
Premature Termination Codon in 5′ Region of Desmoplakin and Plakoglobin Genes May Escape Nonsense-Mediated Decay through the Reinitiation of Translation


Abstract

1. Introduction
2. Results
2.1. PTC Clones and Genotype Distribution
2.2. Genomic Context of 5′ Region for Reinitiation
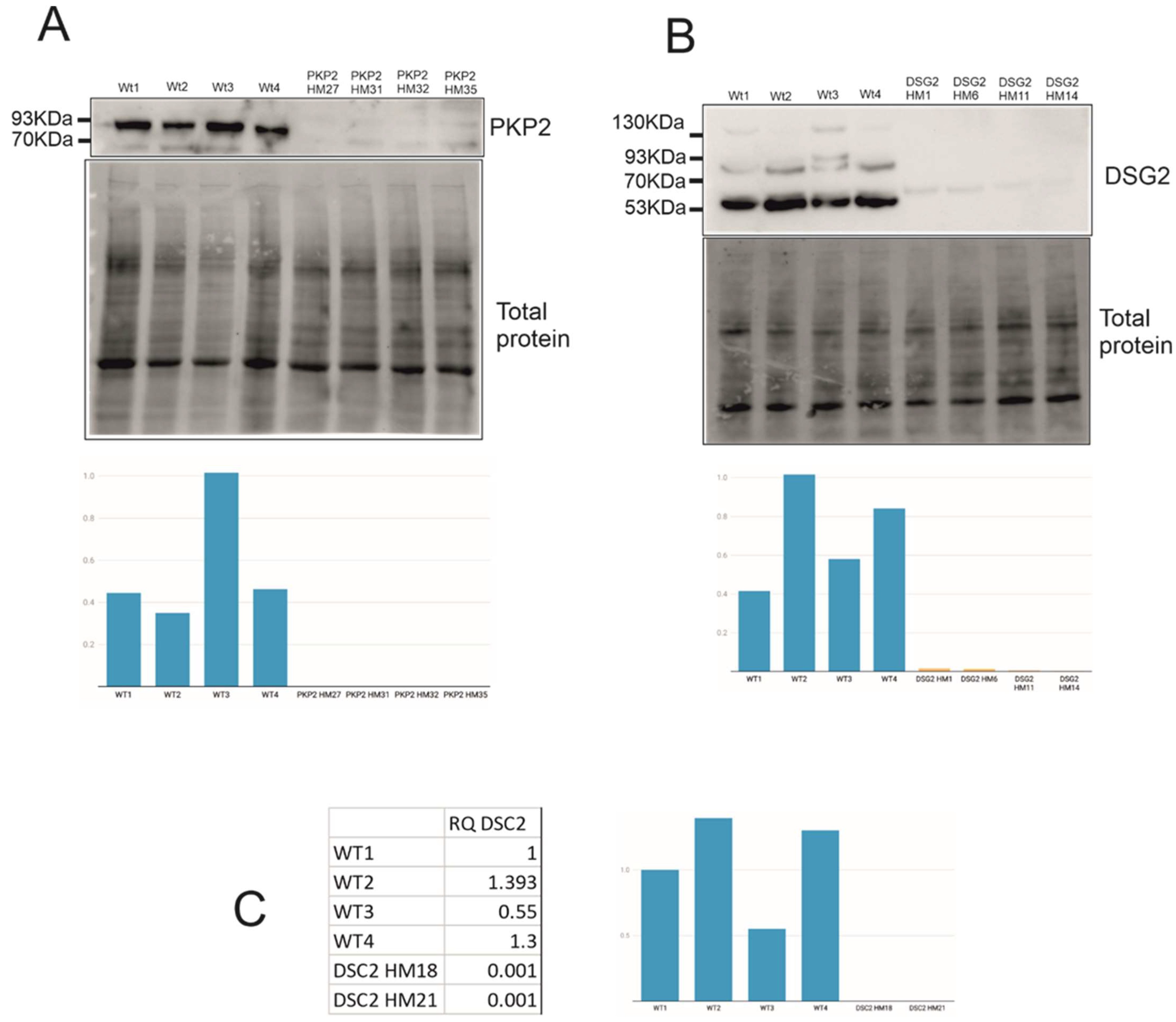
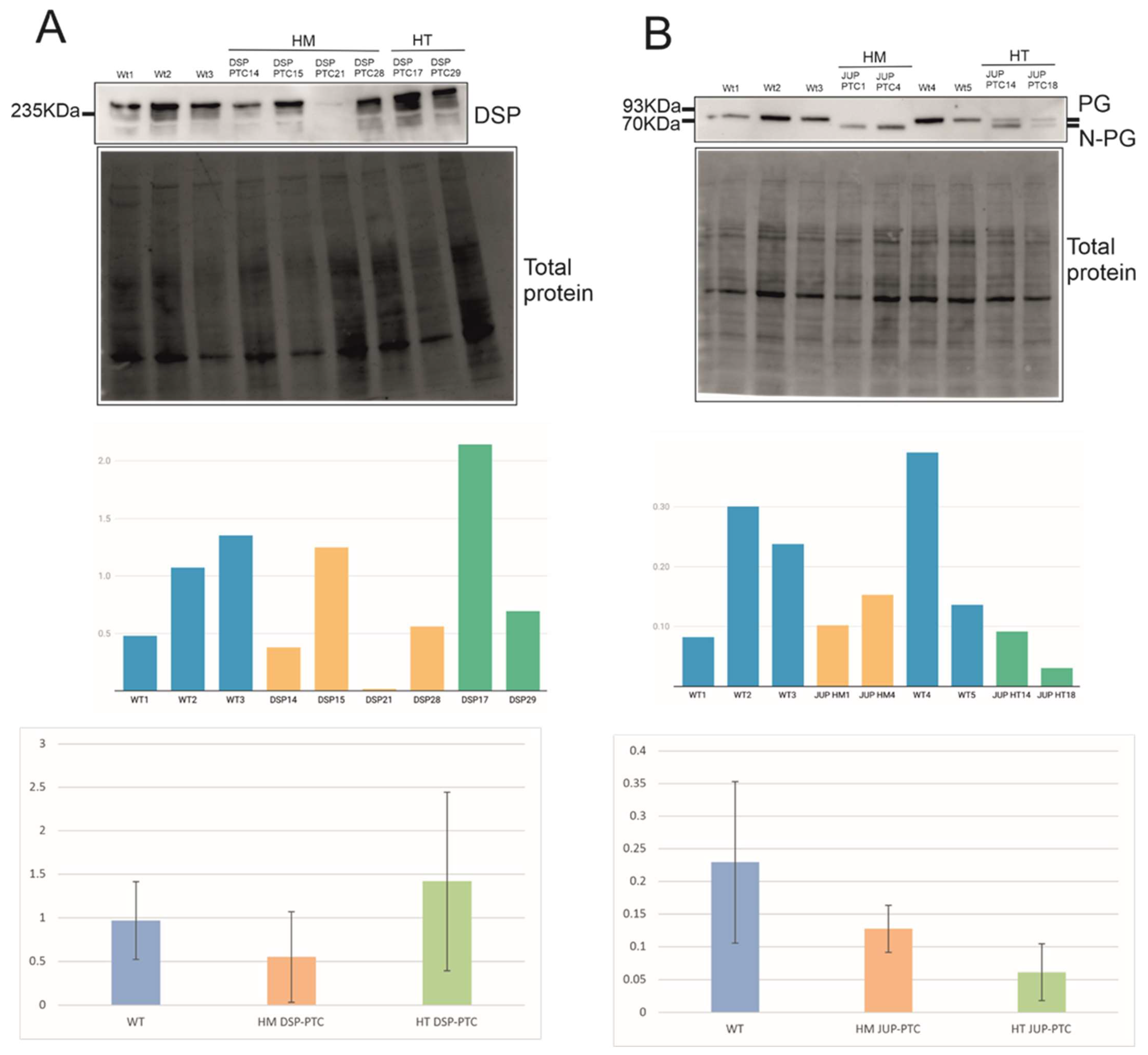
2.3. Expression Levels of PTC Clones and Mechanisms Triggered
2.4. Effect of N-Truncated DSP and JUP on ACM-Related Gene Expression
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Basso, C.; Corrado, D.; Marcus, F.I.; Nava, A.; Thiene, G. Arrhythmogenic Right Ventricular Cardiomyopathy. Lancet 2009, 373, 1289–1300. [Google Scholar] [CrossRef]
- Karmouch, J.; Protonotarios, A.; Syrris, P. Genetic Basis of Arrhythmogenic Cardiomyopathy. Mol. Genet. 2018, 33, 6. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef] [PubMed]
- Norman, M.; Simpson, M.; Mogensen, J.; Shaw, A.; Hughes, S.; Syrris, P.; Sen-Chowdhry, S.; Rowland, E.; Crosby, A.; McKenna, W.J. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation 2005, 112, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Basso, C.; Pilichou, K.; Bauce, B.; Corrado, D.; Thiene, G. Diagnostic Criteria, Genetics, and Molecular Basis of Arrhythmogenic Cardiomyopathy. Heart Fail. Clin. 2018, 14, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Najor, N.A. Desmosomes in Human Disease. Mech. Dis. 2018, 13, 51–70. [Google Scholar] [CrossRef]
- Vermij, S.H.; Abriel, H.; van Veen, T.A.B. Refining the Molecular Organization of the Cardiac Intercalated Disc. Cardiovasc. Res. 2017, 113, 259–275. [Google Scholar] [CrossRef]
- Pilichou, K. Arrhythmogenic Cardiomyopathy. Orphanet J. Rare Dis. 2016, 11, 33. [Google Scholar] [CrossRef]
- Carruth, E.D.; Young, W.; Beer, D.; James, C.A.; Calkins, H.; Jing, L.; Raghunath, S.; Hartzel, D.N.; Leader, J.B.; Kirchner, H.L.; et al. Prevalence and Electronic Health Record-Based Phenotype of Loss-of-Function Genetic Variants in Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Genes. Circ. Genom. Precis. Med. 2019, 12, e002579. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD®): Optimizing Its Use in a Clinical Diagnostic or Research Setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- van der Zwaag, P.A.; Jongbloed, J.D.H.; van den Berg, M.P.; van der Smagt, J.J.; Jongbloed, R.; Bikker, H.; Hofstra, R.M.W.; van Tintelen, J.P. A Genetic Variants Database for Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Hum. Mutat. 2009, 30, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Maquat, L.E. Nonsense-Mediated MRNA Decay in Humans at a Glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef]
- Dyle, M.C.; Kolakada, D.; Cortazar, M.A.; Jagannathan, S. How to Get Away with Nonsense: Mechanisms and Consequences of Escape from Nonsense-mediated RNA Decay. WIREs RNA 2020, 11, e1560. [Google Scholar] [CrossRef]
- Van Damme, P.; Gawron, D.; van Criekinge, W.; Menschaert, G. N-Terminal Proteomics and Ribosome Profiling Provide a Comprehensive View of the Alternative Translation Initiation Landscape in Mice and Men. Mol. Cell. Proteom. 2014, 13, 1245–1261. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of Translation via MRNA Structure in Prokaryotes and Eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Hernández, G.; Osnaya, V.G.; Pérez-Martínez, X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends Biochem. Sci. 2019, 44, 1009–1021. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The Mechanism of Eukaryotic Translation Initiation and Principles of Its Regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef]
- Cohen, S.; Kramarski, L.; Levi, S.; Deshe, N.; Ben David, O.; Arbely, E. Nonsense Mutation-Dependent Reinitiation of Translation in Mammalian Cells. Nucleic Acids Res. 2019, 47, 6330–6338. [Google Scholar] [CrossRef]
- Neu-Yilik, G.; Amthor, B.; Gehring, N.H.; Bahri, S.; Paidassi, H.; Hentze, M.W.; Kulozik, A.E. Mechanism of Escape from Nonsense-Mediated MRNA Decay of Human -Globin Transcripts with Nonsense Mutations in the First Exon. RNA 2011, 17, 843–854. [Google Scholar] [CrossRef]
- Jagannathan, S.; Bradley, R.K. Translational Plasticity Facilitates the Accumulation of Nonsense Genetic Variants in the Human Population. Genome Res. 2016, 26, 1639–1650. [Google Scholar] [CrossRef]
- Rasmussen, T.B.; Nissen, P.H.; Palmfeldt, J.; Gehmlich, K.; Dalager, S.; Jensen, U.B.; Kim, W.Y.; Heickendorff, L.; Mølgaard, H.; Jensen, H.K.; et al. Truncating Plakophilin-2 Mutations in Arrhythmogenic Cardiomyopathy Are Associated with Protein Haploinsufficiency in Both Myocardium and Epidermis. Circ. Cardiovasc. Genet. 2014, 7, 230–240. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.P.J.; Daubert, J.P.; et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: Proposed Modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef]
- Vallverdú-Prats, M.; Alcalde, M.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Fernandez-Falgueras, A.; Coll, M.; Pérez-Serra, A.; Puigmulé, M.; Iglesias, A.; et al. Rare Variants Associated with Arrhythmogenic Cardiomyopathy: Reclassification Five Years Later. J. Pers. Med. 2021, 11, 162. [Google Scholar] [CrossRef]
- Kochetov, A.V.; Allmer, J.; Klimenko, A.I.; Zuraev, B.S.; Matushkin, Y.G.; Lashin, S.A. AltORFev Facilitates the Prediction of Alternative Open Reading Frames in Eukaryotic MRNAs. Bioinformatics 2016, 3, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and Homozygous Loss-of-Function Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. Int. J. Mol. Sci. 2021, 22, 3786. [Google Scholar] [CrossRef] [PubMed]
- Maruthappu, T.; Posafalvi, A.; Castelletti, S.; Delaney, P.J.; Syrris, P.; O’Toole, E.A.; Green, K.J.; Elliott, P.M.; Lambiase, P.D.; Tinker, A.; et al. Loss-of-function Desmoplakin I and II Mutations Underlie Dominant Arrhythmogenic Cardiomyopathy with a Hair and Skin Phenotype. Br. J. Dermatol. 2019, 180, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Pigors, M.; Kiritsi, D.; Krümpelmann, S.; Wagner, N.; He, Y.; Podda, M.; Kohlhase, J.; Hausser, I.; Bruckner-Tuderman, L.; Has, C. Lack of Plakoglobin Leads to Lethal Congenital Epidermolysis Bullosa: A Novel Clinico-Genetic Entity. Hum. Mol. Genet. 2011, 20, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Syrris, P.; Ward, D.; Evans, A.; Asimaki, A.; Gandjbakhch, E.; Sen-Chowdhry, S.; McKenna, W.J. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Associated with Mutations in the Desmosomal Gene Desmocollin-2. Am. J. Hum. Genet. 2006, 79, 978–984. [Google Scholar] [CrossRef]
- Buzina, A.; Shulman, M.J. Infrequent Translation of a Nonsense Codon Is Sufficient to Decrease MRNA Level. Mol. Biol. Cell 1999, 10, 515–524. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Denecke, J.; Kranz, C.; Kemming, D.; Koch, H.-G.; Marquardt, T. An Activated 5? Cryptic Splice Site in the Human ALG3 Gene Generates a Premature Termination Codon Insensitive to Nonsense-Mediated MRNA Decay in a New Case of Congenital Disorder of Glycosylation Type Id (CDG-Id). Hum. Mutat. 2004, 23, 477–486. [Google Scholar] [CrossRef]
- Perrin-Vidoz, L. The Nonsense-Mediated MRNA Decay Pathway Triggers Degradation of Most BRCA1 MRNAs Bearing Premature Termination Codons. Hum. Mol. Genet. 2002, 11, 2805–2814. [Google Scholar] [CrossRef]
- Inácio, Â.; Silva, A.L.; Pinto, J.; Ji, X.; Morgado, A.; Almeida, F.; Faustino, P.; Lavinha, J.; Liebhaber, S.A.; Romão, L. Nonsense Mutations in Close Proximity to the Initiation Codon Fail to Trigger Full Nonsense-Mediated MRNA Decay. J. Biol. Chem. 2004, 279, 32170–32180. [Google Scholar] [CrossRef]
- Nickless, A.; Bailis, J.M.; You, Z. Control of Gene Expression through the Nonsense-Mediated RNA Decay Pathway. Cell Biosci. 2017, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Karamyshev, A.L.; Karamysheva, Z.N. Lost in Translation: Ribosome-Associated MRNA and Protein Quality Controls. Front. Genet. 2018, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Cheedipudi, S.M.; Hu, J.; Fan, S.; Yuan, P.; Karmouch, J.; Czernuszewicz, G.; Robertson, M.J.; Coarfa, C.; Hong, K.; Yao, Y.; et al. Exercise Restores Dysregulated Gene Expression in a Mouse Model of Arrhythmogenic Cardiomyopathy. Cardiovasc. Res. 2020, 116, 1199–1213. [Google Scholar] [CrossRef]
- Garcia-Gras, E. Suppression of Canonical Wnt/ -Catenin Signaling by Nuclear Plakoglobin Recapitulates Phenotype of Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef]
- Gerull, B.; Brodehl, A. Genetic Animal Models for Arrhythmogenic Cardiomyopathy. Front. Physiol. 2020, 11, 624. [Google Scholar] [CrossRef] [PubMed]
- Gerull, B.; Heuser, A.; Wichter, T.; Paul, M.; Basson, C.T.; McDermott, D.A.; Lerman, B.B.; Markowitz, S.M.; Ellinor, P.T.; MacRae, C.A.; et al. Mutations in the Desmosomal Protein Plakophilin-2 Are Common in Arrhythmogenic Right Ventricular Cardiomyopathy. Nat. Genet. 2004, 36, 1162–1164. [Google Scholar] [CrossRef]
- van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.P.; Wilde, A.A.M.; van der Smagt, J.; Boven, L.G.; Mannens, M.M.A.M.; van Langen, I.M.; et al. Plakophilin-2 Mutations Are the Major Determinant of Familial Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A Novel Mutation in NFKBIA/IKBA Results in a Degradation- Resistant N-Truncated Protein and Is Associated with Ectodermal Dysplasia With Immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef]
- McDonald, D.R.; Mooster, J.L.; Reddy, M.; Bawle, E.; Secord, E.; Geha, R.S. Heterozygous N-Terminal Deletion of IκBα Results in Functional Nuclear Factor ΚB Haploinsufficiency, Ectodermal Dysplasia, and Immune Deficiency. J. Allergy Clin. Immunol. 2007, 120, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, M.; Lund, C.; Akram, Z.; Winther, J.R.; Horn, N.; Møller, L.B. Evidence That Translation Reinitiation Leads to a Partially Functional Menkes Protein Containing Two Copper-Binding Sites. Am. J. Hum. Genet. 2006, 79, 214–229. [Google Scholar] [CrossRef]
- Puel, A.; Reichenbach, J.; Bustamante, J.; Ku, C.-L.; Feinberg, J.; Döffinger, R.; Bonnet, M.; Filipe-Santos, O.; de Beaucoudrey, L.; Durandy, A.; et al. The NEMO Mutation Creating the Most-Upstream Premature Stop Codon Is Hypomorphic Because of a Reinitiation of Translation. Am. J. Hum. Genet. 2006, 78, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Witcher, L.L.; Collins, R.; Puttagunta, S.; Mechanic, S.E.; Munson, M.; Gumbiner, B.; Cowin, P. Desmosomal Cadherin Binding Domains of Plakoglobin. J. Biol. Chem. 1996, 271, 10904–10909. [Google Scholar] [CrossRef]
- Hatsell, S.; Medina, L.; Merola, J.; Haltiwanger, R.; Cowin, P. Plakoglobin Is O-Glycosylated Close to the N-Terminal Destruction Box. J. Biol. Chem. 2003, 278, 37745–37752. [Google Scholar] [CrossRef]
- Sacco, P.A.; McGranahan, T.M.; Wheelock, M.J.; Johnson, K.R. Identification of Plakoglobin Domains Required for Association with N-Cadherin and α-Catenin. J. Biol. Chem. 1995, 270, 20201–20206. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Bari, O.; Skillman, S.; Lah, M.D.; Haggstrom, A.N. Compound Heterozygous Mutations in Desmoplakin Associated with Skin Fragility, Follicular Hyperkeratosis, Alopecia, and Nail Dystrophy. Pediatr. Dermatol. 2018, 35, e218–e220. [Google Scholar] [CrossRef]
- Cabral, R.M.; Liu, L.; Hogan, C.; Dopping-Hepenstal, P.J.C.; Winik, B.C.; Asial, R.A.; Dobson, R.; Mein, C.A.; Baselaga, P.A.; Mellerio, J.E.; et al. Homozygous Mutations in the 5′ Region of the JUP Gene Result in Cutaneous Disease but Normal Heart Development in Children. J. Investig. Dermatol. 2010, 130, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 Cells: A Cardiac Muscle Cell Line That Contracts and Retains Phenotypic Characteristics of the Adult Cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Image Lab. Software. Available online: https://www.bio-rad.com/es-es/product/image-lab-software?ID=KRE6P5E8Z (accessed on 10 August 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Desmosomal Gene | Clones | Variants | New Frames | PTC Positions (Codons) | |
|---|---|---|---|---|---|
| PKP2 | 27 | A1 | c.325ins1 | −2 | 93 |
| A2 | c.324del2 | −2 | 93 | ||
| 31 | A1 | c.326ins1 | −2 | 93 | |
| A2 | c.322del2 | −2 | 93 | ||
| 32 | A1 | c.325ins1 | −2 | 93 | |
| A2 | c.324del1 | −1 | 115 | ||
| 35 | A1 | c.325ins1 | −2 | 93 | |
| A2 | c.325del4 | −1 | 115 | ||
| DSP | 28 | A1 | c.329del2 | −2 | 75 |
| A2 | c.331del5 | −2 | 75 | ||
| 21 | A1 | c.332del2 | −2 | 75 | |
| A2 | c.332del4 | −1 | 17 | ||
| 14 | A1 | c.324del29 | −2 | 75 | |
| A2 | NI | - | - | ||
| 15 | A1 | c.326del31 | −1 | 17 | |
| A2 | NI | - | - | ||
| 17 | A1 | WT | 0 | - | |
| A2 | c.324del26 | −2 | 75 | ||
| 29 | A1 | WT | 0 | - | |
| A2 | c.314del32 | −2 | 75 | ||
| DSG2 | 1 | A1 | c.226del4 | −1 | 31 |
| A2 | c.229ins1 | −2 | 38 | ||
| 6 | A1 | c.220del 30 + ins7 | −2 | 38 | |
| A2 | NI | - | - | ||
| 11 | A1 | c.220del7 | −1 | 31 | |
| A2 | c.228del1 | −1 | 31 | ||
| 14 | A1 | c.221del10 | −1 | 31 | |
| A2 | c.229ins1 | −2 | 38 | ||
| DSC2 | 18 | A1 | c.324del4 | −1 | 20 |
| A2 | NI | ||||
| 21 | A1 | c.325del1 | −1 | 20 | |
| A2 | NI | ||||
| JUP | 1 | A1 | c.135del13 | −1 | 37 |
| A2 | c.142del4 | −1 | 37 | ||
| 4 | A1 | c.142ins1 | −2 | 44 | |
| A2 | NI | - | - | ||
| 14 | A1 | c.136del6 | 0 | - | |
| A2 | c.142ins1 | −2 | 44 | ||
| 18 | A1 | WT | 0 | - | |
| A2 | c.135del10 | −1 | 37 |
| PKP2 | tctgcggctttgcgggcaggtcctggcagtccctcctggtcactccgccgacgcgATGGCCGTCCCCGGCTCACTGGCCGAGTGTGGCTACATCCGGACTGTGCTGGGCCAGCAGATCCTGGGTCACCTGGACAGCTCCAGCCTGGCCTTGCCCTCCGAGGCCAGACTGAGGCTGGCCGGCAGCAGCGGCCGCGGCGACCCGGCGGCCCGGAGCCAGCGGATCCAGGAGCAGGTGCAGCAGACCCTGGCCCGCCGGGGCCGGAGCTCTGCGGTCAGCGGGAACCTTCACCGAACCAGCAGTGTCCCTGAGTATGTCTACAAGCTACACGTGGTTGAGAATGACTTTGTTGGACGGCAGTCACCTGTCACTAGGGACTATGACATGCTTAAGGCTGGAATGACTGCCACTTATGGAAGTCGCTGGGGGAGAGCAGCAGCACAGTACAGTTCCCAGAAGTCAGTGGAGGAGAGATCCTGGAGGCAGCCTCTGAGGAGACTTGAGATTTCCCCAGATAGCAGCCCGGAGAGAGCCCAC |
| DSP | gaccaggtgtggcctgggcgccgggtgccagcggggaggagactcgcaccgcctcgaccaacaccaacacccaggcgcgacccagctcctctgagccctcgctgccctccgagccacagctccactccggttcccgcgcctagccagtcgccgtccccgtctccgccctgctggagcgctgagccctcgccagtcctccgcgttccgcgctcctctcccggagtccctcgcgtgctccgaggcgacgcctcgcgtatgccgcggcgctgagcggctctcttgagtgaccgcagacATGAGCTGCAACGGCGGCTCCCACCCGCGGATCAACACGCTGGGTCGCATGACCCGCGCGGAGTCCGGCCCGGACCTGCGCTACGAGATGACCTACAGCGGTGGCGGCGGCGGGGGCGGCGGGGGCGGCGGCGGGGGCACCAGCAGGACGTTCTACTCCCACTCCCGGCGCTGCACCGTCAACGACCAGAACTCCGACGGCTACTGTCAAACCGGCACCATGTCTAGACATCAGAATCAGAACACCATCCAAGAAATGCTGCAAAATTGCTCAGACTGTCTGATGCGGGCGGAGCTGATCGCGCAGCCGGAACTGAAATTCGGAGAAGGGATGCAGCTGGCATGGAACCGAGAGCTGGATGAGTATTTTACACAAGCCAACGATCAGATGGAAATCATAGACGGCTTGATCCGAGAGATGAGGCAGATGGGCCAGCCCTGTGATGCGTATCAGAAAAGACTGCTTCAGCTCCAGGAACAA |
| DSG2 | ctgcctcgtacctccccgcggagcgaacacattcccctcctccatctaggctgtggcccggcccaaggctaccctttctgacccgggcacacctggaaccgcaccccgggtcccgcagagtcagagaagggcggccccgggagggacctgcccaggaggatccgcagggcgccggcgaggcccggaggcgagggcgcggcggatcgaggcgATGGCGCGGAGCCCGGGTGACCGGTGCGCCCTGCTGCTGCTGGTGCAGCTGCTGGCGGTGGTCTGCTTGGACTTTGGAAACGGACTTCACTTAGAGGTCTTCAGCCCAAGAAATGAAGGCAAACCGTTCCCTAAGCACACTCACTTGGTTCGTCAAAAGAGGGCCTGGATCACTGCCCCTGTGGCTCTGCGGGAGGGCGAAGACCTGTCCAGAAAGAACCCGATTGCCAAGATACACTCTGACCTTGCAGAAGAAAAAGGGATAAAAATCACGTACAAGTACACTGGGAAGGGAATTACAGAACCGCCTTTCGGCATATTCGTCTTTGATAGAAACACAGGAGAACTGAACATCACTAGCATTCTTGACCGGGAAGAAACACCATATTTTCTGCTGACAGGCTATGCATTGGACTCCAGAGGAAACAACCTGGAAAAGCCCTTGGAACTACGCATCAAAGTTCTGGACATCAATGACAAC |
| DSC2 | ggggggtgacaagggacatataggtggcctctgctggtgagaaatacctagtacaggtgaaagggtggcggccagagggagttcccaccggttgaattcttaaagtcatgaagactcaagaaaaataacaaggagtggccgattcgagtcttttggacttgcccagagctccaccctcggacagaggaaaagcccctggggacccaggcgggagcatcagacaagcgcgagaaaagcgcctgtgtgcgcgctccacctctgcgcagcgggtgcggggcggtgacctgtcccttgagctggccATGGCGGCTGTGGGATCTATGCGCTCCGGGAGCCCTGCCTTCGGCCTGGGACACCTGTTGACCCTTGCGATCCTTGCACTTGCCTCTGATGCCTGTAAAGAAGTCGTCCTCCAGGTCCCCTCTGAACTACCTGCCGAGAAATTTGTTGGCAGAGTGAACCTGATGGACTGCCTTAAATCAGCAGACATAGTTCATCTGAGTGATCCTGACTTCCAAGTCTTAGAAGATGGTTCTGTGTACACAACCAGTTCTGTTGTTTTGTCCTCGGGGCAAAGAAGCTTTACTATATGGCTTTTTAGCACAGACAGCCAAGAAGAAAGGGAGATATCTGTCCATTTAGAGGGCCCAGTAGAGGTACTAAATAAAAGACCGCATACAGAGAAGGTTCTCAGCCGTGCCAAGAGAAGATGGGCTCCTATCCCTTGTTCCATGCTAGAGAATTCATTGGGTCCCTTCCCACTTTTCCTTCAACAGATCCAG |
| JUP | gccagagtccggagcagccgccgcccgagtgcgccgagctcagttcgctgcccgcgccggctccctcccggccagacccgaccccgattcggctcagcccggctccacgctcagcagccaccATGGAGGTGATGAACCTTATTGAGCAGCCCATCAAGGTGACAGAGTGGCAACAGACATACACCTACGACTCGGGCATCCACTCCGGCGTCAATACCTGTGTGCCCTCTGTAAGCAGCAAGGGCATCATGGATGAGGATGATGCCTGCGGCAGACAGTACACACTCAAGAAGACTACCACCTATACACAAGGGGTGCCACAGAACCAAGGTGACCTGGAATACCAGATGTCCACAACGGCCAGAGCCAAGCGGGTGCGGGAGGCCATGTGTCCAGGGGTCTCAGGCGAGGACAGTTCTCTACTGCTGGCCACCCAGGTGGAGGGGCAGACAACCAATCTGCAGCGCCTGGCCGAACCATCCCAGTTGCTCAAGTCGGCCATCGTCCATCTCATCAACTACCAGGATGATGCAGAGCTGGCCACCCGGGCTCTGCCTGAGCTCACCAAGCTGCTCAACGATGAGGACCCGGTAGTGGTGACC |
| Name | In-Frame | Remark | Context | Mechanism | Position | |
|---|---|---|---|---|---|---|
| PKP2 | Orf1 | Yes | Null, canonical ATG | W | Linear scanning | 0 |
| Orf2 | No | AltORF2; predicted translation level: moderate | H | Reinitiation starter | 256 | |
| DSP | Orf1 | Yes | Null, canonical ATG | W | Linear scanning | 0 |
| Orf2 | Yes | AltORF2; predicted translation level: weak | W | Leaky scanning | 48 | |
| Orf3 | Yes | AltORF3; predicted translation level: moderate | M | Leaky scanning | 87 | |
| Orf4 | Yes | AltORF4; predicted translation level: moderate | H | Reinitiation starter | 219 | |
| DSG2 | Orf1 | Yes | Null, canonical ATG | W | Linear scanning | 0 |
| Orf2 | No | AltORF2; predicted translation level: moderate | M | Leaky scanning | 112 | |
| Orf3 | No | AltORF3; predicted translation level: moderate | M | Leaky scanning | 403 | |
| Orf4 | No | AltORF4; predicted translation level: weak | W | Leaky scanning | 472 | |
| DSC2 | Orf1 | Yes | Null, canonical ATG | W | Linear scanning | 0 |
| Orf2 | Yes | AltORF2; predicted translation level: weak | W | Leaky scanning | 18 | |
| Orf3 | No | AltORF3; predicted translation level: weak | W | Leaky scanning | 88 | |
| Orf4 | Yes | AltORF4; predicted translation level: moderate | M | Leaky scanning | 162 | |
| Orf5 | No | AltORF5; predicted translation level: moderate | H | Reinitiation starter | 226 | |
| JUP | Orf1 | Yes | Null, canonical ATG | W | Linear scanning | 0 |
| Orf2 | Yes | AltORF2; predicted translation level: moderate | M | Leaky scanning | 9 | |
| Orf3 | Yes | AltORF3; predicted translation level: moderate | H | Reinitiation starter | 126 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallverdú-Prats, M.; Brugada, R.; Alcalde, M. Premature Termination Codon in 5′ Region of Desmoplakin and Plakoglobin Genes May Escape Nonsense-Mediated Decay through the Reinitiation of Translation. Int. J. Mol. Sci. 2022, 23, 656. https://doi.org/10.3390/ijms23020656
Vallverdú-Prats M, Brugada R, Alcalde M. Premature Termination Codon in 5′ Region of Desmoplakin and Plakoglobin Genes May Escape Nonsense-Mediated Decay through the Reinitiation of Translation. International Journal of Molecular Sciences. 2022; 23(2):656. https://doi.org/10.3390/ijms23020656
Chicago/Turabian StyleVallverdú-Prats, Marta, Ramon Brugada, and Mireia Alcalde. 2022. "Premature Termination Codon in 5′ Region of Desmoplakin and Plakoglobin Genes May Escape Nonsense-Mediated Decay through the Reinitiation of Translation" International Journal of Molecular Sciences 23, no. 2: 656. https://doi.org/10.3390/ijms23020656
APA StyleVallverdú-Prats, M., Brugada, R., & Alcalde, M. (2022). Premature Termination Codon in 5′ Region of Desmoplakin and Plakoglobin Genes May Escape Nonsense-Mediated Decay through the Reinitiation of Translation. International Journal of Molecular Sciences, 23(2), 656. https://doi.org/10.3390/ijms23020656