Ye, M. SIRT6 promotes the Warburg effect of papillary thyroid cancer cell BCPAP through reactive oxygen species. Onco. Targets Ther. 2019, 12, 2861–2868. [Google Scholar] [CrossRef]
- Revilla, G.; de Pablo Pons, M.; Baila-Rueda, L.; García-León, A.; Santos, D.; Cenarro, A.; Magalhaes, M.; Blanco, R.M.; Moral, A.; Pérez, J.I.; et al. Cholesterol and 27-hydrocholesterol promote thyroid carcinoma aggressiveness. Sci. Rep. 2019, 9, 10260. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, X.; Pan, Y.; Liu, Y.; Zhang, Y. Pyruvate carboxylase promotes thyroid cancer aggressiveness through fatty acid synthesis. BMC Cancer 2021, 21, 722. [Google Scholar] [CrossRef]
- Chiappetta, G.; Valentino, T.; Vitiello, M.; Pasquinelli, R.; Monaco, M.; Palma, G.; Sepe, R.; Luciano, A.; Pallante, P.; Palmieri, D.; et al. PATZ1 acts as a tumor suppressor in thyroid cancer via targeting p53-dependent genes involved in EMT and cell migration. Oncotarget 2015, 6, 5310–5323. [Google Scholar] [CrossRef]
- Chiappetta, G.; Vinh, J. (CNRS, Paris, France); Paris, D.; Motta, A. (IBC-CNR, Naples, Italy); Fedele, M. (IEOS-CNR, Naples, Italy). Personal communication, 2021.
- Sun, N.Y.; Yang, M.H. Metabolic Reprogramming and Epithelial-Mesenchymal Plasticity: Opportunities and Challenges for Cancer Therapy. Front. Oncol. 2020, 10, 792. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Couto, K.; Jha, A.; Choe, S.; Wang, A.; Woo, H.K.; Steadman, M.; DeLaBarre, B.; Gross, S.; Driggers, E.; et al. Mesenchymal phenotype predisposes lung cancer cells to impaired proliferation and redox stress in response to glutaminase inhibition. PLoS ONE 2014, 9, e115144. [Google Scholar] [CrossRef]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797. [Google Scholar] [CrossRef]
- Lim, J.C.W.; Kwan, Y.P.; Tan, M.S.; Teo, M.H.Y.; Chiba, S.; Wahli, W.; Wang, X. The role of PPARbeta/delta in melanoma metastasis. Int. J. Mol. Sci. 2018, 19, 2860. [Google Scholar] [CrossRef]
- Lin, S.J.; Yang, D.R.; Wang, N.; Jiang, M.; Miyamoto, H.; Li, G.; Chang, C. TR4 nuclear receptor enhances prostate cancer initiation via altering the stem cell population and EMT signals in the PPARG-deleted prostate cells. Oncoscience 2015, 2, 142–150. [Google Scholar] [CrossRef]
- Liu, H.; Ma, Y.; He, H.W.; Zhao, W.L.; Shao, R.G. SPHK1 (sphingosine kinase 1) induces epithelial-mesenchymal transition by promoting the autophagy-linked lysosomal degradation of CDH1/E-cadherin in hepatoma cells. Autophagy 2017, 13, 900–913. [Google Scholar] [CrossRef]
- Zeng, Y.; Yao, X.; Chen, L.; Yan, Z.; Liu, J.; Zhang, Y.; Feng, T.; Wu, J.; Liu, X. Sphingosine-1-phosphate induced epithelial-mesenchymal transition of hepatocellular carcinoma via anMMP-7/syndecan-1/TGF-beta autocrine loop. Oncotarget 2016, 7, 63324–63337. [Google Scholar] [CrossRef]
- Lin, J.L.; Wang, M.J.; Lee, D.; Liang, C.C.; Lin, S. Hypoxia-inducible factor-1alpha regulates matrix metalloproteinase-1 activity in human bone marrow-derived mesenchymal stem cells. FEBS Lett. 2008, 582, 2615–2619. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, Y.L.; Liang, X.H. EMT: A new vision of hypoxia promoting cancer progression. Cancer Biol. Ther. 2011, 11, 714–723. [Google Scholar] [CrossRef]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef]
- Suzuki, A.; Maeda, T.; Baba, Y.; Shimamura, K.; Kato, Y. Acidic extracellular pH promotes epithelial mesenchymal transition in Lewis lung carcinoma model. Cancer Cell Int. 2014, 14, 129. [Google Scholar] [CrossRef]
- Lee, S.H.; McIntyre, D.; Honess, D.; Hulikova, A.; Pacheco-Torres, J.; Cerdán, S.; Swietach, P.; Harris, A.L.; Griffiths, J.R. Carbonic anhydrase IX is a pH-stat that sets an acidic tumour extracellular pH in vivo. Br. J. Cancer 2018, 119, 622–630. [Google Scholar] [CrossRef]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Answar, M.S.; Billingham, M.; et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef]
- Vergara, D.; Ravaioli, S.; Fonzi, E.; Adamo, L.; Damato, M.; Bravaccini, S.; Pirini, F.; Gaballo, A.; Barbano, R.; Pasculli, B.; et al. Carbonic Anhydrase XII Expression Is Modulated during Epithelial Mesenchymal Transition and Regulated through Protein Kinase C Signaling. Int. J. Mol. Sci. 2020, 21, 715. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013, 12, 1791–1801. [Google Scholar] [CrossRef]
- Chafe, S.C.; Vizeacoumar, F.S.; Venkateswaran, G.; Nemirovsky, O.; Awrey, S.; Brown, W.S.; McDonald, P.C.; Carta, F.; Metcalfe, A.; Karasinska, J.M.; et al. Genome-wide synthetic lethal screen unveils novel CAIX-NFS1/xCT axis as a targetable vulnerability in hypoxic solid tumors. Sci. Adv. 2021, 7, eabj0364. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chia, S.; Bedard, P.L.; Chu, Q.; Lyle, M.; Tang, L.; Singh, M.; Zhang, Z.; Supuran, C.T.; Renouf, D.J.; et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients With Advanced Solid Tumors. Am. J. Clin. Oncol. 2020, 43, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Chafe, S.C.; McDonald, P.C.; Saberi, S.; Nemirovsky, O.; Venkateswaran, G.; Burugu, S.; Gao, D.; Delaidelli, A.; Kyle, A.H.; Baker, J.H.E.; et al. Targeting Hypoxia-Induced Carbonic Anhydrase IX Enhances Immune-Checkpoint Blockade Locally and Systemically. Cancer Immunol. Res. 2019, 7, 1064–1078. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Brown, W.S.; Saberi, S.; Swayampakula, M.; Venkateswaran, G.; Nemirovsky, O.; Gillespie, J.A.; Karasinska, J.M.; Kalloger, S.E.; et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Libby, C.J.; et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2, e92928. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNA | EMT | Warburg Effect |
|---|---|---|
| PROMOTERS | ||
| HOTAIR | ||
| MALAT1 |
|
|
| H19 |
| |
| UCA1 |
|
|
| TUG1 |
| |
| PVT1 |
|
|
| ANRIL |
|
|
| CRNDE |
|
|
| ROR |
|
|
| CASC9 |
| |
| INHIBITORS | ||
| LincRNA-p21 | ||
| GAS5 |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedele, M.; Sgarra, R.; Battista, S.; Cerchia, L.; Manfioletti, G. The Epithelial–Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. Int. J. Mol. Sci. 2022, 23, 800. https://doi.org/10.3390/ijms23020800
Fedele M, Sgarra R, Battista S, Cerchia L, Manfioletti G. The Epithelial–Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. International Journal of Molecular Sciences. 2022; 23(2):800. https://doi.org/10.3390/ijms23020800
Chicago/Turabian StyleFedele, Monica, Riccardo Sgarra, Sabrina Battista, Laura Cerchia, and Guidalberto Manfioletti. 2022. "The Epithelial–Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression" International Journal of Molecular Sciences 23, no. 2: 800. https://doi.org/10.3390/ijms23020800
APA StyleFedele, M., Sgarra, R., Battista, S., Cerchia, L., & Manfioletti, G. (2022). The Epithelial–Mesenchymal Transition at the Crossroads between Metabolism and Tumor Progression. International Journal of Molecular Sciences, 23(2), 800. https://doi.org/10.3390/ijms23020800