
Computer Simulations of Deep Eutectic Solvents: Challenges, Solutions, and Perspectives

 , , , , , , and
, , , , , , and
Abstract
:
1. Introduction
2. Simulation Methods for DESs
2.1. Quantum Mechanical Methods
2.1.1. DFT-Derived Peculiarities of the Local DES Structure
2.1.2. Relations between DFT and NMR and FTIR Experiments of DESs
2.1.3. Periodic DFT in Studies of the Condensed Phase of DESs
2.2. Molecular Dynamics Simulations
2.3. Polarization and Polarizable Force Fields for Deep Eutectic Solvents
2.3.1. The Polarizable CL&Pol Force Field
2.3.2. The Polarizable SAPT Force Field
2.4. Combinations of Quantum Mechanics and Molecular Dynamics Techniques
2.5. Coarse-Grained Models
2.6. Machine Learning Methods
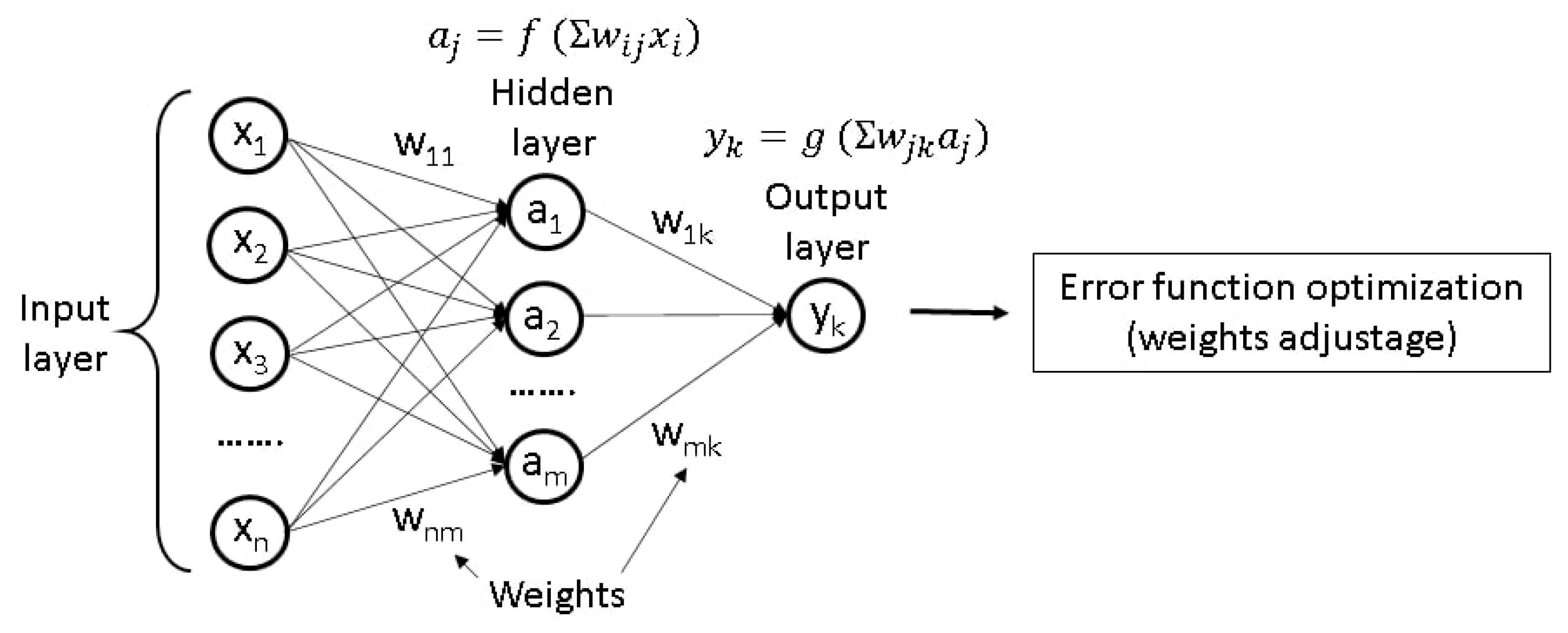
2.6.1. Working Principles of ANNs
2.6.2. DES Property Prediction
2.6.3. Optimization of Experiments using ML
2.6.4. Different Aspects Regarding Application of ML Methods
3. Main Directions of Investigations
3.1. DES Structure
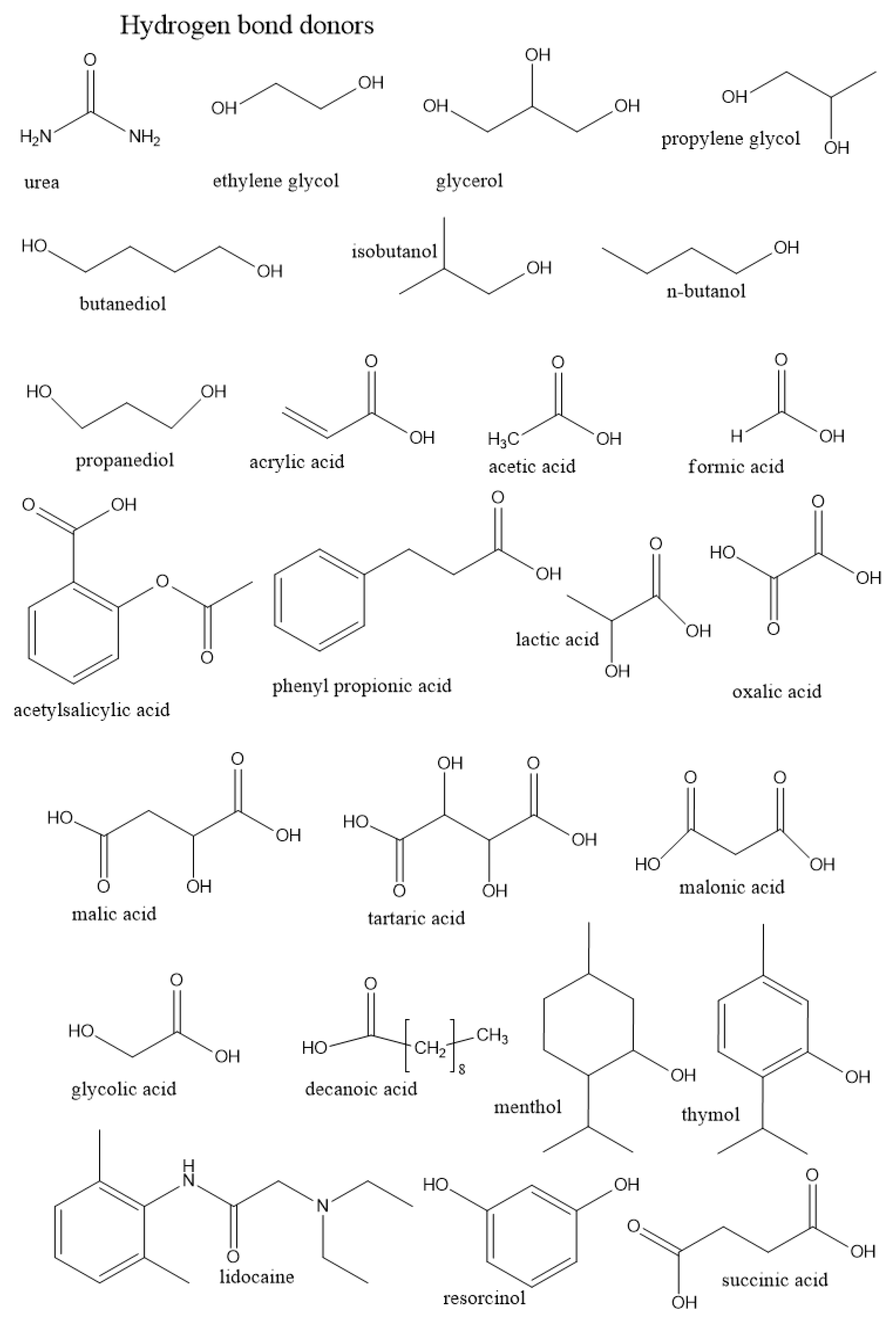
3.1.1. Role of the Hydrogen Bond Donor
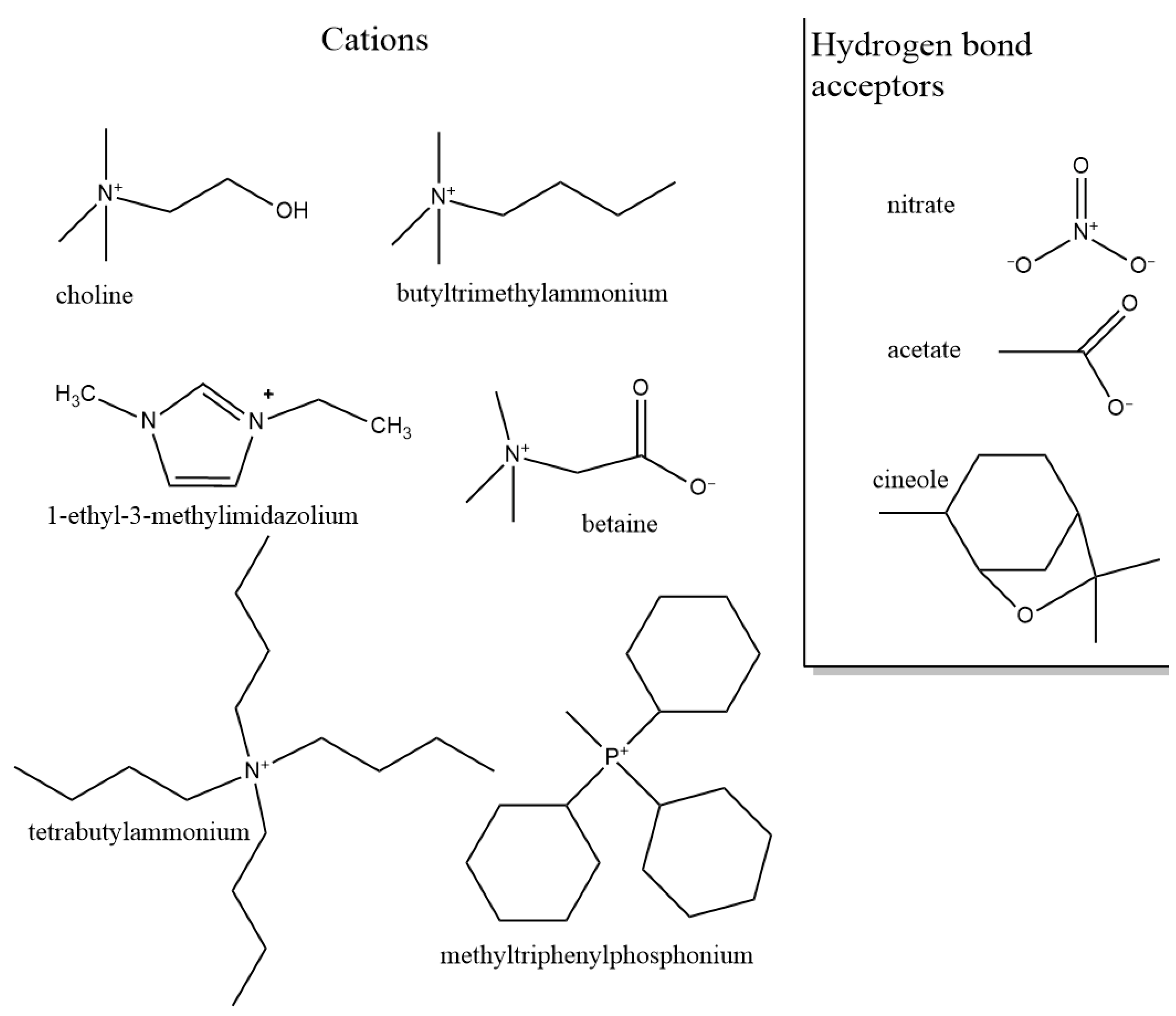
3.1.2. Role of Hydrogen Bond Acceptor (Anion)
3.1.3. Role of Cation
3.1.4. Hydrophobic Deep Eutectic Solvents
3.1.5. Electrolyte-Based DESs
3.1.6. Ternary DES
3.2. Dynamic Properties
3.3. DES for Separations and Gas Capture
3.4. Water Effect on DES
3.4.1. Water Effect on DES Micro- and Nanostructure
3.4.2. Effects of Water on Reline
3.4.3. Water’s Effect on the Properties of DESs
3.4.4. Activity and Stability of Enzymes in DES/Water Mixtures
3.4.5. Hydrophobic DESs
3.4.6. Prediction of DES/Water Mixtures Properties by ANNs
3.5. DES in Nanotechnology
3.6. Biomolecules in DES
3.7. Biomass Pretreatment by DES
4. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
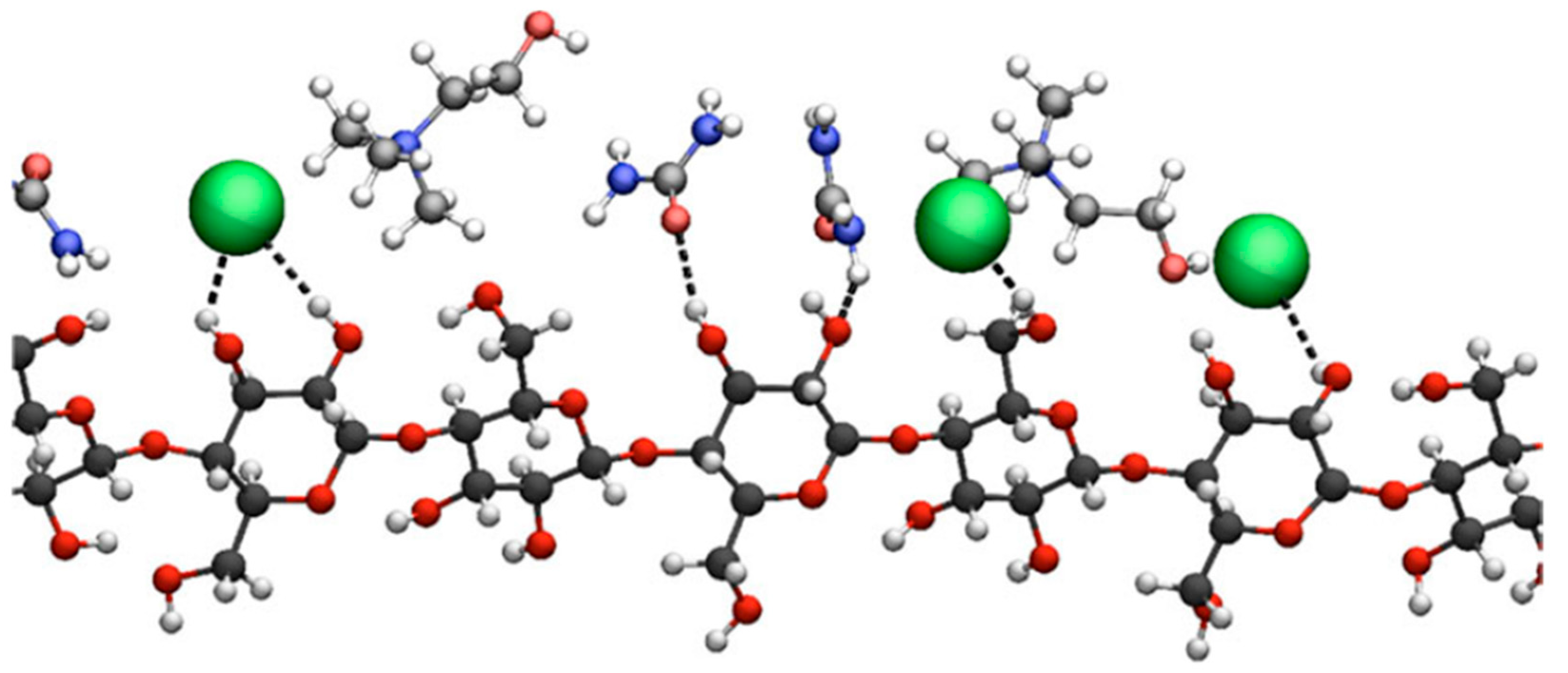{kind=link}
| No | DES | FF/Corrections | Topic of the Publication | Reference |
|---|---|---|---|---|
| 1 | 1-ethyl-3-methylimidazolium chloride:Urea Different molar ratios | CL&P OPLS-AA | Investigation of DES structure | [231] |
| 2 | 1-octyl-3-methylimidazolium: Tetrafluoroborate 1-ethyl-3-methylimidazolium:Tetrafluoroborate Different molar ratios | CL&P | Investigation of DES structure | [370] |
| 3 | Acetamide:LiBr (3.5:1) | CHARMM 27 only/no corrections | Support to experiment | [253] |
| 4 | Acetamide:LiClO4 (3.5:1) | CHARMM 27 + separate parameters for ions | Mechanisms of DES components motion | [249] |
| 5 | Acetamide:LiClO4 (3.5:1) | CHARMM 27 + separate parameters for ions | Mechanism of Acetamide in Deep Eutectic Solvents | [250] |
| 6 | Acetamide:LiClO4 (3.5:1) Acetamide:LiNO3 (3.5:1) Acetamide:LiBr (3.5:1) | CHARMM 27 + separate parameters for ions | Orientation jumps of acetamide molecules | [253] |
| 7 | Acetamide:LiClO4 (4.1:1) Acetamide:LiNO3 (3.5:1) Acetamide:LiBr (3.5:1) | CHARMM 27 + separate parameters for ions | Support to experiment | [252] |
| 8 | Acetamide:LiNO3 (3.5:1) | CHARMM 27 + separate parameters for ions | Mechanisms of DES components motion | [248] |
| 9 | Acetamide:Urea (1.5:1) Acetamide:Urea (2.3:1) | CHARMM + GROMOS96/no corrections | Temperature dependent relaxation dynamics, particle motion characteristics, and heterogeneity aspects of deep eutectic solvents | [254] |
| 10 | Acetamide:Urea (2.3:1) | CHARMM + GROMOS96/no corrections | Structural H-bond relaxation | [255] |
| 11 | Acetamide:Urea (2.3:1) | OPLS-DES/no corrections | Heterogeneity of reorientational relaxation and translational dynamics | [256] |
| 12 | Arginine:Glutamic acid (1:1) Arginine:Oxalic acid (1:1)Arginine:Tartaric acid (1:1) | No name of FF. Parameters are given | Lidocaine in DES | [150] |
| 13 | Arginine:Glutamic acid (1:1) Arginine:Oxalic acid (1:1)Arginine:Tartaric acid (1:1) | No name of FF. Parameters are given | Antibiotics in DES | [151] |
| 14 | Benzene-1,4-diol:Urea Different molar ratios | AMBER99 | Investigation of DES structure and dynamics and water effect on it | [371] |
| 15 | Betaine:Lactic acid (1:1) | No name of FF. Parameters are given | Investigation of DES structure | [153] |
| 16 | Betaine Monohydrate:Glycerol Different molar ratios | GAFF/no corrections | Extraction of palmitic acid by DES | [372] |
| 17 | Betaine Monohydrate:Glycerol (1:2) | GAFF/no corrections | Investigation of DES structure | [373] |
| 18 | Bis(trifluoromethanesulfonyl)imide:Methanesulfonamide Bis(trifluoromethanesulfonyl)imide:Dimethylmethanesulfonamide Different molar ratios | OPLS-AA/no corrections | Characterization of the transport properties of binary DESs | [374] |
| 19 | Caprolactam:Tetrabutylammonium bromide (1:1) Caprolactam:Tetrabutylammonium bromide (1:1) ChCl:Urea (1:2) Methyltriphenylphosphonium bromide:Monoethanolamine (1:6) | Gromos54a7, the optimized forcefield parameters were obtained from the Automated Topology Builder (ATB) database | Natural Gas Desulfurization using DES | [375] |
| 20 | Ceineole:Succinic acid (1:1) Ceineole:Malic acid (1:1) Ceineole:Lactic acid (1:1) | No name of FF. Parameters are given | Investigation of DES structure | [154] |
| 21 | ChCl:Ethylene glycol (1:4) | OPLS-AA/no corrections | DES for gas separation | [276] |
| 22 | ChCl:1,2-ethanediol (1:2) ChCl:1,3-propanediol (1:3) ChCl:1,4-butanediol (1:3) | OPLS and GAFF/no corrections | Dependence of Solvation Dynamics in Alcohol-Based DES | [376] |
| 23 | ChCl:1,2-ethanediol ChCl:1,3-propanediol ChCl:1,4-butanediol ChCl:1,5-pentanediol | Combination of OPLS-AA and Amber parameters | Investigation of DES structure | [377] |
| 24 | ChCl:Acetyl salicylic acid (1:1) | GAFF/no corrections | Investigation of DES structure | [222] |
| 25 | ChCl:Citric acid (1:1) ChCl:Fructose (1:1) ChCl:Malic acid (1:1) ChCl:Lactic acid (1:1) | No name of FF.Parameters are given | Gas Solubility and Rheological Behavior of Natural DES | [54] |
| 26 | ChCl:Ethylene glycol Different molar ratios | OPLS-AA/no corrections | DES for CO2 separation | [274] |
| 27 | ChCl:Ethylene glycol (1:1) ChCl:Ethylene glycol (1:2) | OPLS-AA CL&P/charges scaling for ions is 0.8 | Water effect on DES structure | [294] |
| 28 | ChCl:Ethylene glycol (1:2) | GAFF/ charges scaling for ions is 0.9 | CO2 uptake by a DES in slit nanopores | [311] |
| 29 | ChCl:Ethylene glycol (1:2) | OPLS/no corrections | DES at a solid electrode | [315] |
| 30 | ChCl:Ethylene glycol (1:2) | INTERFACE and CGenFF | DES at solid interfaces | [317] |
| 31 | ChCl:Ethylene glycol (1:2) | GAFF/charges scaling for ions is ±0.9 | Investigation of DES structure | [161] |
| 32 | ChCl:Ethylene glycol (1:2) | CHARMM General Force Field/no corrections | Investigation of DES structure | [213] |
| 33 | ChCl:Ethylene glycol (1:2) | FF developed by authors (0.8 FFM3) based on OPLS | Investigation of DES structure | [214] |
| 34 | ChCl:Ethylene glycol (1:2) | CL&Pol | Force field validation | [137] |
| 35 | ChCl:Ethylene glycol (1:2) | CL&PolCL&P | Force field validation | [140] |
| 36 | ChCl:Ethylene glycol (1:2) | OPLS-AA/no corrections | Water effect on DES physicochemical properties | [302] |
| 37 | ChCl:Ethylene glycol (1:2) | GAFF/no corrections | Solvatochromic parameters, and preferential solvation in aqueous solutions of DES and its components | [293] |
| 38 | ChCl:Ethylene glycol (1:2) | OPLS/no corrections | CO2 absorption in DES | [158] |
| 39 | ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Malonic acid (1:2) | GAFF/charge scaling for ions is 0.9 | Investigation of DES structure | [124] |
| 40 | ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Urea (1:2) | GAFF/no corrections | Solvation dynamics of an ionic probe in DES | [157] |
| 41 | ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Urea (1:2) | GAFF/no corrections | Investigation of separation mechanism of methanol hexane mixtures | [285] |
| 42 | ChCl:Ethylene glycol (1:2) ChCl:Levulinic acid (1:2) | GAFF/charge scaling for ions is 0.9 for systems with ethylene glycol 0.8—for systems with levulinic acid | Fluorinated refrigerants in DESs | [120] |
| 43 | ChCl:Ethylene glycol (1:2) ChCl:Levulinic acid (1:2) | ChCl and Ethylene glycol–GAFF Levulinic acid-HF/6–31G* + AMBER or DFT for optimized clusters | DES for gas separation | [275] |
| 44 | ChCl:Ethylene glycol (1:2) ChCl:Propylene glycol (1:2) ChCl:1,3-propanediol (1:2) ChCl:Glycerol (1:2) | FF developed by authors (0.74 FFM16) based on + OPLS | Investigation of DES structure | [217] |
| 45 | ChCl:Ethylene glycol (1:3) ChCl:Glycerol (1:3) | COMPASS | Silica nanoparticles in DES | [378] |
| 46 | ChCl:Ethylene urea (1:2) ChCl:Thiourea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | “UNIVERSAL” force field Forcite and Blends Module Materials studio package | Desulfurization mechanism by the DES solvents | [280] |
| 47 | ChCl:Glucose (1:3, 1:1, 3:1) | CHARMM 36/no corrections | Investigation of DES structure | [379] |
| 48 | ChCl:Glycerol | OPLS-AA/no corrections | Gels based on DES and cellulose | [127] |
| 49 | ChCl:Glycerol:Resorcinol (1:5:3) | AMBER Parameters are given | Investigation of DES structure | [246] |
| 50 | ChCl:Glycerol (1:2) | CHARMM 36/no corrections | Water effect on DES structure | [287] |
| 51 | ChCl:Glycerol (1:1) | Gromos54a7/no corrections | Effects of a cholinium based DES on function and structure of versatile peroxidase | [380] |
| 52 | ChCl:Glycerol (1:2, 1:3) | GAFF/no corrections | Investigation of DES structure | [215] |
| 53 | ChCl:Glycerol (1:2) | GAFF/charge scaling is 0.9 | Protein in DES | [333] |
| 54 | ChCl:Glycerol (1:2) | GAFF/no corrections | solvation of enzyme in DES | [381] |
| 55 | ChCl:Glycerol (1:2) ChCl:Ethylene glycol (1:2) | GAFF OPLS-DES CGenFF no corrections | Validation of force field for mixtures of DES and water and interaction DES with enzyme | [382] |
| 56 | ChCl:Glycerol (1:2) | GAFF/charge scaling for ions is 0.9 | Effect of water on thermophysical properties DES | [304] |
| 57 | ChCl:Lactic acid (1:1) β-alanine:Lactic acid (1:1) | No name of FF. Parameters are given | Lidocaine in DES | [55] |
| 58 | ChCl:Lactic acid (1:9) | GAFF/no corrections | Lignin dissolution behaviors of DES | [383] |
| 59 | ChCl:Levulinic acid (1:2) | No name of FF. Parameters are given | DES on the metal surface | [318] |
| 60 | ChCl:Levulinic acid (1:2) | No name of FF. Parameters are given | DES for CO2 capture | [52] |
| 61 | ChCl:Malonic acid ChCl:Oxalic acid ChCl:Succinic acid ChCl:Fumaric acid Different molar ratios | GAFF/no corrections | Investigation of DES structure | [221] |
| 62 | ChCl:Malonic acid (1:1, 1:2) | GAFF/no corrections | Separation of nitrogen-containing aromatics by DES | [384] |
| 63 | ChCl:Oxalic acid (1:1) | CHARMM 36/no corrections | Biomass separation | [341] |
| 64 | ChCl:Phenyl propionic acid (1:2) | CHARMM 36/charges scaling is 0.8. | Investigation of DES structure | [224] |
| 65 | ChCl:Phenyl propionic acid Different molar ratios | CHARMM 36/no corrections | Investigation of DES structure | [223] |
| 66 | ChCl:Phenylacetic acid (1:2) | No name of FF. Parameters are given | CO2 absorption with DES | [53] |
| 67 | ChCl:Propylene glycol (1:2) ChCl:Ethylene glycol (1:2) | FF developed by authors (0.74 FFM16) based on OPLS | Investigation of DES structure | [216] |
| 68 | ChCl:Sesamol (1:3) | OPLS-AA/no corrections | Water effect on DES structure | [296] |
| 69 | ChCl:Urea (1:2) | No name of FF. Parameters are given | Investigation of enhanced oil recovery by DES | [385] |
| 70 | ChCl:Urea Different molar ratios | CL&P/charge scaling for ions is 0.9. | Investigation of DES structure | [214] |
| 71 | ChCl:Urea Different molar ratios | GAFF/charge scaling for ions is 0.8 | Investigation of DES structure | [123] |
| 72 | ChCl:Urea (1:2) | GROMOS 96/no corrections | Protein in DES | [330] |
| 73 | ChCl:Urea (1:2) | GAFF/charge scaling is 0.8 | DNA in DES | [335] |
| 74 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Protein in DES | [332] |
| 75 | ChCl:Urea (1:2) | GAFF/charge scaling is 0.8 | DNA in DES | [334] |
| 76 | ChCl:Urea (1:2) | CHARMM 22/no corrections | DES near graphene | [322] |
| 77 | ChCl:Urea (1:2) | No name of FF. Parameters are given | DES at 2D nanomaterial interfaces | [313] |
| 78 | ChCl:Urea (1:2) | No name of FF. Parameters are given | DES with different nanotubes | [324] |
| 79 | ChCl:Urea (1:2) | Combine force field based on CGenFF and CHARMM 36 | DES nanodroplet at carbon material | [323] |
| 80 | ChCl:Urea (1:2) | GAFF/ charges scaling is 0.9 for the ions | DES droplets on ionic substrates | [320] |
| 81 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Dissolution of cellulose in DES | [21] |
| 82 | ChCl:Urea (1:2) | q4-MD force field/no corrections | Molecular encapsulation | [386] |
| 83 | ChCl:Urea (1:2) | Monte Carlo/Empirical potential structure refinement (EPSR) | Investigation of DES structure | [387] |
| 84 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Separation of uranyl Ions | [388] |
| 85 | ChCl:Urea (1:2) | Various force-fields | Force fields comparison | [228] |
| 86 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Flow resistance of DES confined in ionic model nanoslits | [389] |
| 87 | ChCl:Urea (1:2) | CHARMM 36/no corrections | Conformation and Stability of Lysozyme in DES/water mixtures | [291] |
| 88 | ChCl:Urea (1:2) | CHARMM 36/no corrections | Stability and activity of lipase in DES/water mixtures | [305] |
| 89 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Water effect on DES structure | [295] |
| 90 | ChCl:Urea (1:2) | CHARMM 36/no corrections | Water effect on DES structure | [286] |
| 91 | ChCl:Urea (1:2) | CHARMM 36/charge scaling is 0.89 | Water effect on DES structure | [292] |
| 92 | ChCl:Urea (1:2) | OPLS-AA/no corrections | Effect of DES on the water structure | [300] |
| 93 | ChCl:Urea (1:2) | Merck Molecular Force Field (MMFF) | Water effect on DES thermo-physical properties | [36] |
| 94 | ChCl:Urea (1:2) | Gromos54a7/no corrections | Intermolecular interactions between DES and dimethylsulfoxide | [390] |
| 95 | ChCl:Urea (1:2) Butiyltrimetylammonium chloride:Urea (1:2) | Lopes-Padua and OPLS-AA/no corrections | Investigation of DES structure | [211] |
| 96 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) | GAFF based FF/charge scaling for ions is 0.9 for systems with urea 0.8—for systems with ethylene glycol | Water effect on DES properties | [298] |
| 97 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) | Merck Molecular Force Field (MMFF) | Water effect on DES properties | [303] |
| 98 | ChCl:Urea (1:2) ChCl:Ethylene Glycol (1:2) | No name of FF. Aspen plus simulator | CO2 removal from shale gas by DESs | [273] |
| 99 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | GAFF based FF/charge scaling for ions is 0.9 for systems with urea 0.8—for systems with ethylene glycol or glycerol | DES for absorption refrigeration systems | [391] |
| 100 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | GAFF based FF/charge scaling for ions is 0.9 for systems with urea 0.8—for systems with ethylene glycol or glycerol | Water effect on DES thermal conductivity | [299] |
| 101 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | Gromos54a7/no corrections | Water effect on DES structure | [392] |
| 102 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | GAFF/no corrections | Solvatochromic properties and ion solvation structure in DESs | [393] |
| 103 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | GAFF/no corrections | Solvatochromic behavior of dimethyl sulfoxide with DESs | [394] |
| 104 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) and their mixtures | No name of FF. Parameters are given | DESs on 2D materials | [321] |
| 105 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Malonic acid (1:1) ChCl:Oxalic acid (1:1) | OPLS, GAFF/scaling of 1–4 intramolecular interaction energies | Hildebrand and Hansen solubility parameters of DESs | [395] |
| 106 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Oxalic acid (1:1) ChCl:Malonic acid (1:1) ChCl:Glutaric acid (1:1) ChCl:Malic acid (1:1) ChCl:Citric acid (1:1) ChCl:Levulinic acid (1:2) ChCl:Phenyl acetic acid (1:2) ChCl:Acetamide (1:2) | Combine force field based on CGenFF and CHARMM 36 | Lipid membrane in DES | [336] |
| 107 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Propylene glycol (1:2) | GAFF/no corrections | extraction mechanism of 1-butanol separation from alkanol azeotropic system using choline-based DES | [284] |
| 108 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Levulinic acid (1:2) Betaine:Levulinic acid (1:2) | Modified AMBER force field | Mercury Capture from Petroleum using DES | [159] |
| 109 | ChCl:Urea (1:2) ChCl:Ethylene Glycole (1:2) ChCl:Glycerol (1:2) ChCl:2-aminoethan-1-ol (1:6) Allyltriphenyl phosphonium bromid:Ethylene glycol (1:4) Allyltriphenyl phosphonium bromid:Triethylene Glycol (1:4) Allyltriphenyl phosphonium bromid:Levulinic acid (1:4) | No name of FF. Forcite module, from Materials Studio software | Molecular interaction between the different DESs and CO2 | [277] |
| 110 | ChCl:Urea (1:2) ChCl:Glycerol (1:2) ChCl:Levulinic acid (1:2) ChCl:Malonic acid (1:1) ChCl:Phenylacetic acid (1:2) | No name of FF. Parameters are given Charges are calculated for clusters. | Investigation of DES structure | [396] |
| 111 | ChCl:Urea (1:2) ChCl:Glycerol (1:2) ChCl:Malonic acid (1:1) | No name of FF. MDynaMix v.5.2 molecular modeling software | Mechanisms of acid gases capture in DES | [268] |
| 112 | ChCl:Urea (1:2) ChCl:Glycolic acid (1:1) | OPLS/charge scaling is 0.8 | Stability and activity of alcohol dehydrogenase in DES/water mixtures | [288] |
| 113 | ChCl:Urea (1:2) ChCl:Thiourea (1:2) ChCl:Methyl urea (1:2) ChCl:Dimethyl urea (1:2) ChCl:1,1-dimethylurea (1:2)ChCl:N,N′-ethylene urea (1:2) | GAFF/charges scaling is 0.9 for urea and 0.8 for ChCl | Investigation of DES structure | [118] |
| 114 | ChCl:Urea (1:2) ChF:Urea (1:2) ChNO3:Urea (1:2) Ch acetate:Urea (1:2) | CL&P | Investigation of DES structure | [230] |
| 115 | ChCl: Water | ChCl: LJ parameters (AMBER) + partial charges (DFT) Water: TIP3P | Water structuring in DES | [397] |
| 116 | ChCl:Water (1:3.3) | OPLS-AA/no corrections | Investigation of DES structure | [398] |
| 117 | ChCl:Water | OPLS-AA/no corrections | Effect of ChCl on water structure | [301] |
| 118 | ChCl derivatives consisted of a series of elongated alkyl side chains and elongated alcohol side chains [Ch]+, [C4Ch]+, [C6Ch]+, [C8Ch]+, [(C4)3Ch]+, [ChC4OH]+, [ChC6OH]+, [ChC8OH]+, [ChC10OH]+, [ChC12OH]+) HBD:Ethylene glycol Molar ratio 1:4 | CL&P | Investigation of DES structure | [239] |
| 119 | Choline chloride:Trifluoroacetamide (1:2.5) Chlorocholine chloride:Trifluoroacetamide (1:2.5) Tetrametilammonium chloride:Trifluoroacetamide (1:2.5) Tetraethylammonium chloride:Trifluoroacetamide (1:2.5) Benziltriethylammoniun chloride:Trifluoroacetamide (1:2.5) | GAFF/no corrections | Investigation of DES structure | [240] |
| 120 | Choline iodide:Glycerol (1:3) | GAFF/ charges scaling is 0.9 for for the ions | DES in nanopores | [309] |
| 121 | Decanoic acid:Menthol (1:1, 1:2) Decanoic acid:Lidocain (2:1) Menthol:Lidocain (2:1) Thymol:Lidocaine (1:1, 2:1) Thymol:Menthol (1:1, 2:1) | OPLS-AA/M Parameters are given | Investigation of DES structure | [238] |
| 122 | Menthol:Lauric acid 2:1 Menthol:Decanoic acid 1:1 | GAFF/no corrections | Investigation of DES structure | [19] |
| 123 | dl-menthol:Hexanoic acid (1:1) dl-menthol:Octanoic acid (1:1) dl-menthol:Decanoic acid (1:1) | CHARMM 36/no corrections | Dynamics of hydrogen-bonding and translational dynamics and their dependence on acid tail length | [399] |
| 124 | dl-menthol based DESs HBDs: Acetic acid, Butanoic acid, Hexanoic acid, Octanoic acid, Decanoic acid, Dodecanoic acid, Pyruvic acid, Levulinic acid Tetrabutylammonium chloride based DESs HBDs: Acetic acid, Octanoic acid | GAFF/no corrections | Water Stability of Hydrophobic DES | [219] |
| 125 | Ferric chloride:Tetrabutylphosphonium bromide Different molar ratios | Gromos54A7 | Investigation of DES structure | [400] |
| 126 | L-Arginine:Glutamic acid (1:1) L-Arginine:Oxalic acid (1:1) L-Arginine:Tartaric acid (1:1) | No name of FF. Parameters are given | Nitric oxide solubility in DES | [152] |
| 127 | l-Menthol:Acetic acid (1:1) | AMBER 14 | Structure Elucidation of DES | [155] |
| 128 | LiBr:Acetamide (4.5:1) LiNO3:Acetamide (4.5:1) LiClO4:Acetamide (4.5:1) | OPLS-UA | Investigation of DES structure | [244] |
| 129 | LiBr:Acetamide (4.5:1) LiNO3:Acetamide (4.5:1) LiClO4:Acetamide (4.5:1) | CHARMM 22 for nucleic acids | Investigation of DES structure | [243] |
| 130 | LiCl and LiTFSI based DESs HBDs: Urea, Acetamide:N,N0-dimethylpropyleneurea, 2-imidazolidinone, TetramethylureaMolar ratio 1:5 | COMPASS II | Investigation of DES structure | [245] |
| 131 | LiClO4:Acetamide (1:3.5) | CHARMM 27/no corrections | Water effect on DES properties | [401] |
| 132 | LiClO4:Acetamide (1:5.2) LiClO4:Propion amid (1:5.2) | CHARMM 22/no corrections | Investigation of DES structure | [242] |
| 133 | Litium bis-(trifluoromethanesulfonyl)-imide: Urea Different molar ratios | OPLS-AA/no corrections | Investigation of DES structure | [402] |
| 134 | Litium bis-(trifluoromethanesulfonyl)-imide: Urea Different molar ratios | OPLS-AA/no corrections | Investigation of DES structure | [403] |
| 135 | Methyltriphenylphosphonium bromide:Ethylene glycol (1:4) | GAFF/no corrections | Extraction of benzene from hydrocarbon mixture using a phosphonium based DES | [282] |
| 136 | Methyltriphenylphosphonium bromide:Ethylene glycol (1:4) | GAFF/no corrections | Extraction of a polyaromatic hydrocarbon from fuel oils using DES | [283] |
| 137 | Methyltriphenylphosphonium bromide:Ethyleneglycol (1:4) Methyltriphenylphosphonium bromide:Glycerol (1:4) Tetrabutylammonium bromide:Ethyleneglycol (1:4) Tetrabutylammonium bromide:Glycerol (1:4) | GAFF/no corrections | Investigation of DES structure | [232] |
| 138 | Methyltriphenylphosphonium bromide:Monoethanolamine Different molar ratios | No name of FF. Parameters are given | Investigation of DES structure | [233] |
| 139 | Monoethanolamine (MEA) and Methyltriphenylphosphonium bromide (MTPPBr) based DESs. Different molar ratios | Gromos54a7, Ions: MTPP+ (AMBER), MEA and CO2: ATB database | CO2 absorption in DES | [272] |
| 140 | Monoethanolamine hydrochloride:Methyldiethanolamine Diethanolamine hydrochloride:Methyldiethanolamine N-methyl diethanolamine hydrochloride:Methyldiethanolamine Different molar ratios | GAFF/no corrections | CO2 capture performance by DES | [404] |
| 141 | Morpholine and Morpholine based DESs. HBDs: Urea, Diethylene glycol, Carboxylic acid, Thiourea, Methanol Molar ratio 1:4 | OPLS-AA/no corrections | Investigation of DES structure | [405] |
| 142 | Oleic acid:Lidocaine (1:1) | GAFF/no corrections | Phase separation property of a hydrophobic DES | [306] |
| 143 | Proline:Glycolic acid Proline:Malic acid Different molar ratios | Amber-Cornell force field | Investigation of DES structure | [229] |
| 144 | Tetraalkylammonium chloride:Decanoic acid (1:2) Cation alkylchain lengths-4, 7, 8) | GAFF/charges scaling is 0.6–1.0 LJ well-depth scaling factors | Investigation of DES structure | [237] |
| 145 | Tetrabutilamonium chloride:Ethylene glycol (1:3) Tetrabutilamonium chloride:Glycerol (1:5) | OPLS-AA/partial charges scaling for ions is 0.8 | Investigation of DES structure | [406] |
| 146 | Tetrabutylammonium bromide:Formic acid (1:1) | No name of FF. Parameters are given | Oil desulfurization by DES | [149] |
| 147 | Tetrabutylammonium bromide:Imidazole (1:2) Tetrabutylammonium bromide:Ethylene glycol (1:4) Tetrabutylammonium bromide:Glycerol (1:4) | GAFF/no corrections | Dissolution of carbohydrates in DES | [338] |
| 148 | Tetrabutylammonium bromide:Sulfolane Different molar ratios | OPLS-AA and CL&P | Quantification of the total vapor pressures of DESs | [407] |
| 149 | Tetrabutylammonium chloride:Decanoic acid (1:2) Thymol:Decanoic acid (1:2) dl-menthol:Decanoic acid (2:1) | GAFF/charge scaling for ions is 0.833 | Interfacial Properties of Hydrophobic DES | [307] |
| 150 | Tetrabutylammonium chloride:FeCl3: Polyethylene glycol (4:0.05:1) | Gromos54a7/no corrections | Investigation of DES structure | [408] |
| 151 | Tetrabutylammonium chloride:Polyethylene glycol:Ferric chloride (4:1:0.05) | Gromos54a7/no corrections | Mechanism of desulfurization by the DES | [281] |
| 152 | Tetrabutylphosphonium bromide:Phenol (1:4) Tetrabutylphosphonium bromide:Diethylene glycol (1:4) Allyltriphenylphosphonium bromide:Phenol (1:4) Allyltriphenylphosphonium bromide:Phenol (1:6) | AMBER/no corrections | CO2 solubility in DESs | [278] |
| 153 | Triethylammonium acetate:Urea (1:2) | OPLS-AA/no corrections | Protein in DES | [331] |
| No | DES | Method/Basis | Topic of the Publication | Reference |
|---|---|---|---|---|
| 1 | 1-ethyl-3-methylimidazolium chloride: Imidazole Different molar ratios | AIMD The BLYP functional with triple-ζ valence polarization basis set and GTH pseudopotentials. CP2K/QUICKSTEP code DFT B3LYP/6-311+G(2d,2p) | DES as physical solvents for remarkable separation of H2S from CO2 | [409] |
| 2 | 1,8-diazabicyclo [5.4.0]undec-7-enium: methylthiourea (4:1) | DFT M06e2X/6-311++G(d,p) | Dissolution of cellulose in DES | [339] |
| 3 | 24 choline chloride-based DES with molar ratio from 1:4 to 15:1 | DFT B3LYP/6-311G(d,p) B3LYP/def2-SVP def2-SVP/J B3LYP/6-311G(d,p)/def2-TZVP def2-TZVP/J | Biomass separation | [119] |
| 4 | Alanine:Lactic Acid (1:1) Alanine:Malic Acid (1:1) Betaine:Lactic Acid (1:1) ChCl:Malic Acid (1:1) ChCl:Lactic Acid (1:1) ChCl:Fructose (1:1) | DFT B3LYP/6-311++G(d,p) | CO2 absorption by DES | [79] |
| 5 | Alanine:Lactic acid (1:1) Betaine:Lactic acid (1:1) ChCl:Lactic acid (1:1) ChCl:Malic acid (1:1) ChCl:Phenylacetic acid (1:2) | DFT B3LYP/6-311++G(d,p) | High Pressure Methane Solubility in Natural DES | [410] |
| 6 | AlCl3:Urea (1:1, 1.5:1) | AIMD DFT/PAW | Investigation of DES structure | [160] |
| 7 | Arginine:Glutamic acid (1:1) Arginine:Oxalic acid (1:1) Arginine:Tartaric acid (1:1) | DFT B3LYP/6-311++G(d,p) | Lidocaine in DES | [150] |
| 8 | Arginine:Glutamic acid (1:1) Arginine:Oxalic acid (1:1) Arginine:Tartaric acid (1:1) | DFT B3LYP/6-311++G(d,p) | Antibiotics in DES | [151] |
| 9 | Betain:Glycerol (1:2) Betain:dl-lactic acid (1:2) Betain:Levulinic acid (1:2) | DFT GGA/VMN-BP function in DNP 4.4 basis set | Extraction phenolic compounds from oil mixtures by DES | [95] |
| 10 | Betaine:Lactic acid (1:1) | DFT B3LIP/6-311++G(d,p) | Investigation of DES structure | [153] |
| 11 | Camphor:1-decanol Camphor:Decanoic acid Camphor:3,4-xylenol | DFT B3LYP/6-311++G(d,p) | Detoxification of feedstocks using DES | [74] |
| 12 | Caprolactam:Tetrabutylammonium bromide (1:1) Caprolactam:Tetrabutylammonium bromide (1:1) ChCl:Urea (1:2) Methyltriphenylphosphonium bromide:Monoethanolamine (1:6) | DFT B3LYP/6-311++G(d,p) | Natural Gas Desulfurization using DES | [375] |
| 13 | Ceineole:Succinic acid (1:1) Ceineole:Malic acid (1:1) Ceineole:Lactic acid (1:1) | DFT B3LIP/6-311++G(d,p) | Investigation of DES structure | [154] |
| 14 | ChCl:1,2-butanediol (1:4) ChCl:1,3-butanediol (1:4) ChCl:1,4-butanediol (1:4) | DFT DMol3 module with the generalized gradient approximation (GGA)-based Perdew-Wan (PW91) functional | The structure of DES and supercapacitor performance | [88] |
| 15 | ChCl:4-chlorophenol ChCl:4-ethylphenol ChCl:Phenol ChCl:2-methylphenol ChCl:3-methylphenol ChCl:4-methylphenol ChCl:2,6-dimethylphenol Different molar ratios | DFT M06-2x/6–31++G(d,p) | Extractive desulfurization of fuels by DES | [86] |
| 16 | ChCl:Acetyl salicylic acid (1:1) | DFT ωB97XD/6-311++G(d,p) | Investigation of DES structure | [222] |
| 17 | ChCl:Acrylic acid (1:2) | Gaussian software No name of method | Investigation of DES structure | [220] |
| 18 | ChCl:Carboxylic acid (1:2) ChCl:Formic acid (1:2) | DFT B3LYP/DNP | Investigation of DES structure | [68] |
| 19 | ChCl:Citric acid ChCl:Ethylene glycol ChCl:Fructose ChCl:Glycerol ChCl:Lactic acid ChCl:Levulinic acid ChCl:Malic acid ChCl:Phenylacetic acid 1-Butyl-3-methylimidazolium chloride:Acetamide 1-Ethyl-3-methylimidazolium chloride:Acetamide 1-Ethyl-3-methylimidazolium chloride:Ethylene glycol | DFT B3LYP/6-311++G(d,p) | Interactions Between Deep Eutectic Solvents and SO2 | [78] |
| 20 | ChCl:d-(+)-xylose ChCl:d-(−)-ribose ChCl:d-(−)-fructose Different molar ratios | DFT B3LYP/6-311++G(d,p) | DES as absorbents for NH3 capture | [94] |
| 21 | ChCl:Ethylene glycol (1:2) | AIMD GPW/PBE/MOLOPT-TZVP-GTH basis set | Investigation of DES structure | [161] |
| 22 | ChCl:Ethylene glycol (1:2) | AIMD BLIP/DZVP | Investigation of DES structure | [214] |
| 23 | ChCl:Ethylene glycol (1:2) | DFT/PAW | Eutectic-mediated selective synthesis of nanocrystals | [297] |
| 24 | ChCl:Ethylene glycol (1:2) | DFT B3LYP/6-31G(d) TD-DFT CAM-B3LYP/TZVP | Solvatochromic parameters, and preferential solvation in aqueous solutions of DES and its components | [293] |
| 25 | ChCl:Ethylene glycol (1:2) | DFT B3LYP/6–311++G(d,p) | Dissolution of dimethylformamide in DES | [411] |
| 26 | ChCl:Ethylene glycol (1:2) | AIMD MOLOPT-DZVP-SR-GTH with GGA BLYP with GTH pseudopotentials to represent the core electrons | CO2 absorption in DES | [158] |
| 27 | ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Urea (1:2) | DFT B3LYP/6-311++G(d,p) | Solvation dynamics of an ionic probe in DES | [157] |
| 28 | ChCl:Ethylene glycol (1:2) ChCl:Malic acid (1:2) ChCl:Tartaric acid (2:1) ChCl:Oxalic acid (1:2) ChCl:Glycerol (1:2) | HF, M06, B3LYP, CAM-B3LYP or PBEPBE/ 6-31G(d,p) | Investigation of DES structure | [67] |
| 29 | ChCl:Glycerol (1:1) | DFT BLYP/MOLOPT-DZVP-SR-GTH | SO2 solvation in DES | [102] |
| 30 | ChCl:Glycerol (1:2) | DFT M062x/ 6-31++G(d,p) | Investigation of DES structure | [82] |
| 31 | ChCl:Glycerol (1:2) ChCl:Glycerol (1:3) ChCl:Malonic acid (1:1) | DFT B3LYP/6-31+G(d,p) | Mitigation of CO2 in DES | [72] |
| 32 | ChCl:Imidazole:Ethylene glycol (3:7:14) ChCl:Triazole:Ethylene glycol (3:7:14) ChCl:Tetrazole:Ethylene glycol (3:7:14) | AIMD BLYP triple-ζ valence polarization basis set and GTH pseudopotentials DFT 6-31++G(d,p) | DES for Highly Efficient and Reversible Capture of Ammonia | [412] |
| 33 | ChCl:Lactic acid:Ethanol (1:2:1) ChCl:Lactic acid:Ethylene glycol (1:2:1) | Semiempirical method with the PM6 level | Investigation of DES structure | [60] |
| 34 | ChCl:Lactic acid (1:1) β-alanine:Lactic acid (1:1) | DFT B3LYP/6-311++G(d,p) | Lidocaine in DES | [55] |
| 35 | ChCl:Lactic acid (1:9) | DFT M06-2X functional with the standard 6-311+G(d,p)//6-311G(d,p) basis set. | Lignin dissolution behaviors of DES | [383] |
| 36 | ChCl:Levulinic acid (1:2) | DFT B3LYP/6-31+G(d,p) | DES for CO2 capture | [52] |
| 37 | ChCl:Malonic acid:1, 4-butanediol (1:1:1) | DFT B3LYP/3-21G semiempirical method with the PM6 level | Investigation of DES structure | [247] |
| 38 | ChCl:Malonic acid:n-butanol (1:1:1) ChCl:Malonic acid:Iso-butanol (1:1:1) ChCl:Malonic acid:Butandiol (1:1:1) | Semiempirical method with the PM6 level | Investigation of DES structure | [59] |
| 39 | ChCl:Oxalic acid ChCl:Citric acid ChCl:Glycerol ChCl:Ethylene glycol Different molar ratios | 3–21G basis set | Toxicity assessment and enhanced drug solubility profile of green DES derivatives | [413] |
| 40 | ChCl:Phenol (1:2) ChCl:Glycol ethylene (1:2) ChCl:Levulinic acid (1:2) | DFT B3LYP/6-311++G(d,p) | Desulfurization using DES | [71] |
| 41 | ChCl:Phenylacetic acid (1:2) | DFT B3LYP/6-31+G(d,p) | CO2 absorption with DES | [53] |
| 42 | ChCl:R-3-hydroxyl acids Different molar ratios | DFT B3LYP/6-31+G(2d,2p) B3LYP-D3/6-311++G(2d,2p) | Investigation of DES structure | [77] |
| 43 | ChCl:Urea (1:1) ChCl:Ethylene glycol (1:1) | AIMD BLYP/triple-ζ double-polarization basis set and Goedecker–Teter–Hutter pseudopotentials | Solvation structure around CO2 and SO2 in DESs | [90] |
| 44 | ChCl:Urea (1:2) | DFT M06e2X/6-311++G(d,p) | Interaction of Cu, Ag and Au nanoparticles with DES | [87] |
| 45 | ChCl:Urea (1:2) | DFT B3LYP/6-311++G(d,p) | Investigation of DES structure | [76] |
| 46 | ChCl:Urea (1:2) | DFT B3LYP/6-311++G(2d,p) | Investigation of DES structure | [64] |
| 47 | ChCl:Urea (1:2) | periodic DFT B3LYP/6-31+G(d,p) | Investigation of DES structure | [70] |
| 48 | ChCl:Urea (1:2) | AIMD DFT B3LYP/TZ2P | Water effect on DES structure | [109] |
| 49 | ChCl:Urea (1:2) ChCl:Acetylsalicylic acid (1:2) ChCl:Sesamol (1:2) ChCl:Pyrogallol (1:1) | DFT B3LYP/ ug-cc-pVTZ | Solubilization properties and structural characterization in DES | [93] |
| 50 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) | DFT M06-2X/6-31++G(d,p) | Extractive Desulfurization of Fuel with DES | [85] |
| 51 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | AIMD DFTB3 | Investigation of DES structure | [210] |
| 52 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) | DFT B3LYP/cc-pVDZ | Solvatochromic behavior of dimethyl sulfoxide with DESs | [394] |
| 53 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Glycerol (1:2) ChCl:Propylene glycol (1:2) | DFT B3LYP/6-311G(d) | Extraction mechanism of 1-butanol separation from alkanol azeotropic system using choline-based DES | [284] |
| 54 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) ChCl:Levulinic acid (1:2) Betaine:Levulinic acid (1:2) | AIMD BLYP with a triple-ζ, double polarization basis set for nonmetal atoms, and GTH pseudopotentials | Mercury Capture from Petroleum Using DES | [159] |
| 55 | ChCl:Urea (1:2) ChCl:Ethylene glycol (1:2) Choline iodide:Glycerol (1:1) ChCl:Benzoic acid (1:2) | DFT M06-2X/cc-pVDZ level | Adsorption of DESs onto graphene and defective graphene nanoflake | [314] |
| 56 | ChCl:Urea (1:2) ChCl:Glycerol (1:2) ChCl:Malonic acid (1:1) | DFT B3LYP/6-311+G(d) | Mechanisms of acid gases capture at relevant interfaces and at atomistic level in DES | [268] |
| 57 | ChCl:Urea (1:2) ChCl:Thio urea (1:2) ChCl:Ehylene glycol (1:2) ChCl:Glycerol (1:2) | DFT B3LYP/6-31G | Ammonium-based DES for secondary water flooding | [414] |
| 58 | ChCl:Urea (1:2) ChCl:Thio urea (1:2) ChCl:Methyl urea (1:2) ChCl:Dimethyl urea (1:2) ChCl:1,1-dimethylurea (1:2) ChCl:N,N′-ethylene urea (1:2) | AIMD BLYP-D3(BJ) | Investigation of DES structure | [119] |
| 59 | ChCl based DESs HBDs: straight-chain monobasic acids | DFT B3LYP/6-31G(d) | Sulfur removal by DES | [73] |
| 60 | ChCl:Urea (1:2) | BLYP-D2/ DZVP-MOLOPT-SR-GTH | Investigation of DES structure | [209] |
| 61 | DESs based on LiCl and LiTFSI HBD: Urea, Acetamide, N,N0-dimethylpropyleneurea, 2-imidazolidinone, Tetramethylurea Molar ratio 1:5 | DFT B3LIP/6-311++G(d,p) | Investigation of DES structure | [245] |
| 62 | L-Arginine:Glutamic acid (1:1) L-Arginine:Oxalic acid (1:1) L-Arginine:Tartaric acid (1:1) | DFT B3LYP/6-31++G(d,p) | Nitric oxide solubility in DES | [152] |
| 63 | L-Menthol:Acetic acid (1:1) | ωB97XD/6-311G(d,p) | Structure Elucidation of DES | [155] |
| 64 | Lactic acid:Alanine (7:1) Lactic acid:Glycine (7:1) Lactic acid:Histidine (9:1) | DFT B3LYP/6-311G(d,p) | Vapor−liquid equilibria, vapor pressure and water activity for the aqueous solutions of natural deep eutectic solvents (NDESs) | [66] |
| 65 | Litium bis-(trifluoromethanesulfonyl)-imide:Urea Different molar ratios | B3LYP/ def2-TZVPP | Investigation of DES structure | [403] |
| 66 | Methyltriphenylphosphonium bromide:Ethylene glycol (1:4) | DFT B3LYP/6-31G(d) | Extraction of a polyaromatic hydrocarbon from fuel oils using DES | [283] |
| 67 | Methyltriphenylphosphonium bromide:Glycerol (1:2) | HF/6-31G(d) | Adsorption of phenol and crystal violet dye on carbon nanotube functionalized with deep eutectic solvent | [96] |
| 68 | Monoethanolamine hydrochloride:Methyldiethanolamine Diethanolamine hydrochloride:Methyldiethanolamine N-methyl diethanolamine hydrochloride:Methyldiethanolamine Different molar ratios | DFT B3LYP/6-31G(d,p) | CO2 capture performance by DES | [402] |
| 69 | Tetrabutylammonium bromide:Formic acid (1:1) | DFT B3LYP/6-311++G(d,p) | Oil desulfuration by DES | [149] |
| 70 | Tetrabutylammonium bromide:Imidazole (1:2) Tetrabutylammonium bromide:Ethylene glycol (1:4) Tetrabutylammonium bromide:Glycerol (1:4) | DFT B3LYP/6-311+G(d) | Dissolution of carbohydrates in DES | [338] |
| 71 | Tetrabutylphosphonium bromide:Phenol (1:4) Tetrabutylphosphonium bromide:Diethylene glycol (1:4) Allyltriphenylphosphonium bromide:Phenol (1:4) Allyltriphenylphosphonium bromide:Phenol (1:6) | DFT B3LYP/6-31++G(d,p) | CO2 solubility in DESs | [278] |
| 72 | Tetraethylammonium bromide:Ethylene glycol (1:2) Tetraethylammonium chloride:Ethylene glycol (1:2) | DFT B3LYP/6-31G(d) | Influence of Br− and Cl− on DES | [61] |
| 73 | Tetraethylammonium chloride:Lactic acid (1:2) Tetrabutylammonium chloride:Lactic acid (1:2) Benzyltributylammonium chloride:Glycolic acid (1:2) Benzyltributylammonium bromide:Lactic acid (1:2) Dodecyltributylammonium chloride:Lactic acid (1:2) Tetrabutylammonium chloride:Lactic acid (1:2) Benzyltributylammonium chloride:Lactic acid (1:2) | DFT B3LYP/6-31+G(d,p) | DES complex with regulate magnetic (Fe3O4) metal organic framework | [65] |
| 74 | Triethylammonium:Formate Triethylammonium:Acetate Triethylammonium:Propionate Triethylammonium:Butanoate Triethylammonium:Pentanoate Different molar ratios | DFT M06-2X/6-31++G(d,p) | Extractive desulfurization process with DES | [84] |
| No | DES | Models and Methods | Topic of the Publication | Reference |
| 1 | DESs based on ammonium salt and on phosphonium salt | Artificial neural network model. A feed forward back propagation neural network with 9 hidden neurons. The group contribution method applied the modified Lydersen–Joback–Reid, Lee–Kesler and the modified Rackett equations | Prediction of DES densities | [186] |
| 2 | DESs based on ammonium salt and on phosphonium salt | Artificial neural network model 8-4-1. A feed-forward neural network with 4 hidden neurons Levenberg–Marquardt optimization method | Prediction of glycerol removal from palm-oil based biodiesel using DESs Total glycerol content | [187] |
| 3 | DESs based on amine with different HDSs | A combination of multi-linear regression and artificial neural networks methods The stepwise regression algorithm was used for the regression analysis of the experimental viscosity data expressed by s-profile and temperature multi-linear regression model descriptors | Prediction of the viscosity of DESs | [188] |
| 4 | DESs based on ChCl | Response surface methodology and Artificial neural networking | Study of the efficacy of 10 NDES (natural DES), including 3 new NDES, to extract procyanidins and anthocyanins from cranberry pomace | [189] |
| 5 | DESs based on ChCl | Artificial neural network model. A total of two types of neural network to analyze the feed-forward back-propagation and the layer recurrent | Prediction of lead removal from water using DES functionalized CNTs | [190] |
| 6 | N,N-diethylethanolammonium chloride:Glycerol (2:1) | The non-linear autoregressive network with exogenous inputs neural network strategy. The back-propagation training algorithm was selected to update the bias and weight vector values corresponding to the momentum, and the tangent sigmoid transfer function (tansig) was selected as the neuron transfer function for the network | Prediction of arsenic removal from water solution using DES functionalized CNTs | [191] |
| 7 | Benzyltriphenylphosphonium chloride:Glycerol (16:1) | The non-linear autoregressive network with exogenous inputs (NARX) neural network strategy. Three kinetic models were used to identify the adsorption rate and mechanism, and the pseudo-second order best described the adsorption kinetics | Prediction of arsenic removal from water solution using DES functionalized CNTs | [192] |
| 8 | Allyl triphenyl phosphonium bromide:Glycerol | Artificial neural network The non-linear autoregressive exogenous model network Feedforward neural network and layer recurrent neural network | Prediction of mercury removal from water solution using DES functionalized CNTs | [193] |
| 9 | DESs based on acid ChCl and Levulinic acid as HBAs | Particle Swarm Optimization to optimize an Artificial Neural Network Adaptive-network-based fuzzy inference system and particle swarm optimization Least square support vector Multilayer perceptron | Modeling of CO2 solubility in various DESs | [194] |
| 10 | DESs based on ChCl, N,N-diethyl ethanol ammonium chloride, and methyl triphenyl phosphonium bromide salts | Artificial neural network model. Feed-forward back propagation neural network with 8 hidden neurons | Prediction of the electrical conductivity DESs at different temperatures and compositions | [195] |
| 11 | Choline chloride:citric acid 1:1 Choline chloride: monohydrate 1:1 | Artificial neural network model. Artificial neural network and genetic algorithm approach The feed-forward backpropagation neural network algorithm with 4 input layer neurons and 2 output layer neurons for 4 independent and 2 dependent variables. The optimum number of hidden layer neurons was 11 | Experiment design for microwave-assisted extraction of phytochemical compounds from black jamun pulp | [196] |
| 12 | DESs based on allyltriphenylphosphonium bromide: Triethylene glycol with molar ratios of 1:4, 1:10 and 1:16 | Linear and quadratic regression models | Estimation of carbon dioxide solubility in DESs | [198] |
| 13 | DESs based on ChCl: Glycerol ChCl: P-coumaric acid | Principal component analysis Partial least squares Furthermore, based on molecular simulation, the detailed relationships between key variables were further analyzed | Revealing the biomass pretreatment mechanism by evaluating the inner relationships among 42 key process factors | [199] |
| 14 | DESs based on tetraalkylammonium bromide | Principal component analysis Regression analysis | Prediction of DES eutectic temperatures | [200] |
References
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, M.A.R.; Pinho, S.P.; Coutinho, J.A.P. Insights into the Nature of Eutectic and Deep Eutectic Mixtures. J. Solut. Chem. 2019, 48, 962–982. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Munro, H.L.; Rasheed, R.K.; Tambyrajah, V. Preparation of Novel, Moisture-Stable, Lewis-Acidic Ionic Liquids Containing Quaternary Ammonium Salts with Functional Side chainsElectronic Supplementary Information (ESI) Available: Plot of Conductivity vs. Temperature for the Ionic Liquid Formed from Zinc Chloride and Choline Chloride (2:1). See http://www.rsc.org/suppdata/cc/b1/b106357j. Chem. Commun. 2001, 19, 2010–2011. [Google Scholar]
- Meng, X.; Ballerat-Busserolles, K.; Husson, P.; Andanson, J.-M. Impact of Water on the Melting Temperature of Urea + Choline Chloride Deep Eutectic Solvent. New J. Chem. 2016, 40, 4492–4499. [Google Scholar] [CrossRef] [Green Version]
- Amde, M.; Liu, J.-F.; Pang, L. Environmental Application, Fate, Effects, and Concerns of Ionic Liquids: A Review. Environ. Sci. Technol. 2015, 49, 12611–12627. [Google Scholar] [CrossRef]
- Abo-Hamad, A.; Hayyan, M.; AlSaadi, M.A.; Hashim, M.A. Potential Applications of Deep Eutectic Solvents in Nanotechnology. Chem. Eng. J. 2015, 273, 551–567. [Google Scholar] [CrossRef]
- Nkuku, C.A.; LeSuer, R.J. Electrochemistry in Deep Eutectic Solvents. J. Phys. Chem. B 2007, 111, 13271–13277. [Google Scholar] [CrossRef]
- Cunha, S.C.; Fernandes, J.O. Extraction Techniques with Deep Eutectic Solvents. TrAC Trends Anal. Chem. 2018, 105, 225–239. [Google Scholar] [CrossRef]
- Alam, M.A.; Muhammad, G.; Khan, M.N.; Mofijur, M.; Lv, Y.; Xiong, W.; Xu, J. Choline Chloride-Based Deep Eutectic Solvents as Green Extractants for the Isolation of Phenolic Compounds from Biomass. J. Clean. Prod. 2021, 309, 127445. [Google Scholar] [CrossRef]
- Crump, M.R.; Bidinger, S.L.; Pavinatto, F.J.; Gong, A.T.; Sweet, R.M.; MacKenzie, J.D. Sensorized Tissue Analogues Enabled by a 3D-Printed Conductive Organogel. NPJ Flex. Electron. 2021, 5, 1–8. [Google Scholar] [CrossRef]
- Smirnov, M.A.; Fedotova, V.S.; Sokolova, M.P.; Nikolaeva, A.L.; Elokhovsky, V.Y.; Karttunen, M. Polymerizable Choline- and Imidazolium-Based Ionic Liquids Reinforced with Bacterial Cellulose for 3D-Printing. Polymers 2021, 13, 3044. [Google Scholar] [CrossRef] [PubMed]
- Pedro, S.N.; Freire, C.S.R.; Silvestre, A.J.D.; Freire, M.G. Deep Eutectic Solvents and Pharmaceuticals. Encyclopedia 2021, 1, 72. [Google Scholar] [CrossRef]
- Emami, S.; Shayanfar, A. Deep Eutectic Solvents for Pharmaceutical Formulation and Drug Delivery Applications. Pharm. Dev. Technol. 2020, 25, 779–796. [Google Scholar] [CrossRef]
- Svigelj, R.; Dossi, N.; Grazioli, C.; Toniolo, R. Deep Eutectic Solvents (DESs) and Their Application in Biosensor Development. Sensors 2021, 21, 4263. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Z.; Zhang, Z.; Zeng, S.; Li, F.; Zhang, X.; Nie, Y.; Zhang, L.; Zhang, S.; Ji, X. Ionic Liquids/deep Eutectic Solvents for CO2 Capture: Reviewing and Evaluating. Green Energy Environ. 2021, 6, 314–328. [Google Scholar] [CrossRef]
- Pelaquim, F.P.; Barbosa Neto, A.M.; Dalmolin, I.A.L.; Costa, M.C. da Gas Solubility Using Deep Eutectic Solvents: Review and Analysis. Ind. Eng. Chem. Res. 2021, 60, 8607–8620. [Google Scholar] [CrossRef]
- Choi, Y.H.; van Spronsen, J.; Dai, Y.; Verberne, M.; Hollmann, F.; Arends, I.W.C.E.; Witkamp, G.-J.; Verpoorte, R. Are Natural Deep Eutectic Solvents the Missing Link in Understanding Cellular Metabolism and Physiology? Plant Physiol. 2011, 156, 1701–1705. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Mohan, M.; Goud, V.V.; Banerjee, T. Operational Strategies and Comprehensive Evaluation of Menthol Based Deep Eutectic Solvent for the Extraction of Lower Alcohols from Aqueous Media. ACS Sustain. Chem. Eng. 2018, 6, 16920–16932. [Google Scholar] [CrossRef]
- Abranches, D.O.; Martins, M.A.R.; Silva, L.P.; Schaeffer, N.; Pinho, S.P.; Coutinho, J.A.P. Phenolic Hydrogen Bond Donors in the Formation of Non-Ionic Deep Eutectic Solvents: The Quest for Type V DES. Chem. Commun. 2019, 55, 10253–10256. [Google Scholar] [CrossRef] [Green Version]
- Smirnov, M.A.; Tolmachev, D.A.; Glova, A.D.; Sokolova, M.P.; Geydt, P.V.; Lukasheva, N.V.; Lyulin, S.V. Combined Use of Atomic Force Microscopy and Molecular Dynamics in the Study of Biopolymer Systems. Polym. Sci. Ser. C 2021, 63, 256–271. [Google Scholar] [CrossRef]
- Shao, S.; Do, T.N.; Razi, A.; Chitgupi, U.; Geng, J.; Alsop, R.J.; Dzikovski, B.G.; Rheinstädter, M.C.; Ortega, J.; Karttunen, M.; et al. Design of Hydrated Porphyrin-Phospholipid Bilayers with Enhanced Magnetic Resonance Contrast. Small 2017, 13, 1602505. [Google Scholar] [CrossRef]
- Lee, E.H.; Hsin, J.; Sotomayor, M.; Comellas, G.; Schulten, K. Discovery through the Computational Microscope. Structure 2009, 17, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [Green Version]
- Samarov, A.A.; Smirnov, M.A.; Sokolova, M.P.; Popova, E.N.; Toikka, A.M. Choline Chloride Based Deep Eutectic Solvents as Extraction Media for Separation of N-Hexane–ethanol Mixture. Fluid Phase Equilib. 2017, 448, 123–127. [Google Scholar] [CrossRef]
- Samarov, A.A.; Smirnov, M.A.; Toikka, A.M.; Prikhodko, I.V. Study of Deep Eutectic Solvent on the Base Choline Chloride as Entrainer for the Separation Alcohol–Ester Systems. J. Chem. Eng. Data 2018, 63, 1877–1884. [Google Scholar] [CrossRef]
- Wu, J.; Liang, Q.; Yu, X.; Lü, Q.-F.; Ma, L.; Qin, X.; Chen, G.; Li, B. Deep Eutectic Solvents for Boosting Electrochemical Energy Storage and Conversion: A Review and Perspective. Adv. Funct. Mater. 2021, 31, 2011102. [Google Scholar] [CrossRef]
- Vorobiov, V.K.; Smirnov, M.A.; Bobrova, N.V.; Sokolova, M.P. Chitosan-Supported Deep Eutectic Solvent as Bio-Based Electrolyte for Flexible Supercapacitor. Mater. Lett. 2021, 283, 128889. [Google Scholar] [CrossRef]
- Smirnov, M.A.; Nikolaeva, A.L.; Vorobiov, V.K.; Bobrova, N.V.; Abalov, I.V.; Smirnov, A.V.; Sokolova, M.P. Ionic Conductivity and Structure of Chitosan Films Modified with Lactic Acid-Choline Chloride NADES. Polymers 2020, 12, 350. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Lee, K.; Nam, M.W.; Jeong, K.M.; Lee, J.E.; Kim, N.W.; Yin, Y.; Lim, S.Y.; Yoo, D.E.; Lee, J.; et al. Natural Deep Eutectic Solvents as a Storage Medium for Human Interferon-α2: A Green and Improved Strategy for Room-Temperature Biologics. J. Ind. Eng. Chem. 2018, 65, 343–348. [Google Scholar] [CrossRef]
- Lu, C.; Cao, J.; Wang, N.; Su, E. Significantly Improving the Solubility of Non-Steroidal Anti-Inflammatory Drugs in Deep Eutectic Solvents for Potential Non-Aqueous Liquid Administration. Med. Chem. Commun. 2016, 7, 955–959. [Google Scholar] [CrossRef]
- Abbott, A.P.; Ahmed, E.I.; Prasad, K.; Qader, I.B.; Ryder, K.S. Liquid Pharmaceuticals Formulation by Eutectic Formation. Fluid Phase Equilib. 2017, 448, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Chormale, J.H.; Bansal, A.K. Deep Eutectic Systems: An Overview of Fundamental Aspects, Current Understanding and Drug Delivery Applications. Int. J. Pharm. 2021, 610, 121203. [Google Scholar] [CrossRef]
- Di Pietro, M.E.; Hammond, O.; van den Bruinhorst, A.; Mannu, A.; Padua, A.; Mele, A.; Costa Gomes, M. Connecting Chloride Solvation with Hydration in Deep Eutectic Systems. Phys. Chem. Chem. Phys. 2021, 23, 107–111. [Google Scholar] [CrossRef]
- Ma, C.; Laaksonen, A.; Liu, C.; Lu, X.; Ji, X. The Peculiar Effect of Water on Ionic Liquids and Deep Eutectic Solvents. Chem. Soc. Rev. 2018, 47, 8685–8720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.; Mjalli, F.S. Effect of Water on the Thermo-Physical Properties of Reline: An Experimental and Molecular Simulation Based Approach. Phys. Chem. Chem. Phys. 2014, 16, 23900–23907. [Google Scholar] [CrossRef]
- Kovács, A.; Neyts, E.C.; Cornet, I.; Wijnants, M.; Billen, P. Modeling the Physicochemical Properties of Natural Deep Eutectic Solvents. ChemSusChem 2020, 13, 3789–3804. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, I.I.I.; Bahamon, D.; Llovell, F.; Abu-Zahra, M.R.M.; Vega, L.F. Perspectives and Guidelines on Thermodynamic Modelling of Deep Eutectic Solvents. J. Mol. Liq. 2020, 298, 112183. [Google Scholar] [CrossRef]
- Kaur, S.; Kumari, M.; Kashyap, H.K. Microstructure of Deep Eutectic Solvents: Current Understanding and Challenges. J. Phys. Chem. B 2020, 124, 10601–10616. [Google Scholar] [CrossRef]
- Shama, V.M.; Swami, A.R.; Aniruddha, R.; Sreedhar, I.; Reddy, B.M. Process and Engineering Aspects of Carbon Capture by Ionic Liquids. J. CO2 Util. 2021, 48, 101507. [Google Scholar] [CrossRef]
- González de Castilla, A.; Bittner, J.P.; Müller, S.; Jakobtorweihen, S.; Smirnova, I. Thermodynamic and Transport Properties Modeling of Deep Eutectic Solvents: A Review on gE-Models, Equations of State, and Molecular Dynamics. J. Chem. Eng. Data 2020, 65, 943–967. [Google Scholar] [CrossRef]
- Klamt, A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J. Phys. Chem. 1995, 99, 2224–2235. [Google Scholar] [CrossRef]
- Klamt, A.; Jonas, V.; Bürger, T.; Lohrenz, J.C.W. Refinement and Parametrization of COSMO-RS. J. Phys. Chem. A 1998, 102, 5074–5085. [Google Scholar] [CrossRef]
- Chapman, W.G.; Gubbins, K.E.; Jackson, G.; Radosz, M. SAFT: Equation-of-State Solution Model for Associating Fluids. Fluid Phase Equilib. 1989, 52, 31–38. [Google Scholar] [CrossRef]
- Chapman, W.G.; Gubbins, K.E.; Jackson, G.; Radosz, M. New Reference Equation of State for Associating Liquids. Ind. Eng. Chem. Res. 1990, 29, 1709–1721. [Google Scholar] [CrossRef]
- Huang, S.H.; Radosz, M. Equation of State for Small, Large, Polydisperse, and Associating Molecules. Ind. Eng. Chem. Res. 1990, 29, 2284–2294. [Google Scholar] [CrossRef]
- Huang, S.H.; Radosz, M. Equation of State for Small, Large, Polydisperse, and Associating Molecules: Extension to Fluid Mixtures. Ind. Eng. Chem. Res. 1991, 30, 1994–2005. [Google Scholar] [CrossRef]
- Mohan, M.; Huang, K.; Pidatala, V.; Simmons, B.; Singh, S.; Sale, K.L.; Gladden, J. Prediction of Solubility Parameters of Lignin and Ionic Liquids Using Multi-Resolution Simulation Approaches. Green Chem. 2021. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Sazonova, A.Y.; Frolkova, A.K.; Zaitsau, D.H.; Prikhodko, I.V.; Held, C. Separation Performance of BioRenewable Deep Eutectic Solvents. Ind. Eng. Chem. Res. 2015, 54, 3498–3504. [Google Scholar] [CrossRef]
- Mahmoudabadi, S.Z.; Pazuki, G. A Predictive PC-SAFT EOS Based on COSMO for Pharmaceutical Compounds. Sci. Rep. 2021, 11, 6405. [Google Scholar] [CrossRef]
- González-Miquel, M.; Díaz, I. Green Solvent Screening Using Modeling and Simulation. Curr. Opin. Green Sustain. Chem. 2021, 29, 100469. [Google Scholar] [CrossRef]
- Ullah, R.; Atilhan, M.; Anaya, B.; Khraisheh, M.; García, G.; ElKhattat, A.; Tariq, M.; Aparicio, S. A Detailed Study of Cholinium Chloride and Levulinic Acid Deep Eutectic Solvent System for CO2 Capture via Experimental and Molecular Simulation Approaches. Phys. Chem. Chem. Phys. 2015, 17, 20941–20960. [Google Scholar] [CrossRef] [Green Version]
- Altamash, T.; Atilhan, M.; Aliyan, A.; Ullah, R.; García, G.; Aparicio, S. Insights into Choline Chloride–phenylacetic Acid Deep Eutectic Solvent for CO2 Absorption. RSC Adv. 2016, 6, 109201–109210. [Google Scholar] [CrossRef]
- Altamash, T.; Nasser, M.S.; Elhamarnah, Y.; Magzoub, M.; Ullah, R.; Anaya, B.; Aparicio, S.; Atilhan, M. Gas Solubility and Rheological Behavior of Natural Deep Eutectic Solvents (NADES) via Combined Experimental and Molecular Simulation Techniques. ChemistrySelect 2017, 2, 7278–7295. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Atilhan, M.; Aparicio, S. A Theoretical Study on Lidocaine Solubility in Deep Eutectic Solvents. Phys. Chem. Chem. Phys. 2018, 20, 27464–27473. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Bernales, V.S.; Marenich, A.V.; Contreras, R.; Cramer, C.J.; Truhlar, D.G. Quantum Mechanical Continuum Solvation Models for Ionic Liquids. J. Phys. Chem. B 2012, 116, 9122–9129. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Performance of SM6, SM8, and SMD on the SAMPL1 Test Set for the Prediction of Small-Molecule Solvation Free Energies. J. Phys. Chem. B 2009, 113, 4538–4543. [Google Scholar] [CrossRef] [Green Version]
- Jangir, A.K.; Patel, D.; More, R.; Parmar, A.; Kuperkar, K. New Insight into Experimental and Computational Studies of Choline Chloride-Based “green” Ternary Deep Eutectic Solvent (TDES). J. Mol. Struct. 2019, 1181, 295–299. [Google Scholar] [CrossRef]
- Jangir, A.K.; Mandviwala, H.; Patel, P.; Sharma, S.; Kuperkar, K. Acumen into the Effect of Alcohols on Choline Chloride: L-Lactic Acid-Based Natural Deep Eutectic Solvent (NADES): A Spectral Investigation Unified with Theoretical and Thermophysical Characterization. J. Mol. Liq. 2020, 317, 113923. [Google Scholar] [CrossRef]
- Wu, D.; Xu, L.H.; Feng, H.J.; Zhu, Y.W.; Chen, X.Y.; Cui, P. Design and Theoretical Study of Novel Deep Eutectic Solvents: The Effects of Bromine and Chloride Anions on Solvation Structure and Supercapacitor Performance. J. Power Sources 2021, 492, 229634. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules in External Fields. J. Chem. Phys. 1989, 91, 6989–7001. [Google Scholar] [CrossRef]
- Sowlati-Hashjin, S.; Karttunen, M.; Matta, C.F. Manipulation of Diatomic Molecules with Oriented External Electric Fields: Linear Correlations in Atomic Properties Lead to Nonlinear Molecular Responses. J. Phys. Chem. A 2020, 124, 4720–4731. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Li, H.; Zhu, W.; Jiang, W.; Wang, C.; Wu, P.; Zhang, Q.; Li, H. Vibrational Analysis and Formation Mechanism of Typical Deep Eutectic Solvents: An Experimental and Theoretical Study. J. Mol. Graph. Model. 2016, 68, 158–175. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Y.; Chen, J.; Xu, F.; Liu, Z.; He, X.; Li, H.; Zhou, Y. Adsorption of Pharmaceuticals and Personal Care Products by Deep Eutectic Solvents-Regulated Magnetic Metal-Organic Framework Adsorbents: Performance and Mechanism. Chem. Eng. J. 2020, 392, 124808. [Google Scholar] [CrossRef]
- Faraji, S.; Mokhtarpour, M.; Behboudi, E.; Sadrmousavi, A.; Shekaari, H.; Zafarani-Moattar, M.T. Vapor–Liquid Equilibria and Computational Study for Aqueous Solutions of Novel Deep Eutectic Solvents (Amino Acid/Lactic Acid) at 298.15 K. J. Chem. Eng. Data 2020, 65, 3262–3269. [Google Scholar] [CrossRef]
- Naseem, Z.; Shehzad, R.A.; Ihsan, A.; Iqbal, J.; Zahid, M.; Pervaiz, A.; Sarwari, G. Theoretical Investigation of Supramolecular Hydrogen-Bonded Choline Chloride-Based Deep Eutectic Solvents Using Density Functional Theory. Chem. Phys. Lett. 2021, 769, 138427. [Google Scholar] [CrossRef]
- Gautam, R.; Kumar, N.; Lynam, J.G. Theoretical and Experimental Study of Choline Chloride-Carboxylic Acid Deep Eutectic Solvents and Their Hydrogen Bonds. J. Mol. Struct. 2020, 1222, 128849. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, Y.; Jing, Y.; Wang, H.; Hong, K. Main Chemical Species and Molecular Structure of Deep Eutectic Solvent Studied by Experiments with DFT Calculation: A Case of Choline Chloride and Magnesium Chloride Hexahydrate. J. Mol. Model. 2014, 20, 2374. [Google Scholar] [CrossRef]
- Araujo, C.F.; Coutinho, J.A.P.; Nolasco, M.M.; Parker, S.F.; Ribeiro-Claro, P.J.A.; Rudić, S.; Soares, B.I.G.; Vaz, P.D. Inelastic Neutron Scattering Study of Reline: Shedding Light on the Hydrogen Bonding Network of Deep Eutectic Solvents. Phys. Chem. Chem. Phys. 2017, 19, 17998–18009. [Google Scholar] [CrossRef] [Green Version]
- Słupek, E.; Makoś, P. Absorptive Desulfurization of Model Biogas Stream Using Choline Chloride-Based Deep Eutectic Solvents. Sustain. Sci. Pract. Policy 2020, 12, 1619. [Google Scholar] [CrossRef] [Green Version]
- García, G.; Atilhan, M.; Aparicio, S. A Theoretical Study on Mitigation of CO2 through Advanced Deep Eutectic Solvents. Int. J. Greenh. Gas Control 2015, 39, 62–73. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, C.; Li, H.; Wu, P.; Xun, S.; Jiang, W.; Chen, Z.; Zhao, Z.; Li, H. One-Pot Extraction Combined with Metal-Free Photochemical Aerobic Oxidative Desulfurization in Deep Eutectic Solvent. Green Chem. 2015, 17, 2464–2472. [Google Scholar] [CrossRef]
- Makoś, P.; Słupek, E.; Gębicki, J. Extractive Detoxification of Feedstocks for the Production of Biofuels Using New Hydrophobic Deep Eutectic Solvents–Experimental and Theoretical Studies. J. Mol. Liq. 2020, 308, 113101. [Google Scholar] [CrossRef]
- Sinclair, N.S.; Poe, D.; Savinell, R.F.; Maginn, E.J.; Wainright, J.S. A Nitroxide Containing Organic Molecule in a Deep Eutectic Solvent for Flow Battery Applications. J. Electrochem. Soc. 2021, 168, 020527. [Google Scholar] [CrossRef]
- Ashworth, C.R.; Matthews, R.P.; Welton, T.; Hunt, P.A. Doubly Ionic Hydrogen Bond Interactions within the Choline Chloride-Urea Deep Eutectic Solvent. Phys. Chem. Chem. Phys. 2016, 18, 18145–18160. [Google Scholar] [CrossRef] [Green Version]
- Archer, L.; Jachimska, B.; Krzan, M.; Szaleniec, M.; Hebda, E.; Radzik, P.; Pielichowski, K.; Guzik, M. Physical Properties of Biomass-Derived Novel Natural Deep Eutectic Solvents Based on Choline Chloride and (R)-3-Hydroxyacids. J. Mol. Liq. 2020, 315, 113680. [Google Scholar] [CrossRef]
- Atilhan, M.; Altamash, T.; Aparicio, S. Quantum Chemistry Insight into the Interactions Between Deep Eutectic Solvents and SO2. Molecules 2019, 24, 2963. [Google Scholar] [CrossRef] [Green Version]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of Hydrogen Bond Donors and Acceptors on CO2 Absorption by Deep Eutectic Solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Miguel, Á.; Fornari, R.P.; García, N.; Bhowmik, A.; Carrasco-Busturia, D.; García-Lastra, J.M.; Tiemblo, P. Understanding the Molecular Structure of the Elastic and Thermoreversible AlCl3: Urea/Polyethylene Oxide Gel Electrolyte. ChemSusChem 2020, 13, 5523–5530. [Google Scholar] [CrossRef]
- da Costa Lopes, A.M.; Gomes, J.R.B.; Coutinho, J.A.P.; Silvestre, A.J.D. Novel Insights into Biomass Delignification with Acidic Deep Eutectic Solvents: A Mechanistic Study of β-O-4 Ether Bond Cleavage and the Role of the Halide Counterion in the Catalytic Performance. Green Chem. 2020, 22, 2474–2487. [Google Scholar] [CrossRef]
- Wagle, D.V.; Baker, G.A.; Mamontov, E. Differential Microscopic Mobility of Components within a Deep Eutectic Solvent. J. Phys. Chem. Lett. 2015, 6, 2924–2928. [Google Scholar] [CrossRef] [PubMed]
- Pisano, P.L.; Espino, M.; de Fernández, M.L.Á.; Silva, M.F.; Olivieri, A.C. Structural Analysis of Natural Deep Eutectic Solvents. Theoretical and Experimental Study. Microchem. J. 2018, 143, 252–258. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, W.; Zhu, W.; Li, H.; Yin, S.; Chang, Y.; Li, H. A Simple and Cost-Effective Extractive Desulfurization Process with Novel Deep Eutectic Solvents. RSC Adv. 2016, 6, 30345–30352. [Google Scholar] [CrossRef]
- Wagle, D.V.; Zhao, H.; Deakyne, C.A.; Baker, G.A. Quantum Chemical Evaluation of Deep Eutectic Solvents for the Extractive Desulfurization of Fuel. ACS Sustain. Chem. Eng. 2018, 6, 7525–7531. [Google Scholar] [CrossRef]
- Makoś, P.; Boczkaj, G. Deep Eutectic Solvents Based Highly Efficient Extractive Desulfurization of Fuels–Eco-Friendly Approach. J. Mol. Liq. 2019, 296, 111916. [Google Scholar] [CrossRef]
- Ghenaatian, H.R.; Shakourian-Fard, M.; Kamath, G. Interaction of Cun, Agn and Aun (n = 1–4) Nanoparticles with ChCl:Urea Deep Eutectic Solvent. J. Mol. Graph. Model. 2021, 105, 107866. [Google Scholar] [CrossRef]
- Xu, L.H.; Wu, D.; Zhong, M.; Wang, G.B.; Chen, X.Y.; Zhang, Z.J. The Construction of a New Deep Eutectic Solvents System Based on Choline Chloride and Butanediol: The Influence of the Hydroxyl Position of Butanediol on the Structure of Deep Eutectic Solvent and Supercapacitor Performance. J. Power Sources 2021, 490, 229365. [Google Scholar] [CrossRef]
- Li, C.; Lu, D.; Wu, C. A Theoretical Study on Screening Ionic Liquids for SO2 Capture under Low SO2 Partial Pressure and High Temperature. J. Ind. Eng. Chem. 2021, 98, 161–167. [Google Scholar] [CrossRef]
- Malik, A.; Dhattarwal, H.S.; Kashyap, H.K. Distinct Solvation Structures of CO2 and SO2 in Reline and Ethaline Deep Eutectic Solvents Revealed by AIMD Simulations. J. Phys. Chem. B 2021, 125, 1852–1860. [Google Scholar] [CrossRef]
- McGaughy, K.; Reza, M.T. Systems Analysis of SO2-CO2 Co-Capture from a Post-Combustion Coal-Fired Power Plant in Deep Eutectic Solvents. Energies 2020, 13, 438. [Google Scholar] [CrossRef] [Green Version]
- Sanjari, R.; Kazemipour, M.; Zeidabadinejad, L.; Ansari, M. Computational Modeling, Fabrication, and Characterization of the Deep Eutectic Solvent-Based Green Molecular Cage for Selective Metronidazole Extraction from Plasma Followed by UHPLC with Diode Array Detector Determination. J. Sep. Sci. 2021, 44, 3268–3278. [Google Scholar] [CrossRef] [PubMed]
- Migliorati, V.; Fazio, G.; Pollastri, S.; Gentili, A.; Tomai, P.; Tavani, F.; D’Angelo, P. Solubilization Properties and Structural Characterization of Dissociated HgO and HgCl2 in Deep Eutectic Solvents. J. Mol. Liq. 2021, 329, 115505. [Google Scholar] [CrossRef]
- Li, Z.-L.; Zhong, F.-Y.; Huang, J.-Y.; Peng, H.-L.; Huang, K. Sugar-Based Natural Deep Eutectic Solvents as Potential Absorbents for NH3 Capture at Elevated Temperatures and Reduced Pressures. J. Mol. Liq. 2020, 317, 113992. [Google Scholar] [CrossRef]
- Li, G.; Xie, Q.; Liu, Q.; Liu, J.; Wan, C.; Liang, D.; Zhang, H. Separation of Phenolic Compounds from Oil Mixtures by Betaine-based Deep Eutectic Solvents. Asia-Pac. J. Chem. Eng. 2020, 15, e2515. [Google Scholar] [CrossRef]
- Lawal, I.A.; Lawal, M.M.; Azeez, M.A.; Ndungu, P. Theoretical and Experimental Adsorption Studies of Phenol and Crystal Violet Dye on Carbon Nanotube Functionalized with Deep Eutectic Solvent. J. Mol. Liq. 2019, 288, 110895. [Google Scholar] [CrossRef]
- Lopes, M.M.; Barrulas, R.V.; Paiva, T.G.; Ferreira, A.S.D.; Zanatta, M.; Corvo, M.C. Molecular Interactions in Ionic Liquids: The NMR Contribution towards Tailored Solvents. In Nuclear Magnetic Resonance; IntechOpen: London, UK, 2019. [Google Scholar]
- Barich, D.H.; Nicholas, J.B.; Haw, J.F. Gauge-Including Atomic Orbital Proton Chemical Shifts of Strong Hydrogen Bonds: The Importance of Electron Correlation. J. Phys. Chem. A 2001, 105, 4708–4715. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational Density-Functional Perturbation Theory for Dielectrics and Lattice Dynamics. Phys. Rev. B Condens. Matter 2006, 73, 155114. [Google Scholar] [CrossRef] [Green Version]
- Milman, V.; Perlov, A.; Refson, K.; Clark, S.J.; Gavartin, J.; Winkler, B. Structural, Electronic and Vibrational Properties of Tetragonal Zirconia under Pressure: A Density Functional Theory Study. J. Phys. Condens. Matter 2009, 21, 485404. [Google Scholar] [CrossRef]
- Ratcliff, L.E.; Mohr, S.; Huhs, G.; Deutsch, T.; Masella, M.; Genovese, L. Challenges in Large Scale Quantum Mechanical Calculations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1290. [Google Scholar] [CrossRef] [Green Version]
- Korotkevich, A.; Firaha, D.S.; Padua, A.A.H.; Kirchner, B. Ab initio molecular dynamics simulations of SO2 solvation in choline chloride/glycerol deep eutectic solvent. Fluid Phase Equilib. 2017, 448, 59–68. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B Condens. Matter 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Fetisov, E.O.; Harwood, D.B.; Kuo, I.-F.W.; Warrag, S.E.E.; Kroon, M.C.; Peters, C.J.; Siepmann, J.I. First-Principles Molecular Dynamics Study of a Deep Eutectic Solvent: Choline Chloride/Urea and Its Mixture with Water. J. Phys. Chem. B 2018, 122, 1245–1254. [Google Scholar] [CrossRef]
- Zahn, S.; Kirchner, B.; Mollenhauer, D. Charge Spreading in Deep Eutectic Solvents. Chemphyschem 2016, 17, 3354–3358. [Google Scholar] [CrossRef] [PubMed]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular Dynamics Simulations: Advances and Applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar]
- Wong-ekkabut, J.; Karttunen, M. The Good, the Bad and the User in Soft Matter Simulations. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2016, 1858, 2529–2538. [Google Scholar] [CrossRef] [PubMed]
- Hub, J.S.; de Groot, B.L.; Grubmüller, H.; Groenhof, G. Quantifying Artifacts in Ewald Simulations of Inhomogeneous Systems with a Net Charge. J. Chem. Theory Comput. 2014, 10, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, M.; Kulig, W.; Javanainen, M.; Tynkkynen, J.; Hensen, U.; Müller, D.J.; Rog, T.; Vattulainen, I. How To Minimize Artifacts in Atomistic Simulations of Membrane Proteins, Whose Crystal Structure Is Heavily Engineered: β₂-Adrenergic Receptor in the Spotlight. J. Chem. Theory Comput. 2015, 11, 3432–3445. [Google Scholar] [CrossRef]
- Bedrov, D.; Piquemal, J.-P.; Borodin, O.; MacKerell, A.D.; Roux, B.; Schröder, C. Molecular Dynamics Simulations of Ionic Liquids and Electrolytes Using Polarizable Force Fields. Chem. Rev. 2019, 119, 7940–7995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, B.; Acevedo, O. OPLS Force Field for Choline Chloride-Based Deep Eutectic Solvents. J. Phys. Chem. B 2018, 122, 9982–9993. [Google Scholar] [CrossRef]
- Avula, N.V.S.; Karmakar, A.; Kumar, R.; Balasubramanian, S. Efficient Parametrization of Force Field for the Quantitative Prediction of the Physical Properties of Ionic Liquid Electrolytes. J. Chem. Theory Comput. 2021, 17, 4274–4290. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Atilhan, M.; Aparicio, S. The Impact of Charges in Force Field Parameterization for Molecular Dynamics Simulations of Deep Eutectic Solvents. J. Mol. Liq. 2015, 211, 506–514. [Google Scholar] [CrossRef]
- Shayestehpour, O.; Zahn, S. Molecular Features of Reline and Homologous Deep Eutectic Solvents Contributing to Nonideal Mixing Behavior. J. Phys. Chem. B 2020, 124, 7586–7597. [Google Scholar] [CrossRef]
- Abedin, R.; Shen, Y.; Flake, J.C.; Hung, F.R. Deep Eutectic Solvents Mixed with Fluorinated Refrigerants for Absorption Refrigeration: A Molecular Simulation Study. J. Phys. Chem. B 2020, 124, 4536–4550. [Google Scholar] [CrossRef]
- Reddy, T.D.N.; Mallik, B.S. Reciprocity between Ion-Dipole and Hydrogen Bond Interactions in the Binary Mixtures of N,N-Dimethylformamide with Ionic Liquids. J. Mol. Liq. 2020, 301, 112487. [Google Scholar] [CrossRef]
- Tolmachev, D.A.; Boyko, O.S.; Lukasheva, N.V.; Martinez-Seara, H.; Karttunen, M. Overbinding and Qualitative and Quantitative Changes Caused by Simple Na+ and K+ Ions in Polyelectrolyte Simulations: Comparison of Force Fields with and without NBFIX and ECC Corrections. J. Chem. Theory Comput. 2020, 16, 677–687. [Google Scholar] [CrossRef]
- Celebi, A.T.; Dawass, N.; Moultos, O.A.; Vlugt, T.J.H. How Sensitive Are Physical Properties of Choline Chloride-Urea Mixtures to Composition Changes: Molecular Dynamics Simulations and Kirkwood-Buff Theory. J. Chem. Phys. 2021, 154, 184502. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.L.; Painter, P.; Colina, C.M. Experimental and Computational Studies of Choline Chloride-Based Deep Eutectic Solvents. J. Chem. Eng. Data 2014, 59, 3652–3662. [Google Scholar] [CrossRef]
- Smirnov, M.A.; Sokolova, M.P.; Tolmachev, D.A.; Vorobiov, V.K.; Kasatkin, I.A.; Smirnov, N.N.; Klaving, A.V.; Bobrova, N.V.; Lukasheva, N.V.; Yakimansky, A.V. Green Method for Preparation of Cellulose Nanocrystals Using Deep Eutectic Solvent. Cellulose 2020, 27, 4305–4317. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Yethiraj, A. Influence of Electronic Polarization on the Structure of Ionic Liquids. J. Phys. Chem. Lett. 2018, 9, 4765–4770. [Google Scholar] [CrossRef]
- Smith, C.J., 2nd; Wagle, D.V.; Bhawawet, N.; Gehrke, S.; Hollóczki, O.; Pingali, S.V.; O’Neill, H.; Baker, G.A. Combined Small-Angle Neutron Scattering, Diffusion NMR, and Molecular Dynamics Study of a Eutectogel: Illuminating the Dynamical Behavior of Glyceline Confined in Bacterial Cellulose Gels. J. Phys. Chem. B 2020, 124, 7647–7658. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, J.G.; Yethiraj, A. Understanding the Properties of Ionic Liquids: Electrostatics, Structure Factors, and Their Sum Rules. J. Phys. Chem. B 2019, 123, 3499–3512. [Google Scholar] [CrossRef]
- Son, C.Y.; McDaniel, J.G.; Schmidt, J.R.; Cui, Q.; Yethiraj, A. First-Principles United Atom Force Field for the Ionic Liquid BMIM(+)BF4(-): An Alternative to Charge Scaling. J. Phys. Chem. B 2016, 120, 3560–3568. [Google Scholar] [CrossRef] [PubMed]
- Chaumont, A.; Engler, E.; Schurhammer, R. Is Charge Scaling Really Mandatory When Developing Fixed-Charge Atomistic Force Fields for Deep Eutectic Solvents? J. Phys. Chem. B 2020, 124, 7239–7250. [Google Scholar] [CrossRef]
- Rick, S.W.; Stuart, S.J. Potentials and Algorithms for Incorporating Polarizability in Computer Simulations. Rev. Comput. Chem. 2002, 18, 89–146. [Google Scholar]
- Patel, S.; Mackerell, A.D., Jr.; Brooks, C.L., 3rd. CHARMM Fluctuating Charge Force Field for Proteins: II Protein/solvent Properties from Molecular Dynamics Simulations Using a Nonadditive Electrostatic Model. J. Comput. Chem. 2004, 25, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Borodin, O. Polarizable Force Field Development and Molecular Dynamics Simulations of Ionic Liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Huang, J.; Roux, B.; MacKerell, A.D., Jr. An Empirical Polarizable Force Field Based on the Classical Drude Oscillator Model: Development History and Recent Applications. Chem. Rev. 2016, 116, 4983–5013. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; van Gunsteren, W.F. Accounting for Polarization in Molecular Simulation. Comput. Phys. Commun. 2005, 172, 69–85. [Google Scholar] [CrossRef]
- Olano, L.R.; Rick, S.W. Fluctuating Charge Normal Modes: An Algorithm for Implementing Molecular Dynamics Simulations with Polarizable Potentials. J. Comput. Chem. 2005, 26, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Schmollngruber, M.; Lesch, V.; Schröder, C.; Heuer, A.; Steinhauser, O. Comparing Induced Point-Dipoles and Drude Oscillators. Phys. Chem. Chem. Phys. 2015, 17, 14297–14306. [Google Scholar] [CrossRef] [PubMed]
- Goloviznina, K.; Gong, Z.; Costa Gomes, M.F.; Pádua, A.A.H. Extension of the CL&Pol Polarizable Force Field to Electrolytes, Protic Ionic Liquids, and Deep Eutectic Solvents. J. Chem. Theory Comput. 2021, 17, 1606–1617. [Google Scholar] [PubMed]
- Goloviznina, K.; Gong, Z.; Padua, A.A.H. The CL &Pol Polarizable Force Field for the Simulation of Ionic Liquids and Eutectic Solvents. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, e1572. [Google Scholar] [CrossRef]
- Maglia de Souza, R.; Karttunen, M.; Ribeiro, M.C.C. Fine-Tuning the Polarizable CL&Pol Force Field for the Deep Eutectic Solvent Ethaline. J. Chem. Inf. Model. 2021, 61, 5938–5947. [Google Scholar] [CrossRef]
- Jeong, K.-J.; McDaniel, J.G.; Yethiraj, A. Deep Eutectic Solvents: Molecular Simulations with a First-Principles Polarizable Force Field. J. Phys. Chem. B 2021, 125, 7177–7186. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.; Pádua, A.A.H. CL&P: A Generic and Systematic Force Field for Ionic Liquids Modeling. Theor. Chem. Acc. 2012, 131, 1–11. [Google Scholar] [CrossRef]
- Goloviznina, K.; Canongia Lopes, J.N.; Costa Gomes, M.; Pádua, A.A.H. Transferable, Polarizable Force Field for Ionic Liquids. J. Chem. Theory Comput. 2019, 15, 5858–5871. [Google Scholar] [CrossRef] [PubMed]
- Szalewicz, K. Symmetry-Adapted Perturbation Theory of Intermolecular Forces. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Tang, K.T.; Toennies, J.P. An Improved Simple Model for the van Der Waals Potential Based on Universal Damping Functions for the Dispersion Coefficients. J. Chem. Phys. 1984, 80, 3726–3741. [Google Scholar] [CrossRef]
- Thole, B.T. Molecular Polarizabilities Calculated with a Modified Dipole Interaction. Chem. Phys. 1981, 59, 341–350. [Google Scholar] [CrossRef]
- Szabadi, A.; Elfgen, R.; Macchieraldo, R.; Kearns, F.L.; Lee Woodcock, H.; Kirchner, B.; Schröder, C. Comparison between Ab Initio and Polarizable Molecular Dynamics Simulations of 1-Butyl-3-Methylimidazolium Tetrafluoroborate and Chloride in Water. J. Mol. Liq. 2021, 337, 116521. [Google Scholar] [CrossRef]
- McDaniel, J.G.; Schmidt, J.R. Physically-Motivated Force Fields from Symmetry-Adapted Perturbation Theory. J. Phys. Chem. A 2013, 117, 2053–2066. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Atilhan, M.; Aparicio, S. Theoretical Study of Oil Desulfuration by Ammonium-Based Deep Eutectic Solvents. Energy Fuels 2018, 32, 7497–7507. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Aparicio, S.; Atilhan, M. Design of Arginine-Based Therapeutic Deep Eutectic Solvents as Drug Solubilization Vehicles for Active Pharmaceutical Ingredients. Phys. Chem. Chem. Phys. 2019, 21, 10621–10634. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Atilhan, M.; Aparicio, S. Behavior of Antibiotics in Natural Deep Eutectic Solvents. J. Chem. Eng. Data 2020, 65, 4669–4683. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. A Nanoscopic Explanation of Nitric Oxide Solubility in Natural Deep Eutectic Solvents. J. Mol. Liq. 2021, 324, 114673. [Google Scholar] [CrossRef]
- Gutiérrez, A.; Alcalde, R.; Atilhan, M.; Aparicio, S. Insights on Betaine + Lactic Acid Deep Eutectic Solvent. Ind. Eng. Chem. Res. 2020, 59, 11880–11892. [Google Scholar] [CrossRef]
- Rozas, S.; Alomari, N.; Atilhan, M.; Aparicio, S. Theoretical Insights into the Cineole-Based Deep Eutectic Solvents. J. Chem. Phys. 2021, 154, 184504. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Rahman, M.S.; Roy, R.; Gambill, P.; Raynie, D.E.; Halim, M.A. Structure Elucidation of Menthol-Based Deep Eutectic Solvent Using Experimental and Computational Techniques. J. Phys. Chem. A 2021, 125, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Fulfer, K.D.; Ma, J.; Weldeghiorghis, T.K.; Kuroda, D.G. Solvation Dynamics of an Ionic Probe in Choline Chloride-Based Deep Eutectic Solvents. Phys. Chem. Chem. Phys. 2016, 18, 31471–31479. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, R.; Hemmateenejad, B.; Safavi, A.; Shojaeifard, Z.; Shahsavar, A.; Mohajeri, A.; Heydari Dokoohaki, M.; Zolghadr, A.R. Deep Eutectic–water Binary Solvent Associations Investigated by Vibrational Spectroscopy and Chemometrics. Phys. Chem. Chem. Phys. 2018, 20, 18463–18473. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, V.; Esser, L.; Kirchner, B. How Is CO2 Absorbed into a Deep Eutectic Solvent? J. Chem. Phys. 2021, 154, 094503. [Google Scholar] [CrossRef]
- Warrag, S.E.E.; Fetisov, E.O.; van Osch, D.J.G.P.; Harwood, D.B.; Kroon, M.C.; Siepmann, J.I.; Peters, C.J. Mercury Capture from Petroleum Using Deep Eutectic Solvents. Ind. Eng. Chem. Res. 2018, 57, 9222–9230. [Google Scholar] [CrossRef]
- Carrasco-Busturia, D.; Lysgaard, S.; Jankowski, P.; Vegge, T.; Bhowmik, A.; García-Lastra, J.M. Ab Initio Molecular Dynamics Investigations of the Speciation and Reactivity of Deep Eutectic Electrolytes in Aluminum Batteries. ChemSusChem 2021, 14, 2034–2041. [Google Scholar] [CrossRef]
- Zhang, Y.; Poe, D.; Heroux, L.; Squire, H.; Doherty, B.W.; Long, Z.; Dadmun, M.; Gurkan, B.; Tuckerman, M.E.; Maginn, E.J. Liquid Structure and Transport Properties of the Deep Eutectic Solvent Ethaline. J. Phys. Chem. B 2020, 124, 5251–5264. [Google Scholar] [CrossRef]
- Joshi, S.Y.; Deshmukh, S.A. A Review of Advancements in Coarse-Grained Molecular Dynamics Simulations. Mol. Simul. 2021, 47, 786–803. [Google Scholar] [CrossRef]
- Sanbonmatsu, K.Y.; Tung, C.-S. High Performance Computing in Biology: Multimillion Atom Simulations of Nanoscale Systems. J. Struct. Biol. 2007, 157, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Murtola, T.; Bunker, A.; Vattulainen, I.; Deserno, M.; Karttunen, M. Multiscale Modeling of Emergent Materials: Biological and Soft Matter. Phys. Chem. Chem. Phys. 2009, 11, 1869–1892. [Google Scholar] [CrossRef] [PubMed]
- Faller, R.; Schmitz, H.; Biermann, O.; Müller-Plathe, F. Automatic Parameterization of Force Fields for Liquids by Simplex Optimization. J. Comput. Chem. 1999, 20, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of Effective Interaction Potentials from Radial Distribution Functions: A Reverse Monte Carlo Approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini Model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitjanon, J.; Khuntawee, W.; Phongphanphanee, S.; Sutthibutpong, T.; Chattham, N.; Karttunen, M.; Wong-ekkabut, J. Nanocomposite of Fullerenes and Natural Rubbers: MARTINI Force Field Molecular Dynamics Simulations. Polymers 2021, 13, 4044. [Google Scholar] [CrossRef]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules 2021, 11, 1347. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. Int. J. Mol. Sci. 2019, 20, 3774. [Google Scholar] [CrossRef] [Green Version]
- Dhamankar, S.; Webb, M.A. Chemically Specific Coarse-graining of Polymers: Methods and Prospects. J. Polym. Sci. 2021, 59, 2613–2643. [Google Scholar] [CrossRef]
- de Souza, R.M.; Ratochinski, R.H.; Karttunen, M.; Dias, L.G. Self-Assembly of Phosphocholine Derivatives Using the ELBA Coarse-Grained Model: Micelles, Bicelles, and Reverse Micelles. J. Chem. Inf. Model. 2020, 60, 522–536. [Google Scholar] [CrossRef]
- Krishna, V.; Noid, W.G.; Voth, G.A. The Multiscale Coarse-Graining Method. IV. Transferring Coarse-Grained Potentials between Temperatures. J. Chem. Phys. 2009, 131, 024103. [Google Scholar] [CrossRef] [Green Version]
- de Souza, R.M.; Lourenço, T.C.; Amaral de Siqueira, L.J.; Karttunen, M.; Da Silva, J.L.F.; Dias, L.G. Development of Coarse-Grained Force Field to Investigate Sodium-Ion Transport Mechanisms in Cyanoborate-Based Ionic Liquid. J. Mol. Liq. 2021, 338, 116648. [Google Scholar] [CrossRef]
- Español, P.; Warren, P. Statistical Mechanics of Dissipative Particle Dynamics. EPL 1995, 30, 191. [Google Scholar] [CrossRef] [Green Version]
- Groot, R.D.; Warren, P.B. Dissipative Particle Dynamics: Bridging the Gap between Atomistic and Mesoscopic Simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Hu, L.; Yan, Z.; Zhang, J.; Peng, X.; Mo, X.; Wang, A.; Chen, L. Surfactant Aggregates within Deep Eutectic Solvent-Assisted Synthesis of Hierarchical ZIF-8 with Tunable Porosity and Enhanced Catalytic Activity. J. Mater. Sci. 2019, 54, 11009–11023. [Google Scholar] [CrossRef]
- Fan, T.; Chen, L.; Qiu, S.; Yang, C.; Hu, L.; Peng, X.; Zhang, J.; Yan, Z. Synthesis of Hierarchical Porous ZIF-8/3DCNTs Composite Sensor for Ultrasensitive Detection of DA and DFT Studies. J. Electroanal. Chem. 2020, 878, 114541. [Google Scholar] [CrossRef]
- Fan, T.; Chen, L.; Xia, X.; Wu, Y.; Zhang, J.; Yin, K.; Liu, F.; Yan, Z. Dissipative Particle Dynamics Quantitative Simulation of the Formation Mechanism and Emulsification Driving Force of Deep Eutectic Solvent-Based Surfactant-Free and Water-Free Microemulsion. Ind. Eng. Chem. Res. 2021, 60, 3249–3258. [Google Scholar] [CrossRef]
- Vazquez-Salazar, L.I.; Selle, M.; de Vries, A.H.; Marrink, S.J.; Souza, P.C.T. Martini Coarse-Grained Models of Imidazolium-Based Ionic Liquids: From Nanostructural Organization to Liquid–liquid Extraction. Green Chem. 2020, 22, 7376–7386. [Google Scholar] [CrossRef]
- Vainikka, P.; Thallmair, S.; Souza, P.C.T.; Marrink, S.J. Martini 3 Coarse-Grained Model for Type III Deep Eutectic Solvents: Thermodynamic, Structural, and Extraction Properties. ACS Sustain. Chem. Eng. 2021, 9, 17338–17350. [Google Scholar] [CrossRef]
- Kubat, M. An Introduction to Machine Learning; Springer: Cham, Germany, 2017; ISBN 9783319639123. [Google Scholar]
- Bishop, C.M. Pattern Recognition and Machine Learning by Christopher, M. Bishop; Springer Science+Business Media, LLC.: Berlin, Germany, 2006. [Google Scholar]
- Koutsoukos, S.; Philippi, F.; Malaret, F.; Welton, T. A Review on Machine Learning Algorithms for the Ionic Liquid Chemical Space. Chem. Sci. 2021, 12, 6820–6843. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References; John Wiley & Sons: Hoboken, NJ, USA, 2009; ISBN 9783527628773. [Google Scholar]
- Shahbaz, K.; Baroutian, S.; Mjalli, F.S.; Hashim, M.A.; AlNashef, I.M. Densities of Ammonium and Phosphonium Based Deep Eutectic Solvents: Prediction Using Artificial Intelligence and Group Contribution Techniques. Thermochim. Acta 2012, 527, 59–66. [Google Scholar] [CrossRef]
- Shahbaz, K.; Baroutian, S.; Mjalli, F.S.; Hashim, M.A.; AlNashef, I.M. Prediction of Glycerol Removal from Biodiesel Using Ammonium and Phosphunium Based Deep Eutectic Solvents Using Artificial Intelligence Techniques. Chemom. Intellig. Lab. Syst. 2012, 118, 193–199. [Google Scholar] [CrossRef]
- Benguerba, Y.; Alnashef, I.M.; Erto, A.; Balsamo, M.; Ernst, B. A Quantitative Prediction of the Viscosity of Amine Based DESs Using Sσ-Profile Molecular Descriptors. J. Mol. Struct. 2019, 1184, 357–363. [Google Scholar] [CrossRef]
- Alrugaibah, M.; Yagiz, Y.; Gu, L. Use Natural Deep Eutectic Solvents as Efficient Green Reagents to Extract Procyanidins and Anthocyanins from Cranberry Pomace and Predictive Modeling by RSM and Artificial Neural Networking. Sep. Purif. Technol. 2021, 255, 117720. [Google Scholar] [CrossRef]
- Fiyadh, S.S.; AlSaadi, M.A.; AlOmar, M.K.; Fayaed, S.S.; Hama, A.R.; Bee, S.; El-Shafie, A. The Modelling of Lead Removal from Water by Deep Eutectic Solvents Functionalized CNTs: Artificial Neural Network (ANN) Approach. Water Sci. Technol. 2017, 76, 2413–2426. [Google Scholar] [CrossRef] [PubMed]
- Fiyadh, S.S.; AlSaadi, M.A.; AlOmar, M.K.; Fayaed, S.S.; El-Shafie, A. Arsenic Removal from Water Using N,N-Diethylethanolammonium Chloride Based DES-Functionalized CNTs: (NARX) Neural Network Approach. J. Water Supply Res. Technol.-Aqua 2018, 67, 531–542. [Google Scholar] [CrossRef]
- Saadi, F.S.; Abdulhakim, A.M.; Khalid, A.M.; Saadi, F.S.; Mjalli Farouq, S.; Ahmed, E.-S. BTPC-Based DES-Functionalized CNTs for As3+ Removal from Water: NARX Neural Network Approach. J. Environ. Eng. 2018, 144, 04018070. [Google Scholar] [CrossRef]
- Fiyadh, S.S.; AlOmar, M.K.; Binti Jaafar, W.Z.; AlSaadi, M.A.; Fayaed, S.S.; Binti Koting, S.; Lai, S.H.; Chow, M.F.; Ahmed, A.N.; El-Shafie, A. Artificial Neural Network Approach for Modelling of Mercury Ions Removal from Water Using Functionalized CNTs with Deep Eutectic Solvent. Int. J. Mol. Sci. 2019, 20, 4206. [Google Scholar] [CrossRef] [Green Version]
- Dashti, A.; Raji, M.; Amani, P.; Baghban, A.; Mohammadi, A.H. Insight into the Estimation of Equilibrium CO2 Absorption by Deep Eutectic Solvents Using Computational Approaches. Sep. Sci. Technol. 2021, 56, 2351–2368. [Google Scholar] [CrossRef]
- Bagh, F.S.G.; Shahbaz, K.; Mjalli, F.S.; AlNashef, I.M.; Hashim, M.A. Electrical Conductivity of Ammonium and Phosphonium Based Deep Eutectic Solvents: Measurements and Artificial Intelligence-Based Prediction. Fluid Phase Equilib. 2013, 356, 30–37. [Google Scholar] [CrossRef]
- Sharma, M.; Dash, K.K. Deep Eutectic Solvent-based Microwave-assisted Extraction of Phytochemical Compounds from Black Jamun Pulp. J. Food Process Eng. 2021, 44, e13750. [Google Scholar] [CrossRef]
- Stupar, A.; Šeregelj, V.; Ribeiro, B.D.; Pezo, L.; Cvetanović, A.; Mišan, A.; Marrucho, I. Recovery of β-Carotene from Pumpkin Using Switchable Natural Deep Eutectic Solvents. Ultrason. Sonochem. 2021, 76, 105638. [Google Scholar] [CrossRef] [PubMed]
- Ghaedi, H.; Ayoub, M.; Sufian, S.; Murshid, G.; Farrukh, S.; Shariff, A.M. Investigation of Various Process Parameters on the Solubility of Carbon Dioxide in Phosphonium-Based Deep Eutectic Solvents and Their Aqueous Mixtures: Experimental and Modeling. Int. J. Greenhouse Gas Control 2017, 66, 147–158. [Google Scholar] [CrossRef]
- Xu, H.; Kong, Y.; Peng, J.; Song, X.; Liu, Y.; Su, Z.; Li, B.; Gao, C.; Tian, W. Comprehensive Analysis of Important Parameters of Choline Chloride-Based Deep Eutectic Solvent Pretreatment of Lignocellulosic Biomass. Bioresour. Technol. 2021, 319, 124209. [Google Scholar] [CrossRef] [PubMed]
- Kollau, L.J.B.M.; Tuinier, R.; Verhaak, J.; den Doelder, J.; Filot, I.A.W.; Vis, M. Design of Nonideal Eutectic Mixtures Based on Correlations with Molecular Properties. J. Phys. Chem. B 2020, 124, 5209–5219. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.D.; Batra, R.; Chapman, J.; Krishnan, S.; Chen, L.; Ramprasad, R. A Universal Strategy for the Creation of Machine Learning-Based Atomistic Force Fields. NPJ Comput. Mater. 2017, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Botu, V.; Ramprasad, R. Adaptive Machine Learning Framework to Accelerate Ab Initio Molecular Dynamics. Int. J. Quantum Chem. 2015, 115, 1074–1083. [Google Scholar] [CrossRef]
- Häse, F.; Fdez Galván, I.; Aspuru-Guzik, A.; Lindh, R.; Vacher, M. How Machine Learning Can Assist the Interpretation of Ab Initio Molecular Dynamics Simulations and Conceptual Understanding of Chemistry. Chem. Sci. 2019, 10, 2298–2307. [Google Scholar] [CrossRef] [Green Version]
- Korolev, V.; Mitrofanov, A.; Korotcov, A.; Tkachenko, V. Graph Convolutional Neural Networks as “General-Purpose” Property Predictors: The Universality and Limits of Applicability. J. Chem. Inf. Model. 2020, 60, 22–28. [Google Scholar] [CrossRef] [Green Version]
- Jha, D.; Choudhary, K.; Tavazza, F.; Liao, W.-K.; Choudhary, A.; Campbell, C.; Agrawal, A. Enhancing Materials Property Prediction by Leveraging Computational and Experimental Data Using Deep Transfer Learning. Nat. Commun. 2019, 10, 5316. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, R.; Dral, P.O.; Rupp, M.; von Lilienfeld, O.A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci. Data 2014, 1, 140022. [Google Scholar] [CrossRef] [PubMed]
- Kirklin, S.; Saal, J.E.; Meredig, B.; Thompson, A.; Doak, J.W.; Aykol, M.; Rühl, S.; Wolverton, C. The Open Quantum Materials Database (OQMD): Assessing the Accuracy of DFT Formation Energies. NPJ Comput. Mater. 2015, 1, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Rimsza, J.M.; Corrales, L.R. Adsorption Complexes of Copper and Copper Oxide in the Deep Eutectic Solvent 2:1 Urea–choline Chloride. Comput. Theor. Chem. 2012, 987, 57–61. [Google Scholar] [CrossRef]
- Zahn, S. Deep Eutectic Solvents: Similia Similibus Solvuntur? Phys. Chem. Chem. Phys. 2017, 19, 4041–4047. [Google Scholar] [CrossRef]
- Stefanovic, R.; Ludwig, M.; Webber, G.B.; Atkin, R.; Page, A.J. Nanostructure, Hydrogen Bonding and Rheology in Choline Chloride Deep Eutectic Solvents as a Function of the Hydrogen Bond Donor. Phys. Chem. Chem. Phys. 2017, 19, 3297–3306. [Google Scholar] [CrossRef]
- Migliorati, V.; Sessa, F.; D’Angelo, P. Deep Eutectic Solvents: A Structural Point of View on the Role of the Cation. Chem. Phys. Lett. X 2019, 2, 100001. [Google Scholar] [CrossRef]
- Sun, H.; Li, Y.; Wu, X.; Li, G. Theoretical Study on the Structures and Properties of Mixtures of Urea and Choline Chloride. J. Mol. Modeling 2013, 19, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Malik, A.; Kashyap, H.K. Anatomy of Microscopic Structure of Ethaline Deep Eutectic Solvent Decoded through Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 8291–8299. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.S.C.; Voroshylova, I.V.; Pereira, C.M.; Cordeiro, M.N.D.S. Improved Force Field Model for the Deep Eutectic Solvent Ethaline: Reliable Physicochemical Properties. J. Phys. Chem. B 2016, 120, 10124–10137. [Google Scholar] [CrossRef]
- Bonomo, M.; Gontrani, L.; Capocefalo, A.; Sarra, A.; Nucara, A.; Carbone, M.; Postorino, P.; Dini, D. A Combined Electrochemical, Infrared and EDXD Tool to Disclose Deep Eutectic Solvents Formation When One Precursor Is Liquid: Glyceline as Case Study. J. Mol. Liq. 2020, 319, 114292. [Google Scholar] [CrossRef]
- Ferreira, E.S.C.; Voroshylova, I.V.; Figueiredo, N.M.; Pereira, C.M.; Cordeiro, M.N.D.S. Computational and Experimental Study of Propeline: A Choline Chloride Based Deep Eutectic Solvent. J. Mol. Liq. 2020, 298, 111978. [Google Scholar] [CrossRef]
- Ferreira, E.S.C.; Voroshylova, I.V.; Figueiredo, N.M.; Cordeiro, M.N.D.S. Molecular Dynamic Study of Alcohol-Based Deep Eutectic Solvents. J. Chem. Phys. 2021, 155, 064506. [Google Scholar] [CrossRef] [PubMed]
- Alhadid, A.; Mokrushina, L.; Minceva, M. Modeling of Solid–Liquid Equilibria in Deep Eutectic Solvents: A Parameter Study. Molecules 2019, 24, 2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, N.; Naik, P.K.; Ribeiro, B.D.; Gooh Pattader, P.S.; Marrucho, I.M.; Banerjee, T. Molecular Dynamics Insights and Water Stability of Hydrophobic Deep Eutectic Solvents Aided Extraction of Nitenpyram from an Aqueous Environment. J. Phys. Chem. B 2020, 124, 7405–7420. [Google Scholar] [CrossRef] [PubMed]
- Fu, N.; Li, L.; Liu, K.; Kim, C.K.; Li, J.; Zhu, T.; Li, J.; Tang, B. A Choline Chloride-Acrylic Acid Deep Eutectic Solvent Polymer Based on Fe3O4 Particles and MoS2 Sheets (poly(ChCl-AA DES)@Fe3O4@MoS2) with Specific Recognition and Good Antibacterial Properties for β-Lactoglobulin in Milk. Talanta 2019, 197, 567–577. [Google Scholar] [CrossRef]
- Gontrani, L.; Plechkova, N.V.; Bonomo, M. In-Depth Physico-Chemical and Structural Investigation of a Dicarboxylic Acid/Choline Chloride Natural Deep Eutectic Solvent (NADES): A Spotlight on the Importance of a Rigorous Preparation Procedure. ACS Sustain. Chem. Eng. 2019, 7, 12536–12543. [Google Scholar] [CrossRef]
- Saha, M.; Rahman, M.S.; Hossain, M.N.; Raynie, D.E.; Halim, M.A. Molecular and Spectroscopic Insights of a Choline Chloride Based Therapeutic Deep Eutectic Solvent. J. Phys. Chem. A 2020, 124, 4690–4699. [Google Scholar] [CrossRef]
- Bonab, P.J.; Esrafili, M.D.; Ebrahimzadeh, A.R.; Sardroodi, J.J. Molecular Dynamics Simulations of Choline Chloride and Phenyl Propionic Acid Deep Eutectic Solvents: Investigation of Structural and Dynamics Properties. J. Mol. Graph. Model. 2021, 106, 107908. [Google Scholar] [CrossRef]
- Jahanbakhsh Bonab, P.; Rastkar Ebrahimzadeh, A.; Jahanbin Sardroodi, J. Insights into the Interactions and Dynamics of a DES Formed by Phenyl Propionic Acid and Choline Chloride. Sci. Rep. 2021, 11, 6384. [Google Scholar] [CrossRef]
- Stott, P.W.; Williams, A.C.; Barry, B.W. Transdermal Delivery from Eutectic Systems: Enhanced Permeation of a Model Drug, Ibuprofen. J. Control. Release 1998, 50, 297–308. [Google Scholar] [CrossRef]
- Aroso, I.M.; Silva, J.C.; Mano, F.; Ferreira, A.S.D.; Dionísio, M.; Sá-Nogueira, I.; Barreiros, S.; Reis, R.L.; Paiva, A.; Duarte, A.R.C. Dissolution Enhancement of Active Pharmaceutical Ingredients by Therapeutic Deep Eutectic Systems. Eur. J. Pharm. Biopharm. 2016, 98, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Korneev, S.M. Hydrocinnamic Acids: Application and Strategy of Synthesis. Synthesis 2013, 45, 1000–1015. [Google Scholar] [CrossRef]
- Perkins, S.L.; Painter, P.; Colina, C.M. Molecular Dynamic Simulations and Vibrational Analysis of an Ionic Liquid Analogue. J. Phys. Chem. B 2013, 117, 10250–10260. [Google Scholar] [CrossRef] [PubMed]
- van den Bruinhorst, A.; Spyriouni, T.; Hill, J.-R.; Kroon, M.C. Experimental and Molecular Modeling Evaluation of the Physicochemical Properties of Proline-Based Deep Eutectic Solvents. J. Phys. Chem. B 2018, 122, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliorati, V.; D’Angelo, P. Deep Eutectic Solvents: A Structural Point of View on the Role of the Anion. Chem. Phys. Lett. 2021, 777, 138702. [Google Scholar] [CrossRef]
- Cerajewski, U.; Träger, J.; Henkel, S.; Roos, A.H.; Brehm, M.; Hinderberger, D. Nanoscopic Structures and Molecular Interactions Leading to a Dystectic and Two Eutectic Points in [EMIm] [Cl]/urea Mixtures. Phys. Chem. Chem. Phys. 2018, 20, 29591–29600. [Google Scholar] [CrossRef]
- Naik, P.K.; Paul, S.; Banerjee, T. Physiochemical Properties and Molecular Dynamics Simulations of Phosphonium and Ammonium Based Deep Eutectic Solvents. J. Solut. Chem. 2019, 48, 1046–1065. [Google Scholar] [CrossRef]
- Kussainova, D.; Shah, D. Structure of Monoethanolamine Based Type III DESs: Insights from Molecular Dynamics Simulations. Fluid Phase Equilib. 2019, 482, 112–117. [Google Scholar] [CrossRef]
- van Osch, D.J.G.P.; Zubeir, L.F.; van den Bruinhorst, A.; Rocha, M.A.A.; Kroon, M.C. Hydrophobic Deep Eutectic Solvents as Water-Immiscible Extractants. Green Chem. 2015, 17, 4518–4521. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, B.D.; Florindo, C.; Iff, L.C.; Coelho, M.A.Z.; Marrucho, I.M. Menthol-Based Eutectic Mixtures: Hydrophobic Low Viscosity Solvents. ACS Sustain. Chem. Eng. 2015, 3, 2469–2477. [Google Scholar] [CrossRef]
- van Osch, D.J.G.P.; Dietz, C.H.J.T.; Warrag, S.E.E.; Kroon, M.C. The Curious Case of Hydrophobic Deep Eutectic Solvents: A Story on the Discovery, Design, and Applications. ACS Sustain. Chem. Eng. 2020, 8, 10591–10612. [Google Scholar] [CrossRef]
- Salehi, H.S.; Celebi, A.T.; Vlugt, T.J.H.; Moultos, O.A. Thermodynamic, Transport, and Structural Properties of Hydrophobic Deep Eutectic Solvents Composed of Tetraalkylammonium Chloride and Decanoic Acid. J. Chem. Phys. 2021, 154, 144502. [Google Scholar] [CrossRef] [PubMed]
- Abbas, U.L.; Qiao, Q.; Nguyen, M.T.; Shi, J.; Shao, Q. Molecular Dynamics Simulations of Heterogeneous Hydrogen Bond Environment in Hydrophobic Deep Eutectic Solvents. AIChE J. 2022, 68, e17382. [Google Scholar] [CrossRef]
- Alizadeh, V.; Geller, D.; Malberg, F.; Sánchez, P.B.; Padua, A.; Kirchner, B. Strong Microheterogeneity in Novel Deep Eutectic Solvents. Chemphyschem 2019, 20, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Kuroda, D.G. Evidence of Molecular Heterogeneities in Amide-Based Deep Eutectic Solvents. J. Phys. Chem. A 2018, 122, 1185–1193. [Google Scholar] [CrossRef]
- Kaur, S.; Gupta, A.; Kashyap, H.K. Nanoscale Spatial Heterogeneity in Deep Eutectic Solvents. J. Phys. Chem. B 2016, 120, 6712–6720. [Google Scholar] [CrossRef]
- Kaur, S.; Kashyap, H.K. Unusual Temperature Dependence of Nanoscale Structural Organization in Deep Eutectic Solvents. J. Phys. Chem. B 2018, 122, 5242–5250. [Google Scholar] [CrossRef]
- Das, S.; Mukherjee, B.; Biswas, R. Microstructures and Their Lifetimes in Acetamide/electrolyte Deep Eutectics: Anion Dependence. J. Chem. Sci. 2017, 129, 939–951. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghorai, P.K.; Das, S.; Rajbangshi, J.; Biswas, R. Heterogeneous Dynamics, Correlated Time and Length Scales in Ionic Deep Eutectics: Anion and Temperature Dependence. J. Chem. Phys. 2020, 153, 234502. [Google Scholar] [CrossRef]
- Ogawa, H.; Mori, H. Lithium Salt/amide-Based Deep Eutectic Electrolytes for Lithium-Ion Batteries: Electrochemical, Thermal and Computational Study. Phys. Chem. Chem. Phys. 2020, 22, 8853–8863. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ali, M.C.; Yang, Q.; Zhang, Z.; Bao, Z.; Su, B.; Xing, H.; Ren, Q. Hybrid Deep Eutectic Solvents with Flexible Hydrogen-Bonded Supramolecular Networks for Highly Efficient Uptake of NH3. ChemSusChem 2017, 10, 3368–3377. [Google Scholar] [CrossRef] [PubMed]
- Jangir, A.K.; Nain, A.K.; Kuperkar, K. Insight into Structural Properties and Molecular Interactions of Maline (choline Chloride + Malonic Acid) and 1, 4- Butanediol Based Pseudo-Binary Mixture: A Thermophysical, Spectral, and Simulation Portrayal. J. Mol. Liq. 2021, 334, 116050. [Google Scholar] [CrossRef]
- Srinivasan, H.; Sharma, V.K.; Mitra, S.; Biswas, R.; Mukhopadhyay, R. Dynamics in Acetamide+LiNO3 Deep Eutectic Solvents. Phys. B Condens. Matter 2019, 562, 13–16. [Google Scholar] [CrossRef]
- Srinivasan, H.; Sharma, V.K.; Mukhopadhyay, R.; Mitra, S. Solvation and Transport of Lithium Ions in Deep Eutectic Solvents. J. Chem. Phys. 2020, 153, 104505. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, H.; Sharma, V.K.; Sakai, V.G.; Embs, J.P.; Mukhopadhyay, R.; Mitra, S. Transport Mechanism of Acetamide in Deep Eutectic Solvents. J. Phys. Chem. B 2020, 124, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Das, S.; Biswas, R. Fast Fluctuations in Deep Eutectic Melts: Multi-Probe Fluorescence Measurements and All-Atom Molecular Dynamics Simulation Study. Chem. Phys. Lett. 2013, 581, 47–51. [Google Scholar] [CrossRef]
- Guchhait, B.; Das, S.; Daschakraborty, S.; Biswas, R. Interaction and Dynamics of (alkylamide + Electrolyte) Deep Eutectics: Dependence on Alkyl Chain-Length, Temperature, and Anion Identity. J. Chem. Phys. 2014, 140, 104514. [Google Scholar] [CrossRef]
- Das, S.; Biswas, R.; Mukherjee, B. Orientational Jumps in (acetamide + Electrolyte) Deep Eutectics: Anion Dependence. J. Phys. Chem. B 2015, 119, 11157–11168. [Google Scholar] [CrossRef]
- Das, A.; Das, S.; Biswas, R. Density Relaxation and Particle Motion Characteristics in a Non-Ionic Deep Eutectic Solvent (acetamide + Urea): Time-Resolved Fluorescence Measurements and All-Atom Molecular Dynamics Simulations. J. Chem. Phys. 2015, 142, 034505. [Google Scholar] [CrossRef]
- Mukherjee, K.; Das, S.; Tarif, E.; Barman, A.; Biswas, R. Dielectric Relaxation in Acetamide + Urea Deep Eutectics and Neat Molten Urea: Origin of Time Scales via Temperature Dependent Measurements and Computer Simulations. J. Chem. Phys. 2018, 149, 124501. [Google Scholar] [CrossRef]
- Rajbangshi, J.; Mukherjee, K.; Biswas, R. Heterogeneous Orientational Relaxations and Translation-Rotation Decoupling in (choline Chloride + Urea) Deep Eutectic Solvents: Investigation through Molecular Dynamics Simulations and Dielectric Relaxation Measurements. J. Phys. Chem. B 2021, 125, 5920–5936. [Google Scholar] [CrossRef]
- Laage, D.; Hynes, J.T. A Molecular Jump Mechanism of Water Reorientation. Science 2006, 311, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Titantah, J.T.; Karttunen, M. Long-Time Correlations and Hydrophobe-Modified Hydrogen-Bonding Dynamics in Hydrophobic Hydration. J. Am. Chem. Soc. 2012, 134, 9362–9368. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Wiorkiewicz-Kuczera, J.; Karplus, M. An All-Atom Empirical Energy Function for the Simulation of Nucleic Acids. J. Am. Chem. Soc. 1995, 117, 11946–11975. [Google Scholar] [CrossRef]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.P.; Jorgensen, W.L. Halide, Ammonium, and Alkali Metal Ion Parameters for Modeling Aqueous Solutions. J. Chem. Theory Comput. 2006, 2, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Cadena, C.; Maginn, E.J. Molecular Simulation Study of Some Thermophysical and Transport Properties of Triazolium-Based Ionic Liquids. J. Phys. Chem. B 2006, 110, 18026–18039. [Google Scholar] [CrossRef]
- Papoyan, G.; Gu, K.-J.; Wiorkiewicz-Kuczera, J.; Kuczera, K.; Bowman-James, K. Molecular Dynamics Simulations of Nitrate Complexes with Polyammonium Macrocycles: Insight on Phosphoryl Transfer Catalysis. J. Am. Chem. Soc. 1996, 118, 1354–1364. [Google Scholar] [CrossRef]
- Smith, L.J.; Berendsen, H.J.C.; van Gunsteren, W.F. Computer Simulation of Urea−Water Mixtures: A Test of Force Field Parameters for Use in Biomolecular Simulation. J. Phys. Chem. B 2004, 108, 1065–1071. [Google Scholar] [CrossRef]
- Mardani, A.; Streimikiene, D.; Cavallaro, F.; Loganathan, N.; Khoshnoudi, M. Carbon Dioxide (CO2) Emissions and Economic Growth: A Systematic Review of Two Decades of Research from 1995 to 2017. Sci. Total Environ. 2019, 649, 31–49. [Google Scholar] [CrossRef]
- Kumar, M.; Sundaram, S.; Gnansounou, E.; Larroche, C.; Thakur, I.S. Carbon Dioxide Capture, Storage and Production of Biofuel and Biomaterials by Bacteria: A Review. Bioresour. Technol. 2018, 247, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Okolie, J.A.; Abdelrasoul, A.; Niu, C.; Dalai, A.K. Review of Post-Combustion Carbon Dioxide Capture Technologies Using Activated Carbon. J. Environ. Sci. 2019, 83, 46–63. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Atilhan, M.; Aparicio, S. Interfacial Properties of Deep Eutectic Solvents Regarding to CO2 Capture. J. Phys. Chem. C 2015, 119, 21413–21425. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Taghizadeh, A.; Vatanpour, V.; Ganjali, M.R.; Saeb, M.R. Deep Eutectic Solvents in Membrane Science and Technology: Fundamental, Preparation, Application, and Future Perspective. Sep. Purif. Technol. 2021, 258, 118015. [Google Scholar] [CrossRef]
- Karadas, F.; Atilhan, M.; Aparicio, S. Review on the Use of Ionic Liquids (ILs) as Alternative Fluids for CO2 Capture and Natural Gas Sweetening. Energy Fuels 2010, 24, 5817–5828. [Google Scholar] [CrossRef]
- Peters, L.; Hussain, A.; Follmann, M.; Melin, T.; Hägg, M.-B. CO2 Removal from Natural Gas by Employing Amine Absorption and Membrane technology—A Technical and Economical Analysis. Chem. Eng. J. 2011, 172, 952–960. [Google Scholar] [CrossRef]
- Kussainova, D.; Shah, D. Monoethanolamine Based DESs for CO2 Absorption: Insights from Molecular Dynamics Simulations. Sep. Purif. Technol. 2020, 231, 115931. [Google Scholar] [CrossRef]
- Haider, M.B.; Jha, D.; Marriyappan Sivagnanam, B.; Kumar, R. Modelling and Simulation of CO2 Removal from Shale Gas Using Deep Eutectic Solvents. J. Environ. Chem. Eng. 2019, 7, 102747. [Google Scholar] [CrossRef]
- Lin, H.; Gong, K.; Ying, W.; Chen, D.; Zhang, J.; Yan, Y.; Peng, X. CO2 -Philic Separation Membrane: Deep Eutectic Solvent Filled Graphene Oxide Nanoslits. Small 2019, 15, e1904145. [Google Scholar] [CrossRef]
- Shen, Y.; Abedin, R.; Hung, F.R. On the Performance of Confined Deep Eutectic Solvents and Ionic Liquids for Separations of Carbon Dioxide from Methane: Molecular Dynamics Simulations. Langmuir 2019, 35, 3658–3671. [Google Scholar] [CrossRef]
- Lin, H.; Gong, K.; Hykys, P.; Chen, D.; Ying, W.; Sofer, Z.; Yan, Y.; Li, Z.; Peng, X. Nanoconfined Deep Eutectic Solvent in Laminated MXene for Efficient CO2 Separation. Chem. Eng. J. 2021, 405, 126961. [Google Scholar] [CrossRef]
- Alioui, O.; Benguerba, Y.; Alnashef, I.M. Investigation of the CO2-Solubility in Deep Eutectic Solvents Using COSMO-RS and Molecular Dynamics Methods. J. Mol. Liq. 2020, 307, 113005. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, H.; Song, Z.; Chen, L.; Deng, L.; Qi, Z. Carbon Dioxide Solubility in Phosphonium-Based Deep Eutectic Solvents: An Experimental and Molecular Dynamics Study. Ind. Eng. Chem. Res. 2019, 58, 17514–17523. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Li, Z.; Yin, J.; Cui, Y.; Liu, Y.; Yang, G. Extraction Desulfurization of Fuels with “metal Ions” Based Deep Eutectic Solvents (MDESs). Green Chem. 2016, 18, 3789–3795. [Google Scholar] [CrossRef]
- El-hoshoudy, A.N.; Soliman, F.S.; Abd El-Aty, D.M. Extractive Desulfurization Using Choline Chloride-Based DES/molybdate Nanofluids; Experimental and Theoretical Investigation. J. Mol. Liq. 2020, 318, 114307. [Google Scholar] [CrossRef]
- Shah, D.; Gapeyenko, D.; Urakpayev, A.; Torkmahalleh, M. Molecular Dynamics Simulations on Extractive Desulfurization of Fuels by Tetrabutylammonium Chloride Based Deep Eutectic Solvents. J. Mol. Liq. 2019, 274, 254–260. [Google Scholar] [CrossRef]
- Kumar, N.; Naik, P.K.; Banerjee, T. Molecular Modeling Insights in the Extraction of Benzene from Hydrocarbon Stream Using Deep Eutectic Solvent. J. Mol. Liq. 2020, 317, 113909. [Google Scholar] [CrossRef]
- Naik, P.K.; Mohan, M.; Banerjee, T.; Paul, S.; Goud, V.V. Molecular Dynamic Simulations for the Extraction of Quinoline from Heptane in the Presence of a Low-Cost Phosphonium-Based Deep Eutectic Solvent. J. Phys. Chem. B 2018, 122, 4006–4015. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Yao, D.; Ma, Z.; Zhao, J.; Zhang, W.; Cui, P.; Ma, Y.; Zhu, Z.; Wang, Y. Molecular Kinetic Extraction Mechanism Analysis of 1-Butanol from N-Heptane-1-Butanol by Choline-Based DESs as Extractants. J. Mol. Liq. 2021, 322, 114665. [Google Scholar] [CrossRef]
- Liu, X.; Xing, J.; Sun, M.; Su, Z.; Chen, Z.; Wang, Y.; Cui, P. Phase Behavior and Extraction Mechanism of Methanol-N-Hexane Separation Using Choline-Based Deep Eutectic Solvent. J. Mol. Liq. 2022, 345, 118204. [Google Scholar] [CrossRef]
- Kumari, P.; Kaur, S.S.; Kashyap, H.K. Influence of Hydration on the Structure of Reline Deep Eutectic Solvent: A Molecular Dynamics Study. ACS Omega 2018, 3, 15246–15255. [Google Scholar] [CrossRef]
- Weng, L.; Toner, M. Janus-Faced Role of Water in Defining Nanostructure of Choline Chloride/glycerol Deep Eutectic Solvent. Phys. Chem. Chem. Phys. 2018, 20, 22455–22462. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Bittner, J.P.; Domínguez de María, P.; Jakobtorweihen, S.; Kara, S. Modeling Alcohol Dehydrogenase Catalysis in Deep Eutectic Solvent/Water Mixtures. Chembiochem 2020, 21, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Witkamp, G.-J.; Verpoorte, R.; Choi, Y.H. Tailoring Properties of Natural Deep Eutectic Solvents with Water to Facilitate Their Applications. Food Chem. 2015, 187, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Pandey, S. Densities and Viscosities of (choline Chloride+ Urea) Deep Eutectic Solvent and Its Aqueous Mixtures in the Temperature Range 293.15 K to 363.15 K. J. Chem. Eng. Data 2014, 59, 2221–2229. [Google Scholar] [CrossRef]
- Kumari, P.; Kumari, M.; Kashyap, H.K. How Pure and Hydrated Reline Deep Eutectic Solvents Affect the Conformation and Stability of Lysozyme: Insights from Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2020, 124, 11919–11927. [Google Scholar] [CrossRef]
- Sapir, L.; Harries, D. Restructuring a Deep Eutectic Solvent by Water: The Nanostructure of Hydrated Choline Chloride/Urea. J. Chem. Theory Comput. 2020, 16, 3335–3342. [Google Scholar] [CrossRef]
- Aryafard, M.; Karimi, A.; Harifi-Mood, A.R.; Minofar, B. Molecular Dynamics Simulations, Solvatochromic Parameters, and Preferential Solvation in Aqueous Solutions of Ethaline, Ethylene Glycol, and Choline Chloride. J. Chem. Eng. Data 2020, 65, 4556–4566. [Google Scholar] [CrossRef]
- Alizadeh, V.; Malberg, F.; Pádua, A.A.H.; Kirchner, B. Are There Magic Compositions in Deep Eutectic Solvents? Effects of Composition and Water Content in Choline Chloride/Ethylene Glycol from Ab Initio Molecular Dynamics. J. Phys. Chem. B 2020, 124, 7433–7443. [Google Scholar] [CrossRef]
- Bezerra-Neto, J.R.; Bezerra, L.L.; Sousa, N.G.; Dos Santos, L.P.M.; Marinho, E.S.; Monteiro, N.K.V.; Correia, A.N.; de Lima-Neto, P. Molecular Approach about the Effect of Water on the Electrochemical Behaviour of Ag+ Ions in Urea-Choline Chloride-Water Mixture. J. Mol. Model. 2020, 26, 339. [Google Scholar] [CrossRef]
- Busato, M.; Di Lisio, V.; Del Giudice, A.; Tomai, P.; Migliorati, V.; Galantini, L.; Gentili, A.; Martinelli, A.; D’Angelo, P. Transition from Molecular- to Nano-Scale Segregation in a Deep Eutectic Solvent-Water Mixture. J. Mol. Liq. 2021, 331, 115747. [Google Scholar] [CrossRef]
- Ghorpade, U.V.; Suryawanshi, M.P.; Shin, S.W.; Wang, X.; Jo, E.; Bae, H.; Park, K.; Ha, J.-S.; Kolekar, S.S.; Kim, J.H. Eutectic Solvent-Mediated Selective Synthesis of Cu–Sb–S-Based Nanocrystals: Combined Experimental and Theoretical Studies toward Highly Efficient Water Splitting. J. Mater. Chem. A Mater. Energy Sustain. 2018, 6, 19798–19809. [Google Scholar] [CrossRef]
- Celebi, A.T.; Vlugt, T.J.H.; Moultos, O.A. Structural, Thermodynamic, and Transport Properties of Aqueous Reline and Ethaline Solutions from Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 11014–11025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celebi, A.T.; Vlugt, T.J.H.; Moultos, O.A. Thermal Conductivity of Aqueous Solutions of Reline, Ethaline, and Glyceline Deep Eutectic Solvents; a Molecular Dynamics Simulation Study. Mol. Phys. 2021, e1876263. [Google Scholar] [CrossRef]
- Sarkar, S.; Maity, A.; Chakrabarti, R. Microscopic Structural Features of Water in Aqueous-Reline Mixtures of Varying Compositions. Phys. Chem. Chem. Phys. 2021, 23, 3779–3793. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Maity, A.; Chakrabarti, R. In Silico Elucidation of Molecular Picture of Water–Choline Chloride Mixture. J. Phys. Chem. B 2021, 125, 13212–13228. [Google Scholar] [CrossRef]
- Bezerra-Neto, J.R.; Sousa, N.G.; Dos Santos, L.P.M.; Correia, A.N.; de Lima-Neto, P. The Effect of Water on the Physicochemical Properties of an Ethylene Glycol and Choline Chloride Mixture Containing Cu2+ Ions: Electrochemical Results and Dynamic Molecular Simulation Approach. Phys. Chem. Chem. Phys. 2018, 20, 9321–9327. [Google Scholar] [CrossRef] [PubMed]
- Lukaczynska-Anderson, M.; Mamme, M.H.; Ceglia, A.; Van den Bergh, K.; De Strycker, J.; De Proft, F.; Terryn, H.; Ustarroz, J. The Role of Hydrogen Bond Donor and Water Content on the Electrochemical Reduction of Ni2+ from Solvents-an Experimental and Modelling Study. Phys. Chem. Chem. Phys. 2020, 22, 16125–16135. [Google Scholar] [CrossRef]
- Baz, J.; Held, C.; Pleiss, J.; Hansen, N. Thermophysical Properties of Glyceline-Water Mixtures Investigated by Molecular Modelling. Phys. Chem. Chem. Phys. 2019, 21, 6467–6476. [Google Scholar] [CrossRef]
- Shehata, M.; Unlu, A.; Sezerman, U.; Timucin, E. Lipase and Water in a Deep Eutectic Solvent: Molecular Dynamics and Experimental Studies of the Effects of Water-In-Deep Eutectic Solvents on Lipase Stability. J. Phys. Chem. B 2020, 124, 8801–8810. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Mitra, A.; Paul, S. Phase Separation Property of a Hydrophobic Deep Eutectic Solvent-Water Binary Mixture: A Molecular Dynamics Simulation Study. J. Chem. Phys. 2021, 154, 244504. [Google Scholar] [CrossRef]
- Salehi, H.S.; Moultos, O.A.; Vlugt, T.J.H. Interfacial Properties of Hydrophobic Deep Eutectic Solvents with Water. J. Phys. Chem. B 2021, 125, 12303–12314. [Google Scholar] [CrossRef]
- Atilhan, M.; Costa, L.T.; Aparicio, S. Elucidating the Properties of Graphene–Deep Eutectic Solvents Interface. Langmuir 2017, 33, 5154–5165. [Google Scholar] [CrossRef]
- Shen, Y.; He, X.; Hung, F.R. Structural and Dynamical Properties of a Deep Eutectic Solvent Confined Inside a Slit Pore. J. Phys. Chem. C 2015, 119, 24489–24500. [Google Scholar] [CrossRef]
- Mamme, M.H.; Moors, S.L.C.; Terryn, H.; Deconinck, J.; Ustarroz, J.; De Proft, F. Atomistic Insight into the Electrochemical Double Layer of Choline Chloride-Urea Deep Eutectic Solvents: Clustered Interfacial Structuring. J. Phys. Chem. Lett. 2018, 9, 6296–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Hung, F.R. A Molecular Simulation Study of Carbon Dioxide Uptake by a Deep Eutectic Solvent Confined in Slit Nanopores. J. Phys. Chem. C 2017, 121, 24562–24575. [Google Scholar] [CrossRef]
- Safavi, A.; Shekarnoush, M.; Ajamian, M.; Zolghadr, A.R. High-Yield Synthesis, Characterization, Self-Assembly of Extremely Thin Gold Nanosheets in Sugar Based Deep Eutectic Solvents and Their High Electrocatalytic Activity. J. Mol. Liq. 2019, 279, 208–223. [Google Scholar] [CrossRef]
- Rozas, S.; Atilhan, M.; Aparicio, S. Deep Eutectic Solvent Reline at 2D Nanomaterial Interfaces. J. Phys. Chem. B 2020, 124, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Shakourian-Fard, M.; Taimoory, S.M.; Ghenaatian, H.R.; Kamath, G.; Trant, J.F. A DFT Study of the Adsorption of Deep Eutectic Solvents onto Graphene and Defective Graphene Nanoflakes. J. Mol. Liq. 2021, 327, 114850. [Google Scholar] [CrossRef]
- Zec, N.; Mangiapia, G.; Zheludkevich, M.L.; Busch, S.; Moulin, J.-F. Revealing the Interfacial Nanostructure of a Deep Eutectic Solvent at a Solid Electrode. Phys. Chem. Chem. Phys. 2020, 22, 12104–12112. [Google Scholar] [CrossRef]
- Patidar, P.; Kanoje, B.; Bahadur, A.; Kuperkar, K.; Ray, D.; Aswal, V.K.; Wang, M.; Chen, L.-J.; Bahadur, P. Micellar Characteristics of an Amphiphilic Star-Block Copolymer in DES-Water Mixture. Colloid Polym. Sci. 2021, 299, 117–128. [Google Scholar] [CrossRef]
- Elbourne, A.; Meftahi, N.; Greaves, T.L.; McConville, C.F.; Bryant, G.; Bryant, S.J.; Christofferson, A.J. Nanostructure of a Deep Eutectic Solvent at Solid Interfaces. J. Colloid Interface Sci. 2021, 591, 38–51. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. Deep Eutectic Solvents on the Surface of Face Centered Cubic Metals. J. Phys. Chem. C 2016, 120, 10400–10409. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. Molecular Dynamics Simulations of Metal Nanoparticles in Deep Eutectic Solvents. J. Phys. Chem. C 2018, 122, 18029–18039. [Google Scholar] [CrossRef]
- Gao, Q.; Wu, N.; Qin, Y.; Laaksonen, A.; Zhu, Y.; Ji, X.; Lu, X. Molecular Insight into Wetting Behavior of Deep Eutectic Solvent Droplets on Ionic Substrates: A Molecular Dynamics Study. J. Mol. Liq. 2020, 319, 114298. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. Molecular Dynamics Simulations of Mixed Deep Eutectic Solvents and Their Interaction with Nanomaterials. J. Mol. Liq. 2019, 283, 147–154. [Google Scholar] [CrossRef]
- Kaur, S.; Sharma, S.; Kashyap, H.K. Bulk and Interfacial Structures of Reline Deep Eutectic Solvent: A Molecular Dynamics Study. J. Chem. Phys. 2017, 147, 194507. [Google Scholar] [CrossRef]
- Malik, A.; Dhattarwal, H.S.; Kashyap, H.K. Molecular Dynamics Investigation of Wetting-Dewetting Behavior of Reline DES Nanodroplet at Model Carbon Material. J. Chem. Phys. 2020, 153, 164704. [Google Scholar] [CrossRef] [PubMed]
- Rozas, S.; Atilhan, M.; Aparicio, S. Insights on (C, BN, Si, Ge, MoS2) Nanotubes in Reline Deep Eutectic Solvent. J. Phys. Chem. B 2020, 124, 3556–3567. [Google Scholar] [CrossRef]
- Castro, V.I.B.; Craveiro, R.; Silva, J.M.; Reis, R.L.; Paiva, A.; Duarte, A.R.C. Natural Deep Eutectic Systems as Alternative Nontoxic Cryoprotective Agents. Cryobiology 2018, 83, 15–26. [Google Scholar] [CrossRef]
- Das, A.; Mukhopadhyay, C. Urea-Mediated Protein Denaturation: A Consensus View. J. Phys. Chem. B 2009, 113, 12816–12824. [Google Scholar] [CrossRef]
- Lim, W.K.; Rösgen, J.; Englander, S.W. Urea, but Not Guanidinium, Destabilizes Proteins by Forming Hydrogen Bonds to the Peptide Group. Proc. Natl. Acad. Sci. USA 2009, 106, 2595–2600. [Google Scholar] [CrossRef] [Green Version]
- Rossky, P.J. Protein Denaturation by Urea: Slash and Bond. Proc. Natl. Acad. Sci. USA 2008, 105, 16825–16826. [Google Scholar] [CrossRef] [Green Version]
- Gorke, J.T.; Srienc, F.; Kazlauskas, R.J. Hydrolase-Catalyzed Biotransformations in Deep Eutectic Solvents. Chem. Commun. 2008, 1235–1237. [Google Scholar] [CrossRef] [PubMed]
- Monhemi, H.; Housaindokht, M.R.; Moosavi-Movahedi, A.A.; Bozorgmehr, M.R. How a Protein Can Remain Stable in a Solvent with High Content of Urea: Insights from Molecular Dynamics Simulation of Candida Antarctica Lipase B in Urea: Choline Chloride Deep Eutectic Solvent. Phys. Chem. Chem. Phys. 2014, 16, 14882–14893. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ghosh, S.; Chakrabarti, R. Ammonium Based Stabilizers Effectively Counteract Urea-Induced Denaturation in a Small Protein: Insights from Molecular Dynamics Simulations. RSC Adv. 2017, 7, 52888–52906. [Google Scholar] [CrossRef] [Green Version]
- Maity, A.; Sarkar, S.; Theeyancheri, L.; Chakrabarti, R. Choline Chloride as a Nano-Crowder Protects HP-36 from Urea-Induced Denaturation: Insights from Solvent Dynamics and Protein-Solvent Interactions. Chemphyschem 2020, 21, 552–567. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Roy, R.; Paul, S. Potential of a Natural Deep Eutectic Solvent, Glyceline, in the Thermal Stability of the Trp-Cage Mini-Protein. J. Phys. Chem. B 2020, 124, 7598–7610. [Google Scholar] [CrossRef]
- Pal, S.; Paul, S. Effect of Hydrated and Nonhydrated Choline Chloride–Urea Deep Eutectic Solvent (Reline) on Thrombin-Binding G-Quadruplex Aptamer (TBA): A Classical Molecular Dynamics Simulation Study. J. Phys. Chem. C 2019, 123, 11686–11698. [Google Scholar] [CrossRef]
- Pal, S.; Paul, S. Understanding The Role of Reline, a Natural DES, on Temperature-Induced Conformational Changes of C-Kit G-Quadruplex DNA: A Molecular Dynamics Study. J. Phys. Chem. B 2020, 124, 3123–3136. [Google Scholar] [CrossRef] [PubMed]
- Atilhan, M.; Costa, L.T.; Aparicio, S. On the Behaviour of Aqueous Solutions of Deep Eutectic Solvents at Lipid Biomembranes. J. Mol. Liq. 2017, 247, 116–125. [Google Scholar] [CrossRef]
- Chen, Y.; Mu, T. Application of Deep Eutectic Solvents in Biomass Pretreatment and Conversion. Green Energy Environ. 2019, 4, 95–115. [Google Scholar] [CrossRef]
- Mohan, M.; Naik, P.K.; Banerjee, T.; Goud, V.V.; Paul, S. Solubility of Glucose in Tetrabutylammonium Bromide Based Deep Eutectic Solvents: Experimental and Molecular Dynamic Simulations. Fluid Phase Equilib. 2017, 448, 168–177. [Google Scholar] [CrossRef]
- Fu, H.; Wang, X.; Sang, H.; Hou, Y.; Chen, X.; Feng, X. Dissolution Behavior of Microcrystalline Cellulose in DBU-Based Deep Eutectic Solvents: Insights from Spectroscopic Investigation and Quantum Chemical Calculations. J. Mol. Liq. 2020, 299, 112140. [Google Scholar] [CrossRef]
- Sánchez-Badillo, J.A.; Gallo, M.; Rutiaga-Quiñones, J.G.; López-Albarrán, P. Solvent Behavior of an Ionic Liquid Set around a Cellulose Iβ Crystallite Model through Molecular Dynamics Simulations. Cellulose 2021, 28, 6767–6795. [Google Scholar] [CrossRef]
- Muley, P.D.; Mobley, J.K.; Tong, X.; Novak, B.; Stevens, J.; Moldovan, D.; Shi, J.; Boldor, D. Rapid Microwave-Assisted Biomass Delignification and Lignin Depolymerization in Deep Eutectic Solvents. Energy Convers. Manag. 2019, 196, 1080–1088. [Google Scholar] [CrossRef]
- Provatas, N.; Elder, K. Phase-Field Methods in Materials Science and Engineering; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010. [Google Scholar]
- Steinbach, I. Phase-Field Models in Materials Science. Model. Simul. Mater. Sci. Eng. 2009, 17, 073001. [Google Scholar] [CrossRef]
- Boettinger, W.J.; Warren, J.A.; Beckermann, C.; Karma, A. Phase-Field Simulation of Solidification. Annu. Rev. Mater. Res. 2002, 32, 163–194. [Google Scholar] [CrossRef]
- Humadi, H.; Ofori-Opoku, N.; Provatas, N.; Hoyt, J.J. Atomistic Modeling of Solidification Phenomena Using the Phase-Field-Crystal Model. JOM 2013, 65, 1103–1110. [Google Scholar] [CrossRef]
- Grossmann, B.; Elder, K.R.; Grant, M.; Kosterlitz, J.M. Directional Solidification in Two and Three Dimensions. Phys. Rev. Lett. 1993, 71, 3323–3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, K.R.; Katakowski, M.; Haataja, M.; Grant, M. Modeling Elasticity in Crystal Growth. Phys. Rev. Lett. 2002, 88, 245701. [Google Scholar] [CrossRef] [Green Version]
- Alster, E.; Elder, K.R.; Voorhees, P.W. Displacive Phase-Field Crystal Model. Phys. Rev. Mater. 2020, 4. [Google Scholar] [CrossRef]
- Courtemanche, M. Complex Spiral Wave Dynamics in a Spatially Distributed Ionic Model of Cardiac Electrical Activity. Chaos 1996, 6, 579–600. [Google Scholar] [CrossRef] [PubMed]
- Efimov, I.R.; Krinsky, V.I.; Jalife, J. Dynamics of Rotating Vortices in the Beeler-Reuter Model of Cardiac Tissue. Chaos Solitons Fractals 1995, 5, 513–526. [Google Scholar] [CrossRef]
- Elder, K.; Grant, M. Modeling Elastic and Plastic Deformations in Nonequilibrium Processing Using Phase Field Crystals. Phys. Rev. E 2004, 70, 051605. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, G.; Li, Y.; Hong, Z.; Wang, D.; Shi, S. Application of Phase-Field Method in Rechargeable Batteries. NPJ Comput. Mater. 2020, 6, 176. [Google Scholar] [CrossRef]
- Wheeler, D.; Keller, T.; DeWitt, S.J.; Jokisaari, A.M.; Schwen, D.; Guyer, J.E.; Aagesen, L.K.; Heinonen, O.G.; Tonks, M.R.; Voorhees, P.W.; et al. PFHub: The Phase-Field Community Hub. J. Open Res. Softw. 2019, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, S.; Rudraraju, S.; Montiel, D.; Andrews, W.B.; Thornton, K. PRISMS-PF: A General Framework for Phase-Field Modeling with a Matrix-Free Finite Element Method. NPJ Comput. Mater. 2020, 6, 1–12. [Google Scholar] [CrossRef]
- Permann, C.J.; Gaston, D.R.; Andrš, D.; Carlsen, R.W.; Kong, F.; Lindsay, A.D.; Miller, J.M.; Peterson, J.W.; Slaughter, A.E.; Stogner, R.H.; et al. MOOSE: Enabling Massively Parallel Multiphysics Simulation. SoftwareX 2020, 11, 100430. [Google Scholar] [CrossRef]
- Cimrman, R.; Lukeš, V.; Rohan, E. Multiscale Finite Element Calculations in Python Using SfePy. Adv. Comput. Math. 2019, 45, 1897–1921. [Google Scholar] [CrossRef] [Green Version]
- Guyer, J.E.; Wheeler, D.; Warren, J.A. FiPy: Partial Differential Equations with Python. Comput. Sci. Eng. 2009, 11, 6–15. [Google Scholar] [CrossRef]
- Silber, S.A.; Karttunen, M. SymPhas—General Purpose Software for Phase-Field, Phase-Field Crystal and Reaction-Diffusion Simulations. Adv. Theory Sim. 2021, 2100351. [Google Scholar] [CrossRef]
- Wheeler, A.A.; Boettinger, W.J.; McFadden, G.B. Phase-Field Model for Isothermal Phase Transitions in Binary Alloys. Phys. Rev. A 1992, 45, 7424–7439. [Google Scholar] [CrossRef]
- Elder, K.R.; Drolet, F.; Kosterlitz, J.M.; Grant, M. Stochastic Eutectic Growth. Phys. Rev. Lett. 1994, 72, 677–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karma, A. Phase-Field Model of Eutectic Growth. Phys. Rev. E 1994, 49, 2245–2250. [Google Scholar] [CrossRef]
- Wheeler, A.A.; McFadden, G.B.; Boettinger, W.J. Phase-Field Model for Solidification of a Eutectic Alloy. Proc. R. Soc. Lond. Ser. A Math. Phys. Eng. Sci. 1996, 452, 495–525. [Google Scholar]
- Gyoon Kim, S.; Tae Kim, W.; Suzuki, T.; Ode, M. Phase-Field Modeling of Eutectic Solidification. J. Cryst. Growth 2004, 261, 135–158. [Google Scholar] [CrossRef]
- Choudhury, A.N. Quantitative Phase-Field Model for Phase Transformations in Multi-Component Alloys; KIT Scientific Publishing: Karlsruhe, Germany, 2012. [Google Scholar]
- Choudhury, A.; Nestler, B. Grand-Potential Formulation for Multicomponent Phase Transformations Combined with Thin-Interface Asymptotics of the Double-Obstacle Potential. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2012, 85, 021602. [Google Scholar] [CrossRef]
- Plapp, M. Unified Derivation of Phase-Field Models for Alloy Solidification from a Grand-Potential Functional. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2011, 84, 031601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, B.; Garcke, H.; Stinner, B. Multicomponent Alloy Solidification: Phase-Field Modeling and Simulations. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2005, 71, 041609. [Google Scholar] [CrossRef] [Green Version]
- Steinmetz, P.; Hötzer, J.; Kellner, M.; Genau, A.; Nestler, B. Study of Pattern Selection in 3D Phase-Field Simulations during the Directional Solidification of Ternary Eutectic Al-Ag-Cu. Comput. Mater. Sci. 2018, 148, 131–140. [Google Scholar] [CrossRef]
- Kollau, L.J.B.M.; Vis, M.; van den Bruinhorst, A.; Tuinier, R.; de With, G. Entropy Models for the Description of the Solid–liquid Regime of Deep Eutectic Solutions. J. Mol. Liq. 2020, 302, 112155. [Google Scholar] [CrossRef]
- Cosby, T.; Kapoor, U.; Shah, J.K.; Sangoro, J. Mesoscale Organization and Dynamics in Binary Ionic Liquid Mixtures. J. Phys. Chem. Lett. 2019, 10, 6274–6280. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zhang, C.; Zhang, L.; Wei, H.; Li, Y.; Yu, G. Insights into Hydrotropic Solubilization for Hybrid Ion Redox Flow Batteries. ACS Energy Lett. 2018, 3, 2641–2648. [Google Scholar] [CrossRef]
- Zahrina, I.; Nasikin, M.; Mulia, K.; Prajanto, M.; Yanuar, A. Molecular Interactions between Betaine Monohydrate-Glycerol Deep Eutectic Solvents and Palmitic Acid: Computational and Experimental Studies. J. Mol. Liq. 2018, 251, 28–34. [Google Scholar] [CrossRef]
- Zahrina, I.; Mulia, K.; Yanuar, A.; Nasikin, M. Molecular Interactions in the Betaine Monohydrate-Polyol Deep Eutectic Solvents: Experimental and Computational Studies. J. Mol. Struct. 2018, 1158, 133–138. [Google Scholar] [CrossRef]
- Pauric, A.D.; Halalay, I.C.; Goward, G.R. Combined NMR and Molecular Dynamics Modeling Study of Transport Properties in Sulfonamide Based Deep Eutectic Lithium Electrolytes: LiTFSI Based Binary Systems. Phys. Chem. Chem. Phys. 2016, 18, 6657–6667. [Google Scholar] [CrossRef]
- Karibayev, M.; Shah, D. Comprehensive Computational Analysis Exploring the Formation of Caprolactam-Based Deep Eutectic Solvents and Their Applications in Natural Gas Desulfurization. Energy Fuels 2020, 34, 9894–9902. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ghosh, D.; Haldar, T.; Deb, P.; Sakpal, S.S.; Deshmukh, S.H.; Kashid, S.M.; Bagchi, S. Hydrocarbon Chain-Length Dependence of Solvation Dynamics in Alcohol-Based Deep Eutectic Solvents: A Two-Dimensional Infrared Spectroscopic Investigation. J. Phys. Chem. B 2019, 123, 9355–9363. [Google Scholar] [CrossRef]
- Chatterjee, S.; Haldar, T.; Ghosh, D.; Bagchi, S. Electrostatic Manifestation of Micro-Heterogeneous Solvation Structures in Deep-Eutectic Solvents: A Spectroscopic Approach. J. Phys. Chem. B 2020, 124, 3709–3715. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fang, H.; Liu, X.; Xu, B.; Rao, Z. Novel Silica Filled Deep Eutectic Solvent Based Nanofluids for Energy Transportation. ACS Sustain. Chem. Eng. 2019, 7, 20159–20169. [Google Scholar] [CrossRef]
- Barani Pour, S.; Jahanbin Sardroodi, J.; Rastkar Ebrahimzadeh, A. The Study of Structure and Interactions of Glucose-Based Natural Deep Eutectic Solvents by Molecular Dynamics Simulation. J. Mol. Liq. 2021, 334, 115956. [Google Scholar] [CrossRef]
- Mamashli, F.; Badraghi, J.; Delavari, B.; Lanjanian, H.; Sabbaghian, M.; Hosseini, M.; Saboury, A.A. Improvement of Versatile Peroxidase Activity and Stability by a Cholinium-Based Ionic Liquid. J. Mol. Liq. 2018, 272, 597–608. [Google Scholar] [CrossRef]
- Lehmann, C.; Bocola, M.; Streit, W.R.; Martinez, R.; Schwaneberg, U. Ionic Liquid and Deep Eutectic Solvent-Activated CelA2 Variants Generated by Directed Evolution. Appl. Microbiol. Biotechnol. 2014, 98, 5775–5785. [Google Scholar] [CrossRef] [PubMed]
- Bittner, J.P.; Huang, L.; Zhang, N.; Kara, S.; Jakobtorweihen, S. Comparison and Validation of Force Fields for Deep Eutectic Solvents in Combination with Water and Alcohol Dehydrogenase. J. Chem. Theory Comput. 2021, 17, 5322–5341. [Google Scholar] [CrossRef]
- Ji, H.; Lv, P. Mechanistic Insights into the Lignin Dissolution Behaviors of a Recyclable Acid Hydrotrope, Deep Eutectic Solvent (DES), and Ionic Liquid (IL). Green Chem. 2020, 22, 1378–1387. [Google Scholar] [CrossRef]
- Yan, W.-W.; Zong, Z.-M.; Li, Z.-X.; Li, J.; Liu, G.-H.; Ma, Z.-H.; Zhang, Y.-Y.; Xu, M.-L.; Liu, F.-J.; Wei, X.-Y. Effective Separation and Purification of Nitrogen-Containing Aromatics from the Light Portion of a High-Temperature Coal Tar Using Choline Chloride and Malonic Acid: Experimental and Molecular Dynamics Simulation. ACS Sustain. Chem. Eng. 2020, 8, 9464–9471. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. Molecular Dynamics Study on the Use of Deep Eutectic Solvents for Enhanced Oil Recovery. J. Pet. Sci. Eng. 2022, 209, 109953. [Google Scholar] [CrossRef]
- Triolo, A.; Lo Celso, F.; Russina, O. Structural Features of β-Cyclodextrin Solvation in the Deep Eutectic Solvent, Reline. J. Phys. Chem. B 2020, 124, 2652–2660. [Google Scholar] [CrossRef]
- Hammond, O.S.; Bowron, D.T.; Edler, K.J. Liquid Structure of the Choline Chloride-Urea Deep Eutectic Solvent (reline) from Neutron Diffraction and Atomistic Modelling. Green Chem. 2016, 18, 2736–2744. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Vats, B.; Pandey, A.K.; Sharma, M.K.; Sahu, P.; Yadav, A.K.; Ali, S.M.; Kannan, S. Insight into Speciation and Electrochemistry of Uranyl Ions in Deep Eutectic Solvents. J. Phys. Chem. B 2020, 124, 181–189. [Google Scholar] [CrossRef]
- Zhang, Y.; You, Y.; Gao, Q.; Zhang, C.; Wang, S.; Qin, Y.; Zhu, Y.; Lu, X. Molecular Insight into Flow Resistance of Choline Chloride/urea Confined in Ionic Model Nanoslits. Fluid Phase Equilib. 2021, 533, 112934. [Google Scholar] [CrossRef]
- Shah, D.; Mansurov, U.; Mjalli, F.S. Intermolecular Interactions and Solvation Effects of Dimethylsulfoxide on Type III Deep Eutectic Solvents. Phys. Chem. Chem. Phys. 2019, 21, 17200–17208. [Google Scholar] [CrossRef] [PubMed]
- Abedin, R.; Heidarian, S.; Flake, J.C.; Hung, F.R. Computational Evaluation of Mixtures of Hydrofluorocarbons and Deep Eutectic Solvents for Absorption Refrigeration Systems. Langmuir 2017, 33, 11611–11625. [Google Scholar] [CrossRef]
- Zhekenov, T.; Toksanbayev, N.; Kazakbayeva, Z.; Shah, D.; Mjalli, F.S. Formation of Type III Deep Eutectic Solvents and Effect of Water on Their Intermolecular Interactions. Fluid Phase Equilib. 2017, 441, 43–48. [Google Scholar] [CrossRef]
- Aryafard, M.; Abbasi, M.; Řeha, D.; Harifi-Mood, A.R.; Minofar, B. Experimental and Theoretical Investigation of Solvatochromic Properties and Ion Solvation Structure in DESs of Reline, Glyceline, Ethaline and Their Mixtures with PEG 400. J. Mol. Liq. 2019, 284, 59–67. [Google Scholar] [CrossRef]
- Harifi-Mood, A.R.; Ghobadi, R.; Matić, S.; Minofar, B.; Řeha, D. Solvation Analysis of Some Solvatochromic Probes in Binary Mixtures of Reline, Ethaline, and Glyceline with DMSO. J. Mol. Liq. 2016, 222, 845–853. [Google Scholar] [CrossRef]
- Salehi, H.S.; Ramdin, M.; Moultos, O.A.; Vlugt, T.J.H. Computing Solubility Parameters of Deep Eutectic Solvents from Molecular Dynamics Simulations. Fluid Phase Equilib. 2019, 497, 10–18. [Google Scholar] [CrossRef]
- Atilhan, M.; Aparicio, S. Behavior of Deep Eutectic Solvents under External Electric Fields: A Molecular Dynamics Approach. J. Phys. Chem. B 2017, 121, 221–232. [Google Scholar] [CrossRef]
- Vraneš, M.; Tot, A.; Papović, S.; Panić, J.; Gadžurić, S. Is Choline Kosmotrope or Chaotrope? J. Chem. Thermodyn. 2018, 124, 65–73. [Google Scholar] [CrossRef]
- Triolo, A.; Lo Celso, F.; Brehm, M.; Di Lisio, V.; Russina, O. Liquid Structure of a Choline Chloride-Water Natural Deep Eutectic Solvent: A Molecular Dynamics Characterization. J. Mol. Liq. 2021, 331, 115750. [Google Scholar] [CrossRef]
- Malik, A.; Kashyap, H.K. Multiple Evidences of Dynamic Heterogeneity in Hydrophobic Deep Eutectic Solvents. J. Chem. Phys. 2021, 155, 044502. [Google Scholar] [CrossRef]
- Mjalli, F.S.; Shah, D. Simulation Based Insight into Solvation Properties of Ferric Chloride Based Eutectic Solvent. Chem. Eng. Trans. 2015, 43, 1843–1848. [Google Scholar] [CrossRef]
- Srinivasan, H.; Sharma, V.K.; Mitra, S. Water Accelerates the Hydrogen-Bond Dynamics and Abates Heterogeneity in Deep Eutectic Solvent Based on Acetamide and Lithium Perchlorate. J. Chem. Phys. 2021, 155, 024505. [Google Scholar] [CrossRef]
- Lesch, V.; Heuer, A.; Rad, B.R.; Winter, M.; Smiatek, J. Atomistic Insights into Deep Eutectic Electrolytes: The Influence of Urea on the Electrolyte Salt LiTFSI in View of Electrochemical Applications. Phys. Chem. Chem. Phys. 2016, 18, 28403–28408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandy, A.; Smiatek, J. Mixtures of LiTFSI and Urea: Ideal Thermodynamic Behavior as Key to the Formation of Deep Eutectic Solvents? Phys. Chem. Chem. Phys. 2019, 21, 12279–12287. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Lin, X.; Wang, X.; Xu, J.; Xu, X. Understanding the CO2 Capture Performance by MDEA-Based Deep Eutectics Solvents with Excellent Cyclic Capacity. Fuel 2021, 293, 120466. [Google Scholar] [CrossRef]
- Sanam; Bux, K.; al-Rashida, M.; Alharthy, R.D.; Moin, S.T.; Hameed, A. Morpholinium and Piperidinium Based Deep Eutectic Solvents for Synthesis of pyrazole-5-carbonitriles, Indoles and Tetrazoles: Bulk PropertiesviaMolecular Dynamics Simulations. ChemistrySelect 2018, 3, 12907–12917. [Google Scholar] [CrossRef]
- Panda, D.K.; Bhargava, B.L. Intermolecular Interactions in Tetrabutylammonium Chloride Based Deep Eutectic Solvents: Classical Molecular Dynamics Studies. J. Mol. Liq. 2021, 335, 116139. [Google Scholar] [CrossRef]
- Lima, F.; Dietz, C.H.J.T.; Silvestre, A.J.D.; Branco, L.C.; Canongia Lopes, J.; Gallucci, F.; Shimizu, K.; Held, C.; Marrucho, I.M. Vapor Pressure Assessment of Sulfolane-Based Eutectic Solvents: Experimental, PC-SAFT, and Molecular Dynamics. J. Phys. Chem. B 2020, 124, 10386–10397. [Google Scholar] [CrossRef]
- Gapeyenko, D.; Jamwal, P.; Shah, D. Molecular Dynamics Simulations to Design Novel Solvents for Deep Desulfurization. In Proceedings of the Advanced Materials; Springer International Publishing: Cham, Switzerland, 2018; pp. 119–124. [Google Scholar]
- Wu, X.; Cheng, N.-N.; Jiang, H.; Zheng, W.-T.; Chen, Y.; Huang, K.; Liu, F. 1-Ethyl-3-Methylimidazolium Chloride plus Imidazole Deep Eutectic Solvents as Physical Solvents for Remarkable Separation of H2S from CO2. Sep. Purif. Technol. 2021, 276, 119313. [Google Scholar] [CrossRef]
- Altamash, T.; Amhamed, A.I.; Aparicio, S.; Atilhan, M. Combined Experimental and Theoretical Study on High Pressure Methane Solubility in Natural Deep Eutectic Solvents. Ind. Eng. Chem. Res. 2019, 58, 8097–8111. [Google Scholar] [CrossRef]
- Kalhor, P.; Yarivand, O.; Seifpanahi-Shabani, K. Quantum Chemical Calculations on Dissolution of Dimethylformamide in Ethaline. J. Mol. Graph. Model. 2021, 107, 107966. [Google Scholar] [CrossRef]
- Zhong, F.-Y.; Zhou, L.; Shen, J.; Liu, Y.; Fan, J.-P.; Huang, K. Rational Design of Azole-Based Deep Eutectic Solvents for Highly Efficient and Reversible Capture of Ammonia. ACS Sustain. Chem. Eng. 2019, 7, 14170–14179. [Google Scholar] [CrossRef]
- Jangir, A.K.; Lad, B.; Dani, U.; Shah, N.; Kuperkar, K. In Vitro Toxicity Assessment and Enhanced Drug Solubility Profile of Green Deep Eutectic Solvent Derivatives (DESDs) Combined with Theoretical Validation. RSC Adv. 2020, 10, 24063–24072. [Google Scholar] [CrossRef]
- El-hoshoudy, A.N.; Soliman, F.S.; Mansour, E.M.; Zaki, T.; Desouky, S.M. Experimental and Theoretical Investigation of Quaternary Ammonium-Based Deep Eutectic Solvent for Secondary Water Flooding. J. Mol. Liq. 2019, 294, 111621. [Google Scholar] [CrossRef]
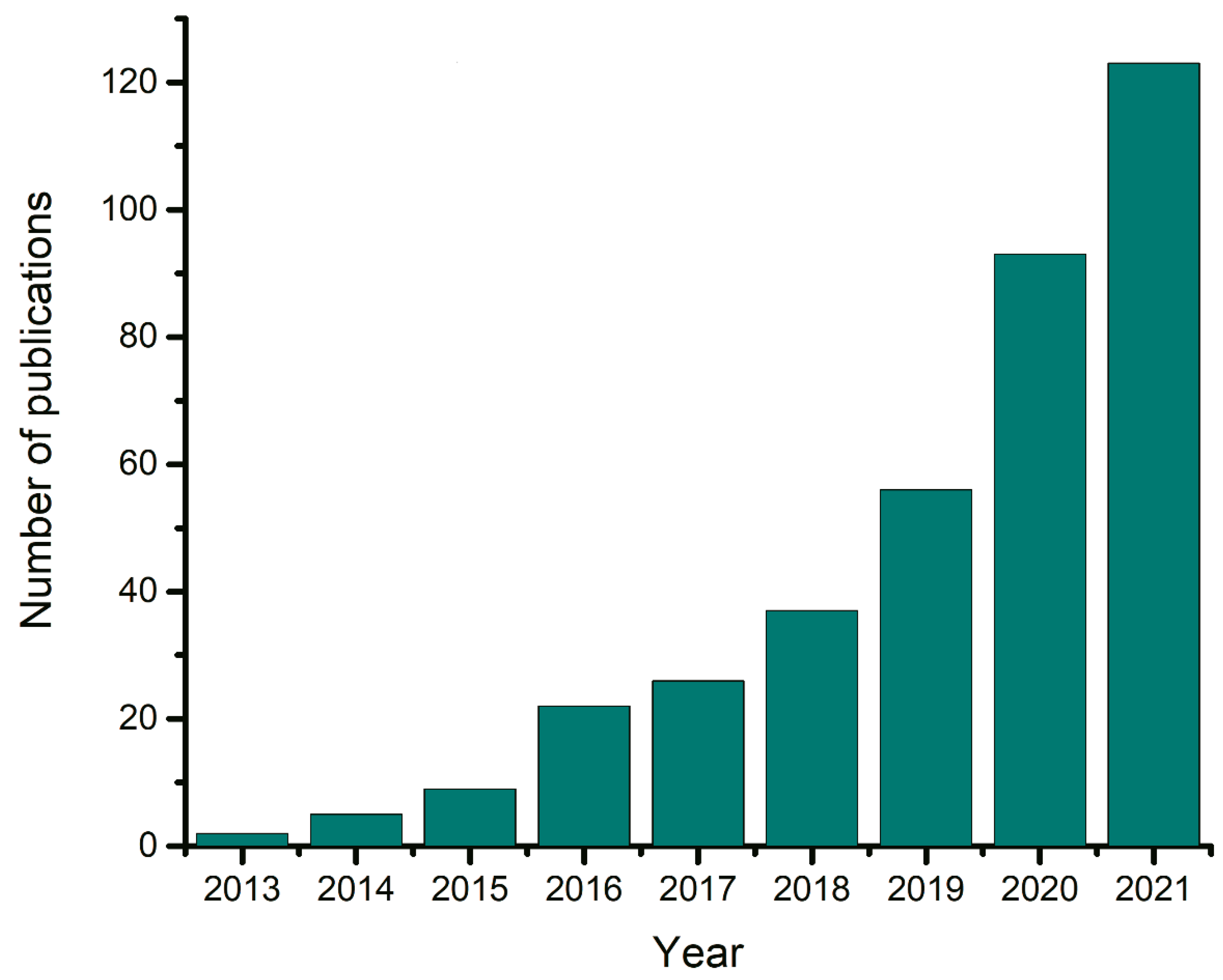
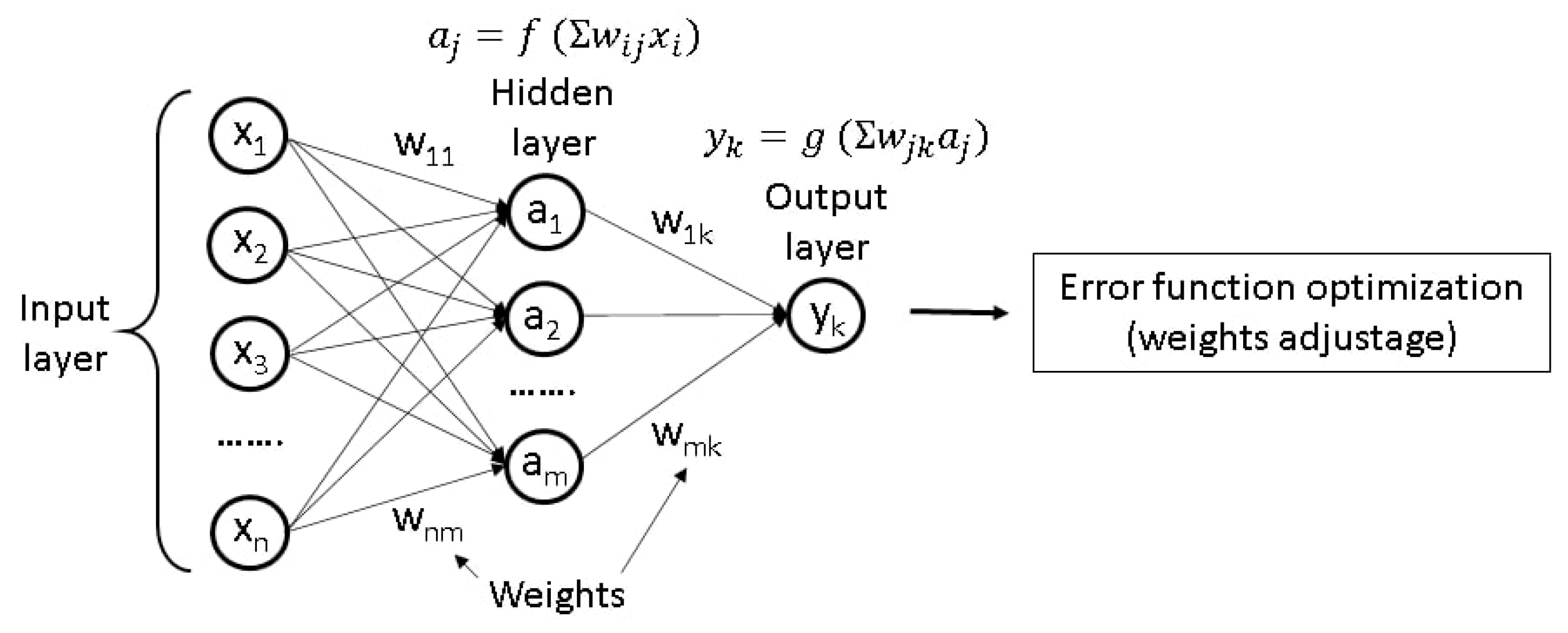
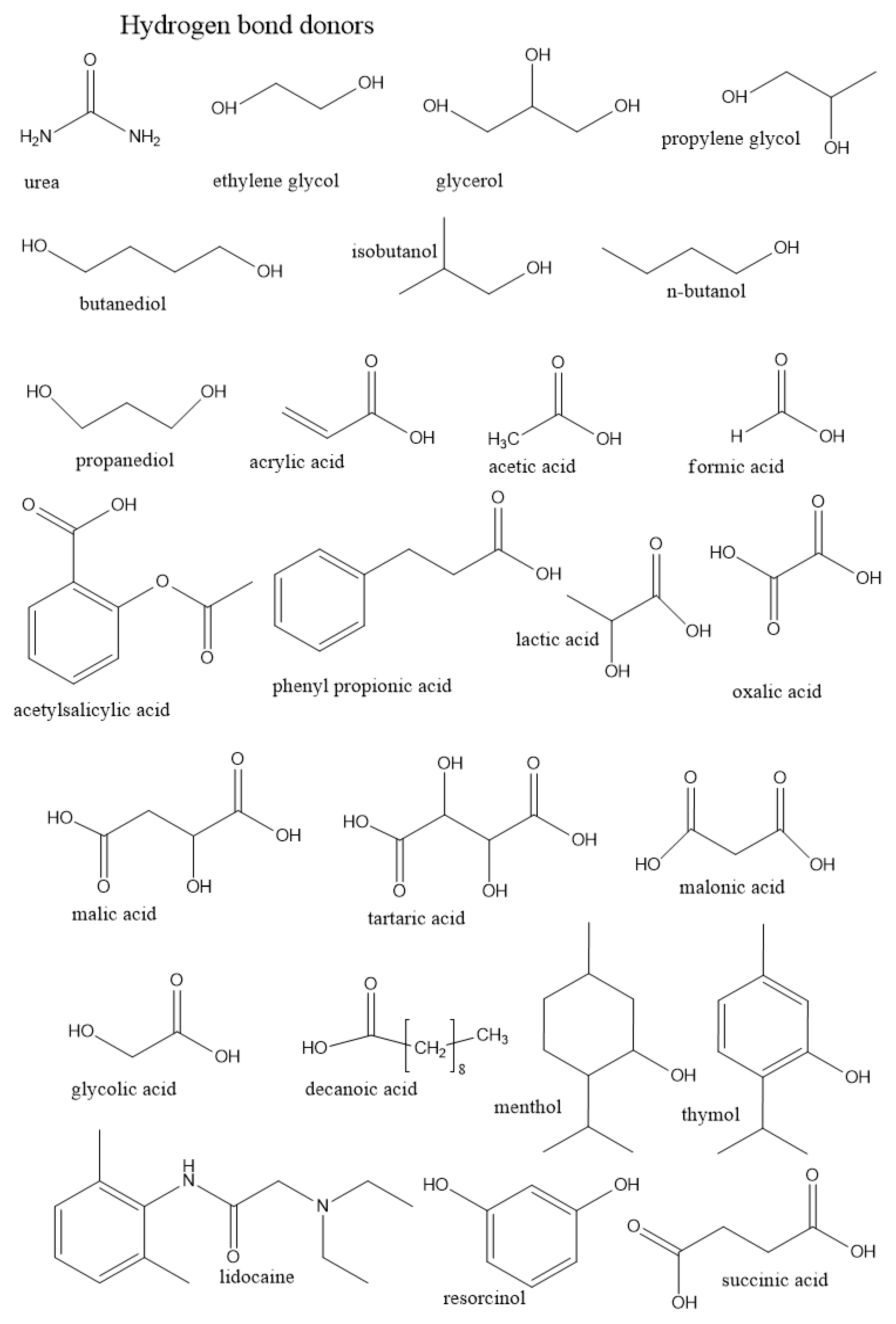
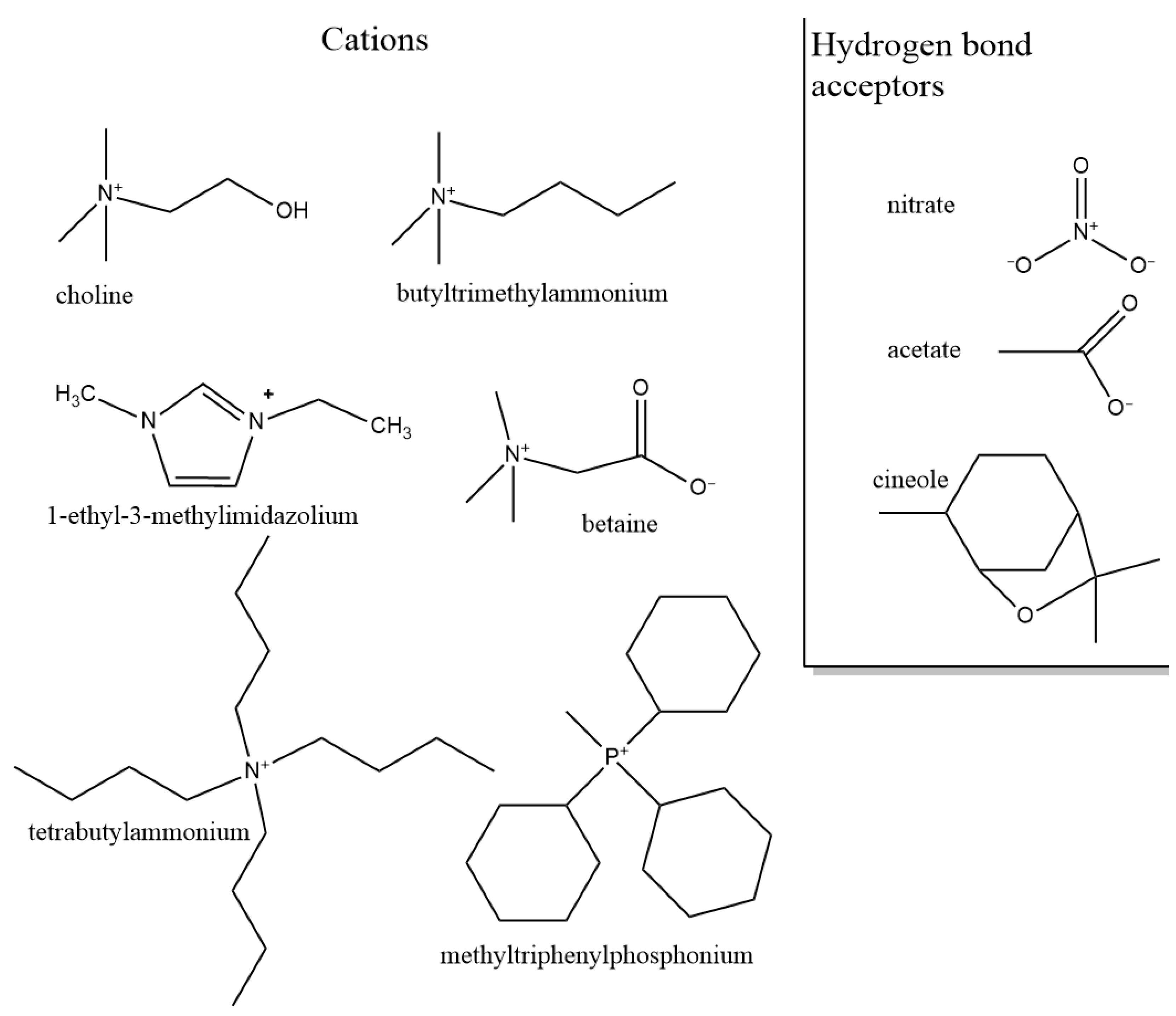
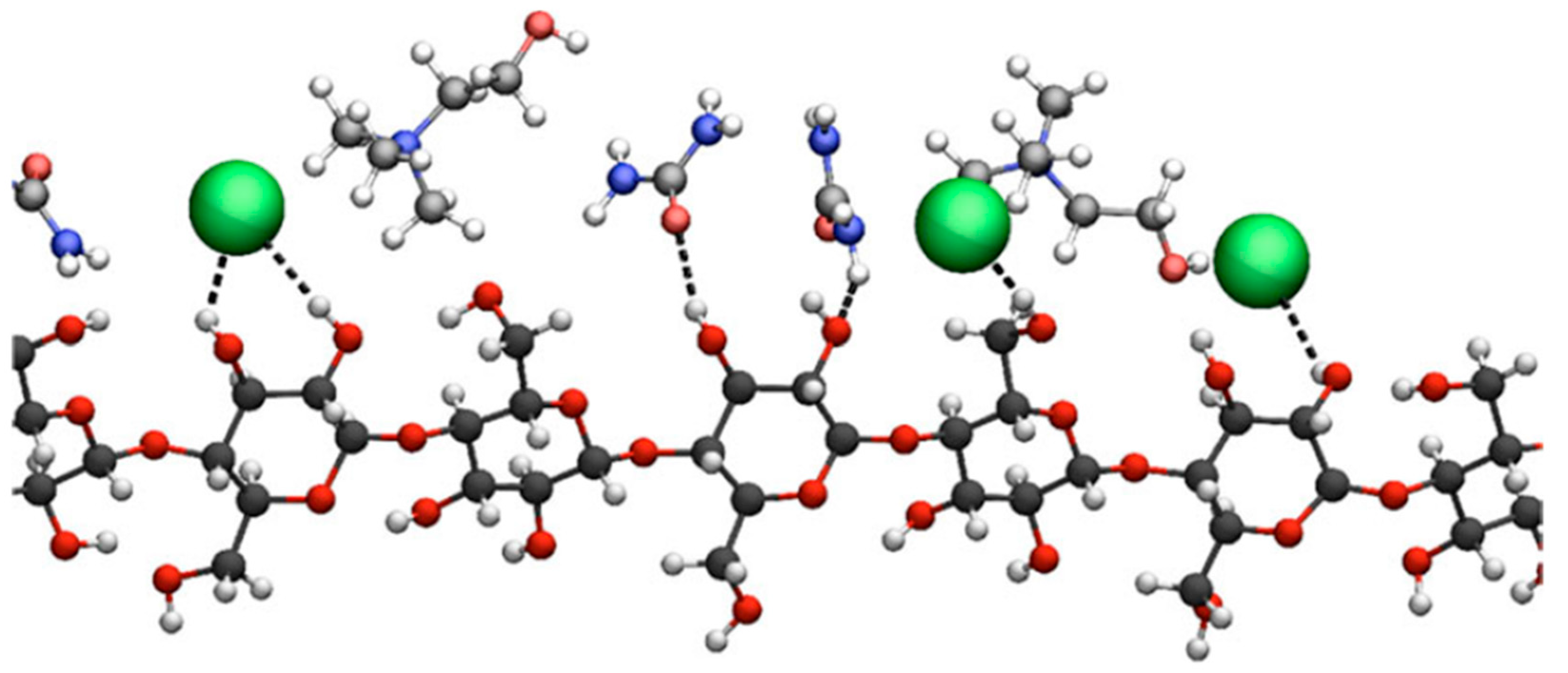
| Type | Composition | Example |
|---|---|---|
| Type I | Organic and metal salts | Choline chloride + Metal halide (SnCl2, ZnCl2, etc.) |
| Type II | Organic and metal salt hydrate | Choline chloride + Metal salt hydrate (CrCl3∙6H2O, etc.) |
| Type III | Organic salt and H-bond donor | Choline chloride + organic compound (urea, carboxylic acids, alcohols, etc.) |
| Type IV | Metal salt and H-bond donor | Metal halide (ZnCl2) + organic compound (urea, carboxylic acids, alcohols, etc.) |
| Type V | Non-ionic DESs. Hydrogen Bond Acceptor and Hydrogen Bond Donor | Thymol + menthol |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolmachev, D.; Lukasheva, N.; Ramazanov, R.; Nazarychev, V.; Borzdun, N.; Volgin, I.; Andreeva, M.; Glova, A.; Melnikova, S.; Dobrovskiy, A.; et al. Computer Simulations of Deep Eutectic Solvents: Challenges, Solutions, and Perspectives. Int. J. Mol. Sci. 2022, 23, 645. https://doi.org/10.3390/ijms23020645
Tolmachev D, Lukasheva N, Ramazanov R, Nazarychev V, Borzdun N, Volgin I, Andreeva M, Glova A, Melnikova S, Dobrovskiy A, et al. Computer Simulations of Deep Eutectic Solvents: Challenges, Solutions, and Perspectives. International Journal of Molecular Sciences. 2022; 23(2):645. https://doi.org/10.3390/ijms23020645
Chicago/Turabian StyleTolmachev, Dmitry, Natalia Lukasheva, Ruslan Ramazanov, Victor Nazarychev, Natalia Borzdun, Igor Volgin, Maria Andreeva, Artyom Glova, Sofia Melnikova, Alexey Dobrovskiy, and et al. 2022. "Computer Simulations of Deep Eutectic Solvents: Challenges, Solutions, and Perspectives" International Journal of Molecular Sciences 23, no. 2: 645. https://doi.org/10.3390/ijms23020645
APA StyleTolmachev, D., Lukasheva, N., Ramazanov, R., Nazarychev, V., Borzdun, N., Volgin, I., Andreeva, M., Glova, A., Melnikova, S., Dobrovskiy, A., Silber, S. A., Larin, S., de Souza, R. M., Ribeiro, M. C. C., Lyulin, S., & Karttunen, M. (2022). Computer Simulations of Deep Eutectic Solvents: Challenges, Solutions, and Perspectives. International Journal of Molecular Sciences, 23(2), 645. https://doi.org/10.3390/ijms23020645