
A Hot Topic: Cancer Immunotherapy and Natural Killer Cells



{kind=link}
Abstract
:1. Introduction
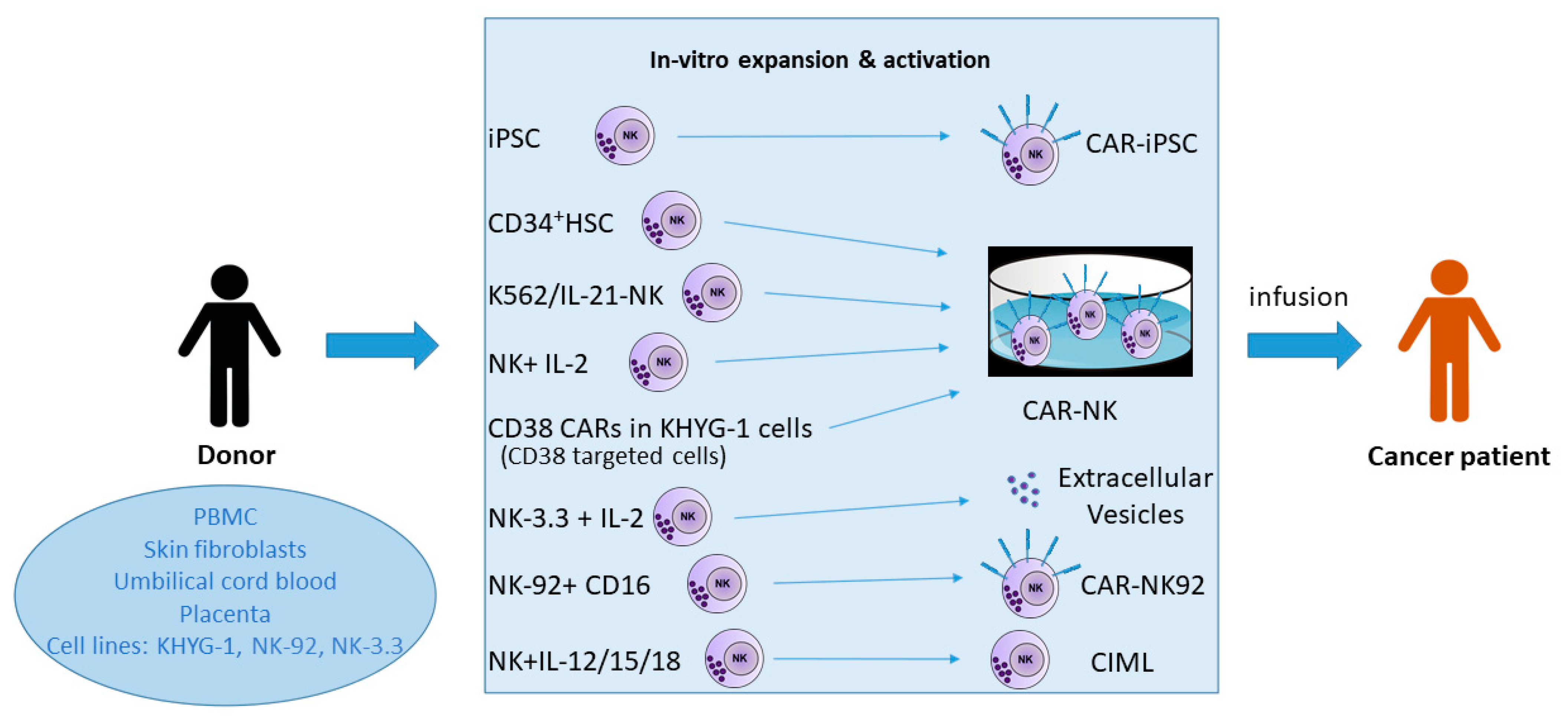
2. Natural Killer Cell Sources for Immunotherapy
3. Methods for the Massive Expansion of NK Cells for Immunotherapy
4. Adoptive Transfer of NK Cells
5. Harnessing of Autologous NK Cells
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Molgora, M.; Cortez, V.S.; Colonna, M. Killing the invaders: NK cell impact in tumors and anti-tumor therapy. Cancers 2021, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hu, Y.; Xiao, W.; Tian, Z. Chimeric antigen receptor- and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell. Mol. Immunol. 2021, 18, 2083–2100. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Guo, F.; Wang, Y.; Cui, J. NK cell therapy: A rising star in cancer treatment. Cancers 2021, 13, 4129. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Oldham, R.K. Natural killer cells: Artifact to reality: An odyssey in biology. Cancer Metast. Rev. 1983, 2, 323–336. [Google Scholar] [CrossRef]
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469. [Google Scholar] [CrossRef]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56bright NK cells: An update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- Amand, M.; Iserentant, G.; Poli, A.; Sleiman, M.; Fievez, V.; Sanchez, I.P.; Sauvageot, N.; Michel, T.; Aouali, N.; Janji, B.; et al. Human CD56dimCD16dim cells as an individualized natural killer cell subset. Front. Immunol. 2017, 8, 699. [Google Scholar] [CrossRef]
- Noschka, R.; Wondany, F.; Kizilsavas, G.; Weil, T.; Weidinger, G.; Walther, P.; Michaelis, J.; Stenger, S. Gran1: A granulysin-derived peptide with potent activity against intracellular Mycobacterium tuberculosis. Int. J. Mol. Sci. 2021, 22, 8392. [Google Scholar] [CrossRef] [PubMed]
- Sojka, D.K.; Plougastel-Douglas, B.; Yang, L.; Pak-Wittel, M.A.; Artyomov, M.N.; Ivanova, Y.; Zhong, C.; Chase, J.M.; Rothman, P.B.; Yu, J.; et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. eLife 2014, 3, e01659. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Mundy-Bosse, B.L.; Yu, J.; Caligiuri, M.A. The broad spectrum of human natural killer cell diversity. Immunity 2017, 47, 820–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogra, P.; Rancan, C.; Ma, W.; Toth, M.; Senda, T.; Carpenter, D.J.; Kubota, M.; Matsumoto, R.; Thapa, P.; Szabo, P.A.; et al. Tissue determinants of human NK cell development, function, and residence. Cell 2020, 180, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Von Andrian, U.H. NK cell memory: Discovery of a mystery. Nat. Immunol. 2021, 22, 669–671. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tian, Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell. Mol. Immunol. 2016, 14, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, J.; Hsu, K.C. Natural killer cell education and the response to infection and cancer therapy: Stay tuned. Trends Immunol. 2018, 39, 222–239. [Google Scholar] [CrossRef]
- Xu, Z.; Zhu, X.; Su, L.; Zou, C.; Chen, X.; Hou, Y.; Gong, C.; Ng, W.; Ni, Z.; Wang, L.; et al. A high-throughput assay for screening natural products that boost NK cell-mediated killing of cancer cells. Pharm. Biol. 2020, 58, 357–366. [Google Scholar] [CrossRef]
- Turin, I.; Delfanti, S.; Ferulli, F.; Brugnatelli, S.; Tanzi, M.; Maestri, M.; Cobianchi, L.; Lisini, D.; Luinetti, O.; Paulli, M.; et al. In vitro killing of colorectal carcinoma cells by autologous activated NK Cells is boosted by anti-epidermal growth factor receptor-induced ADCC regardless of RAS mutation status. J. Immunother. 2018, 41, 190–200. [Google Scholar] [CrossRef]
- Barberi, C.; De Pasquale, C.; Allegra, A.; Sidoti Migliore, G.; Oliveri, D.; Loiacono, F.; Innao, V.; Musolino, C.; Pende, D.; Cantoni, C.; et al. Myeloma cells induce the accumulation of activated CD94low NK cells by cell-to-cell contacts involving CD56 molecules. Blood Adv. 2020, 4, 2297–2307. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y.; et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Mensali, N.; Dillard, P.; Fayzullin, A.; Köksal, H.; Gaudernack, G.; Kvalheim, G.; Inderberg, E.M.; Wälchli, S. “Built-in” PD-1 blocker to rescue NK-92 activity from PD-L1-mediated tumor escape mechanisms. FASEB J. 2021, 35, e21750. [Google Scholar] [CrossRef]
- Biederstädt, A.; Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 2021, 114, 554–571. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Lotze, M.T.; Muul, L.M.; Chang, A.E.; Avis, F.P.; Leitman, S.; Linehan, W.M.; Robertson, C.N.; Lee, R.E.; Rubin, J.T. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N. Engl. J. Med. 1987, 316, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Riley, J.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin. Cancer Res. 2011, 17, 6287–6297. [Google Scholar] [CrossRef] [Green Version]
- Ljunggren, H.G.; Karre, K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol. Today 1990, 11, 237–244. [Google Scholar] [CrossRef]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; Della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Asp. Med. 2021, 80, 100870. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Defor, L.C. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy-advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Tam, Y.K.; Maki, G.; Miyagawa, B.; Hennemann, B.; Tonn, T.; Klingemann, H.G. Characterization of genetically altered, interleukin 2-independent natural killer cell lines suitable for adoptive cellular immunotherapy. Hum. Gene Ther. 1999, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Morillon, Y.M., 2nd; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef] [Green Version]
- Park, H.R.; Ahn, Y.O.; Kim, T.M.; Kim, S.; Kim, S.; Lee, Y.S.; Kim, M.; Keam, B.; Kim, D.W.; Heo, D.S. NK92-CD16 cells are cytotoxic to non-small cell lung cancer cell lines that have acquired resistance to tyrosine kinase inhibitors. Cytotherapy 2019, 21, 603–611. [Google Scholar] [CrossRef]
- Stikvoort, A.S.; van der Schans, J.; Sarkar, S.; Poels, R.; Ruiter, R.; Naik, J.; Yuan, H.; de Bruijn, J.D.; van de Donk, N.W.C.J.; Zweegman, S.; et al. CD38-specific chimeric antigen receptor expressing natural killer KHYG-1 cells: A proof of concept for an “Off the Shelf” therapy for multiple myeloma. Hemasphere 2021, 5, e596. [Google Scholar] [CrossRef] [PubMed]
- Mahle, N.H.; Radcliff, G.; Sevilla, C.L.; Kornbluth, J.; Callewaert, D.M. Kinetics of cellular cytotoxicity mediated by a cloned human natural killer cell line. Immunobiology 1989, 179, 230–243. [Google Scholar] [CrossRef]
- Cochran, A.M.; Kornbluth, J. Extracellular vesicles from the human natural killer cell line NK3.3 have broad and potent anti-tumor activity. Front. Cell Dev. Biol. 2021, 9, 698639. [Google Scholar] [CrossRef]
- Federici, C.; Shahaj, E.; Cecchetti, S.; Camerini, S.; Casella, M.; Iessi, E.; Camisaschi, C.; Paolino, G.; Calvieri, S.; Ferro, S.; et al. Natural-killer-derived extracellular vesicles: Immune sensors and interactors. Front. Immunol. 2020, 11, 262. [Google Scholar] [CrossRef] [Green Version]
- Gunesch, J.T.; Angelo, L.S.; Mahapatra, S.; Deering, R.P.; Kowalko, J.E.; Sleiman, P.; Tobias, J.W.; Monaco-Shawver, L.; Orange, J.S.; Mace, E.M. Genome-wide analyses and functional profiling of human NK cell lines. Mol. Immunol. 2019, 115, 64–75. [Google Scholar] [CrossRef]
- Alici, E.; Sutlu, T.; Björkstrand, B.; Gilljam, M.; Stellan, B.; Nahi, H.; Concha Quezada, H.; Gahrton, G.; Ljunggren, H.G.; Dirac, M.S. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood 2008, 111, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- Harada, H.; Saijo, K.; Watanabe, S.; Tsuboi, K.; Nose, T.; Ishiwata, I.; Ohno, T. Selective expansion of human natural killer cells from peripheral blood mononuclear cells by the cell line, HFWT. JPN J. Cancer Res. 2002, 93, 303–319. [Google Scholar] [CrossRef]
- Harada, H.; Watanabe, S.; Saijo, K.; Ishiwata, I.; Ohno, T. A Wilms tumor cell line, HFWT, can greatly stimulate proliferation of CD56+ human natural killer cells and their novel precursors in blood mononuclear cells. Exp. Hematol. 2004, 32, 614–621. [Google Scholar] [CrossRef]
- Ahn, Y.O.; Kim, S.; Kim, T.M.; Song, E.Y.; Park, M.H.; Heo, D.S. Irradiated and activated autologous PBMCs induce expansion of highly cytotoxic human NK cells in vitro. J. Immunother. 2013, 36, 373–381. [Google Scholar] [CrossRef]
- Klöß, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V. Optimization of human NK cell manufacturing: Fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef]
- Zimmer, J.; Donato, L.; Hanau, D.; Cazenave, J.P.; Tongio, M.M.; Moretta, A.; de la Salle, H. Activity and phenotype of natural killer cells in peptide transporter (TAP)-deficient patients (type I bare lymphocyte syndrome). J. Exp. Med. 1998, 187, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perussia, B.; Ramoni, C.; Anegon, I.; Cuturi, M.C.; Faust, J.; Trinchieri, G. Preferential proliferation of natural killer cells among peripheral blood mononuclear cells cocultured with B lymphoblastoid cell lines. Nat. Immun. Cell Growth Regul. 1987, 6, 171–188. [Google Scholar] [PubMed]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef] [Green Version]
- Shimasaki, N.; Coustan-Smith, E.; Kamiya, T.; Campana, D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy 2016, 18, 1422–1434. [Google Scholar] [CrossRef]
- Somanchi, S.S.; Senyukov, V.V.; Denman, C.J.; Lee, D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2011, 48, 2540. [Google Scholar] [CrossRef] [PubMed]
- Denman, C.J.; Senyukov, V.V.; Somanchi, S.S.; Phatarpekar, P.V.; Kopp, L.M.; Johnson, J.L.; Singh, H.; Hurton, L.; Maiti, S.N.; Huls, M.H.; et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 2012, 7, e30264. [Google Scholar] [CrossRef]
- Ojo, E.O.; Sharma, A.A.; Liu, R.; Moreton, S.; Checkley-Luttge, M.A.; Gupta, K.; Lee, G.; Lee, D.A.; Otegbeye, F.; Sekaly, R.P.; et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci. Rep. 2019, 9, 14916. [Google Scholar] [CrossRef] [Green Version]
- Thangaraj, J.L.; Phan, M.T.; Kweon, S.; Kim, J.; Lee, J.M.; Hwang, I.; Park, J.; Doh, J.; Lee, S.H.; Vo, M.C.; et al. Expansion of cytotoxic natural killer cells in multiple myeloma patients using K562 cells expressing OX40 ligand and membrane-bound IL-18 and IL-21. Cancer Immunol. Immunother. 2021. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Yang, B.; Kim, Y.S.; Park, G.M.; Kim, H.; Kim, H.; Kim, F.J.; Hwang, H.K.; Shin, E.C.; Cho, S. Harnessing novel engineered feeder cells expressing activating molecules for optimal expansion of NK cells with potent antitumor activity. Cell. Mol. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchick, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Vago, L.; Eikema, D.J.; de Wreede, L.C.; Ciceri, F.; Diaz, M.A.; Locatelli, F.; Jindra, P.; Milone, G.; Diez-Martin, J.L.; et al. Natural killer cell alloreactivity in HLA-haploidentical hematopoietic transplantation: A study on behalf of the CTIWP of the EBMT. Bone Marrow Transplant 2021, 56, 1900–1907. [Google Scholar] [CrossRef]
- Shah, N.N.; Baird, K.; Delbrook, C.P.; Fleisher, T.A.; Kohler, M.E.; Rampertaap, S.; Lemberg, K.; Hurley, C.K.; Kleiner, D.E.; Merchant, M.S.; et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood 2015, 125, 784–792. [Google Scholar] [CrossRef]
- Mamo, T.; Williams, S.M.; Kinney, S.; Tessier, K.M.; DeFor, T.E.; Cooley, S.; Miller, J.S.; McKenna, D.H. Infusion reactions in natural killer cell immunotherapy: A retrospective review. Cytotherapy 2021, 23, 627–634. [Google Scholar] [CrossRef]
- Norelli, M.; Camisa, B.; Barbiera, J.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef]
- Lee, D.A. The off-target effects of nonspecific NK cells. Blood 2015, 125, 744–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, A.; Michel, T.; Patil, N.; Zimmer, J. Revisiting the functional impact of NK cells. Trends Immunol. 2018, 39, 460–472. [Google Scholar] [CrossRef]
- Silla, L.; Valim, V.; Pezzi, A.; da Silva, M.; Wilke, I.; Nobrega, J.; Vargas, A.; Armorin, B.; Correa, B.; Zambonato, B.; et al. Adoptive immunotherapy with double-bright (CD56bright/CD16bright expanded natural killer cells in patients with relapsed or refractory acute myeloid leukemia: A proof-of-concept study. Br. J. Haematol. 2021, 195, 710–721. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.M.; Cashen, A.F.; Cubitt, C.C.; Neal, C.C.; Wong, P.; Wagner, J.A.; Foster, M.; Schappe, T.; Desai, S.; McClain, E.; et al. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 2020, 10, 1854–1871. [Google Scholar] [CrossRef]
- Marin, N.D.; Krasnick, B.A.; Becker-Hapak, M.; Conant, L.; Goedegebuure, S.P.; Berrien-Elliott, M.M.; Robbins, K.J.; Foltz, J.A.; Foster, M.; Wong, P. Memory-like differentiation enhances NK cell responses to melanoma. Clin. Cancer Res. 2021, 27, 4859–4869. [Google Scholar] [CrossRef]
- Mikelez-Alonso, I.; Magadán, S.; González-Fernández, Á.; Borrego, F. Natural killer (NK) cell-based immunotherapies and the many faces of NK cell memory: A look into how nanoparticles enhance NK cell activity. Adv. Drug Deliv. Rev. 2021, 176, 113860. [Google Scholar] [CrossRef]
- Van Vliet, A.A.; Georgoudaki, A.M.; Raimo, M.; de Gruijl, T.D.; Spanholtz, J. Adoptive NK cell therapy: A promising treatment prospect for metastatic melanoma. Cancers 2021, 13, 4722. [Google Scholar] [CrossRef]
- Barnes, S.; Schilizzi, O.; Audsley, K.M.; Newnes, H.V.; Foley, B. Deciphering the immunological phenomenon of adaptive natural killer (NK) cells and cytomegalovirus (CMV). Int. J. Mol. Sci. 2020, 21, 8864. [Google Scholar] [CrossRef]
- Merino, A.M.; Kim, H.; Miller, J.S.; Cichocki, F. Unraveling exhaustion in adaptive and conventional NK cells. J. Leukoc. Biol. 2020, 108, 1361–1368. [Google Scholar] [CrossRef]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.; Blazar, B.R.; Miller, J.S. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Hippen, K.L.; Lemire, A.; Hying, S.; Luo, X.; Lenvik, T.; Curtsinger, J.; Davis, Z.; Zhang, B.; Cooley, S.; et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol. Res. 2018, 6, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Q.; Rückert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Liu, L.L.; Beziat, V.; Oei, V.Y.; Pfefferle, A.; Schaffer, M.; Lehmann, S.; Hellström-Lindberg, E.; Söderhäll, S.; Heyman, M.; Grandér, D.; et al. Ex vivo expanded adaptive NK cells effectively kill primary acute lymphoblastic leukemia cells. Cancer Immunol. Res. 2017, 5, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Cichocki, F.; Valamehr, B.; Bjordahl, R.; Zhang, B.; Rezner, B.; Rogers, P.; Gaidarova, S.; Moreno, S.; Tuininga, K.; Dougherty, P.; et al. GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res. 2017, 77, 5664–5675. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, P.; Kim, S.I. iPSC-derived natural killer cells for cancer immunotherapy. Mol. Cells 2021, 44, 541–548. [Google Scholar] [CrossRef]
- Lu, S.J.; Feng, Q. CAR-NK cells from engineered pluripotent stem cells: Off-the-shelf therapeutics for all patients. Stem. Cells Transl. Med. 2021, 10, S10–S17. [Google Scholar] [CrossRef]
- Morgan, M.A.; Kloos, A.; Lenz, D.; Kattre, N.; Nowak, J.; Bentele, M.; Keisker, M.; Dahlke, J.; Zimmermann, K.; Sauer, M.; et al. Improved activity against acute myeloid leukemia with chimeric antigen receptor (CAR)-NK-92 cells designed to target CD123. Viruses 2021, 13, 1365. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Suerth, J.D.; Maetzig, T.; Brugman, M.H.; Heinz, N.; Appelt, J.U.; Kaufmann, K.B.; Schmidt, M.; Grez, M.; Modlich, U.; Baum, C.; et al. Alpharetroviral self-inactivating vectors: Long-term transgene expression in murine hematopoietic cells and low genotoxicity. Mol. Ther. 2012, 20, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zheng, H.; Diao, Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef] [Green Version]
- Vogler, M.; Shanmugalingam, S.; Särchen, V.; Reindl, L.M.; Grèze, V.; Buchinger, L.; Kühn, M.; Ullrich, E. Unleashing the power of NK cells in anticancer immunotherapy. J. Mol. Med. 2021. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Lee, C.C.; Cho, D.Y.; Wang, Y.L.; Chen, C.Y.; Weng, C.Y.; Chiu, S.C.; Hung, M.C.; Wang, S.C. Suppression of breast cancer cells resistant to a pure anti-estrogen with CAR-transduced natural killer cells. Am. J. Cancer Res. 2021, 11, 44569. [Google Scholar]
- Hintz, H.H.; Snyder, K.M.; Wu, J.; Hullsiek, R.; Dahlvang, J.D.; Hart, G.T.; Walcheck, B.; LeBeau, A.M. Simultaneous engagement of tumor and stroma targeting antibodies by engineered NK-92 cells expressing CD64 controls prostate cancer growth. Cancer Immunol. Res. 2021, 9, 1270–1282. [Google Scholar] [CrossRef]
- Lamb, M.G.; Rangarajan, H.G.; Tullius, B.P.; Lee, D.A. Natural killer cell therapy for hematologic malignancies: Successes, challenges, and the future. Stem. Cell Res. Ther. 2021, 12, 211. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G. Anti-CTLA-4 immunotherapy: Uncoupling toxicity and efficacy. Cell Res. 2018, 28, 501–502. [Google Scholar] [CrossRef]
- Liu, B.; Song, Y.; Liu, D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Enqvist, M.; Jacobs, B.; Junlén, H.R.; Schaffer, M.; Melén, C.M.; Friberg, D.; Engelbrekt Wahlin, B.; Malmberg, K.J. Systemic and intra-nodal activation of NK cells after rituximab monotherapy for follicular lymphoma. Front. Immunol. 2019, 10, 2085. [Google Scholar] [CrossRef]
- Goldsmith, S.R.; Foley, N.; Schroeder, M.A. Daratumumab for the treatment of multiple myeloma. Drugs Today 2021, 57, 591–605. [Google Scholar] [CrossRef]
- Pazina, T.; James, A.M.; Colby, K.B.; Yang, Y.; Gale, A.; Jhatakia, A.; Kearney, A.Y.; Graziano, R.F.; Bezman, N.A.; Robbins, M.D.; et al. Enhanced SLAMF7 homotypic interactions by elotuzumab improves NK cell killing of multiple myeloma. Cancer Immunol. Res. 2019, 7, 1633–1646. [Google Scholar] [CrossRef] [Green Version]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Lesokhin, A.; Borrello, I.; Timmerman, J.; Gutierrez, M.; Zhu, L.; McKiver, M.P.; Ansell, S.M. A phase 1b study of dual PD-1 and CTLA-4 or KIR blockade in patients with relapsed/refractory lymphoid malignancies. Leukemia 2021, 35, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Hanna, G.J.; ONeill, A.; Shin, K.Y.; Wong, K.; Jo, V.Y.; Quinn, C.T.; Cuttler, J.M.; Flynn, M.; Lizotte, P.H.; Annino, D.J.; et al. Neoadjuvant and adjuvant nivolumab and lirilumab in patients with recurrent, resectable squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Galot, R.; Le Tourneau, C.; Saada-Bouzid, E.; Daste, A.; Even, C.; Debruyne, P.; Henry, S.; Zanetta, S.; Rutten, A.; Licitra, L.; et al. A phase II study of monalizumab in patients with recurrent/metastatic squamous cell carcinoma of the head and neck: The I1 cohort of the EORTC-HNCG-1559 UPSTREAM trial. Eur. J. Cancer 2021, 158, 17–26. [Google Scholar] [CrossRef]
- Tian, T.; Li, Z. Targeting Tim-3 in cancer with resistance to PD-1/PD-L1 blockade. Front. Oncol. 2021, 11, 731175. [Google Scholar] [CrossRef]
- Rotte, A.; Sahasranaman, S.; Budha, N. Targeting TIGIT for immunotherapy of cancer: Update on clinical development. Biomedicines 2021, 9, 1277. [Google Scholar] [CrossRef]
- Zeng, T.; Cao, Y.; Jin, T.; Tian, Y.; Dai, C.; Xu, F. The CD112R/CD112 axis: A breakthrough in cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 285. [Google Scholar] [CrossRef]
- Buckle, I.; Guillerey, C. Inhibitory receptors and immune checkpoints regulating natural killer cell responses to cancer. Cancers 2021, 13, 4263. [Google Scholar] [CrossRef]
- Esen, F.; Deniz, G.; Aktas, E.C. PD-1, CTLA-4, LAG-3, and TIGIT: The roles of immune checkpoint receptors on the regulation of human NK cell phenotype and functions. Immunol. Lett. 2021, 240, 15–23. [Google Scholar] [CrossRef]
- Tanaka, J.; Miller, J.S. Recent progress and challenges in cellular therapy using NK cells for hematological malignancies. Blood Rev. 2020, 44, 100678. [Google Scholar] [CrossRef]
- Cheng, Y.; Zheng, X.; Wang, X.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z.; Sun, H. Trispecific killer engager 161519 enhances natural killer cell function and provides anti-tumor activity against CD19-positive cancers. Cancer Biol. Med. 2020, 17, 1026–1038. [Google Scholar] [CrossRef] [PubMed]
- Felices, M.; Lenvik, T.R.; Kodal, B.; Lenvik, A.J.; Hinderlie, P.; Bendzick, L.E.; Schirm, D.K.; Kaminski, M.F.; McElmurry, R.T.; Geller, M.A.; et al. Potent cytolytic activity and specific IL15 delivery in a second-generation trispecific killer engager. Cancer Immunol. Res. 2020, 8, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Gauthier, L.; Debroas, G.; Vivier, E. Natural killer cell engagers in cancer immunotherapy: Next generation of immuno-oncology treatments. Eur. J. Immunol. 2021, 51, 1934–1942. [Google Scholar] [CrossRef] [PubMed]
- Rothe, A.; Sasse, S.; Topp, M.S.; Eichenauer, D.A.; Hummel, H.; Reiners, K.S.; Dietlein, M.; Kuhnert, G.; Kessler, J.; Buerkle, C.; et al. A phase I study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood 2015, 125, 4024–4031. [Google Scholar] [CrossRef]
- Kerbauy, L.N.; Marin, N.D.; Kaplan, M.; Banerjee, P.P.; Berrien-Elliott, M.M.; Becker-Hapak, M.; Basar, R.; Foster, M.; Garcia Melo, L.; Neal, C.C.; et al. Combining AFM13, a bispecific CD30/CD16 antibody, with cytokine-activated blood and cord blood-derived NK cells facilitates CAR-like responses against CD30 + malignancies. Clin. Cancer Res. 2021, 27, 3744–3756. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013, 21, 3599–3608. [Google Scholar] [CrossRef]
- Au, K.M.; Park, S.I.; Wang, A.Z. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci. Adv. 2020, 6, eaba8564. [Google Scholar] [CrossRef]
- Gong, L.; Li, Y.; Cui, K.; Chen, Y.; Hong, H.; Li, J.; Li, D.; Yin, Y.; Wu, Z.; Hang, Z. Nanobody-engineered natural killer cell conjugates for solid tumor adoptive immunotherapy. Small 2021, 17, 2103463. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michel, T.; Ollert, M.; Zimmer, J. A Hot Topic: Cancer Immunotherapy and Natural Killer Cells. Int. J. Mol. Sci. 2022, 23, 797. https://doi.org/10.3390/ijms23020797
Michel T, Ollert M, Zimmer J. A Hot Topic: Cancer Immunotherapy and Natural Killer Cells. International Journal of Molecular Sciences. 2022; 23(2):797. https://doi.org/10.3390/ijms23020797
Chicago/Turabian StyleMichel, Tatiana, Markus Ollert, and Jacques Zimmer. 2022. "A Hot Topic: Cancer Immunotherapy and Natural Killer Cells" International Journal of Molecular Sciences 23, no. 2: 797. https://doi.org/10.3390/ijms23020797
APA StyleMichel, T., Ollert, M., & Zimmer, J. (2022). A Hot Topic: Cancer Immunotherapy and Natural Killer Cells. International Journal of Molecular Sciences, 23(2), 797. https://doi.org/10.3390/ijms23020797