
Collagen Type XI Inhibits Lung Cancer-Associated Fibroblast Functions and Restrains the Integrin Binding Site Availability on Collagen Type I Matrix


Abstract
1. Introduction
2. Results
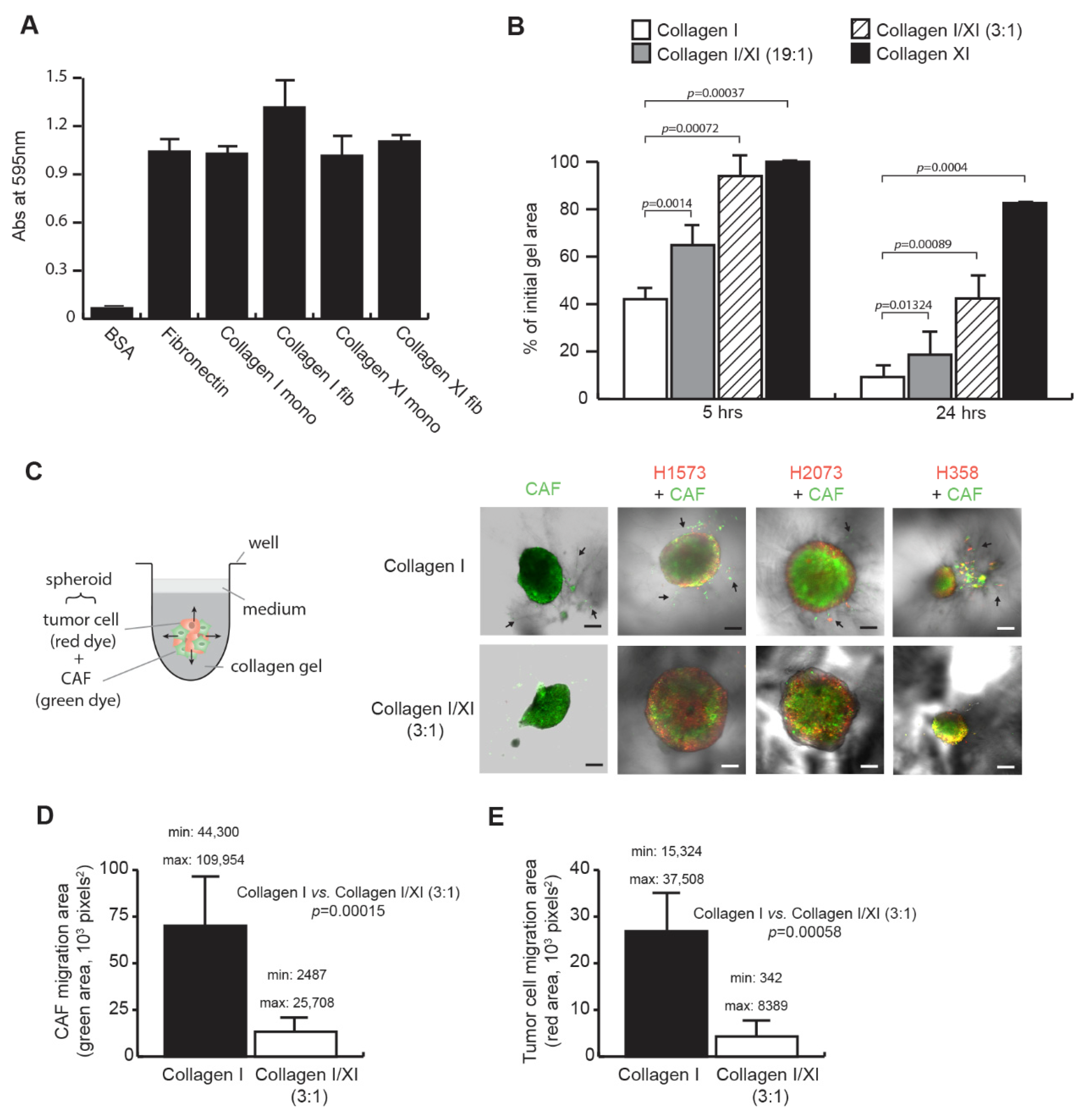
2.1. Collagen Type XI Inhibits Lung CAF Features
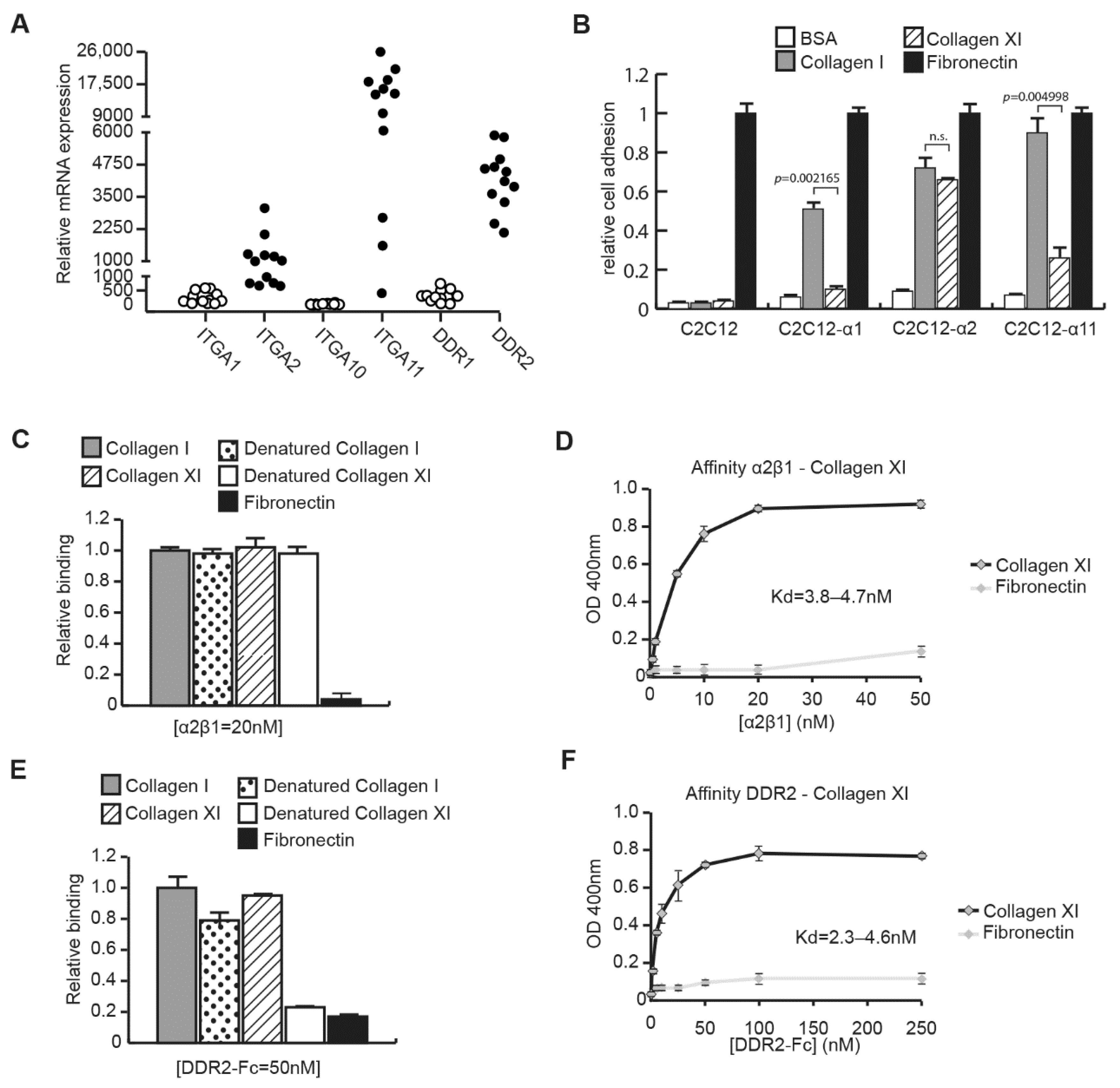
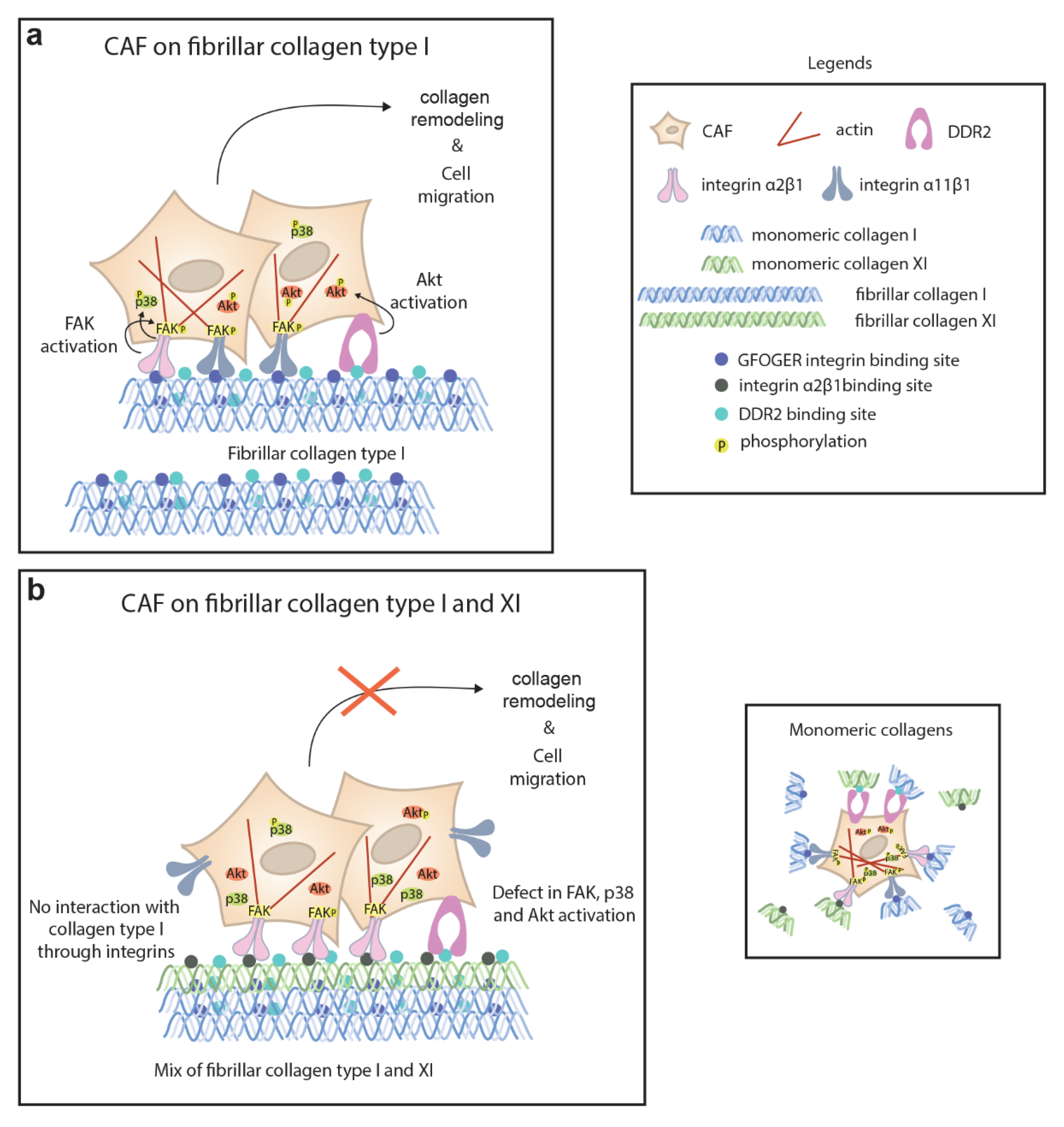
2.2. CAFs Interact with Collagen Type XI through Integrin α2β1 and DDR2
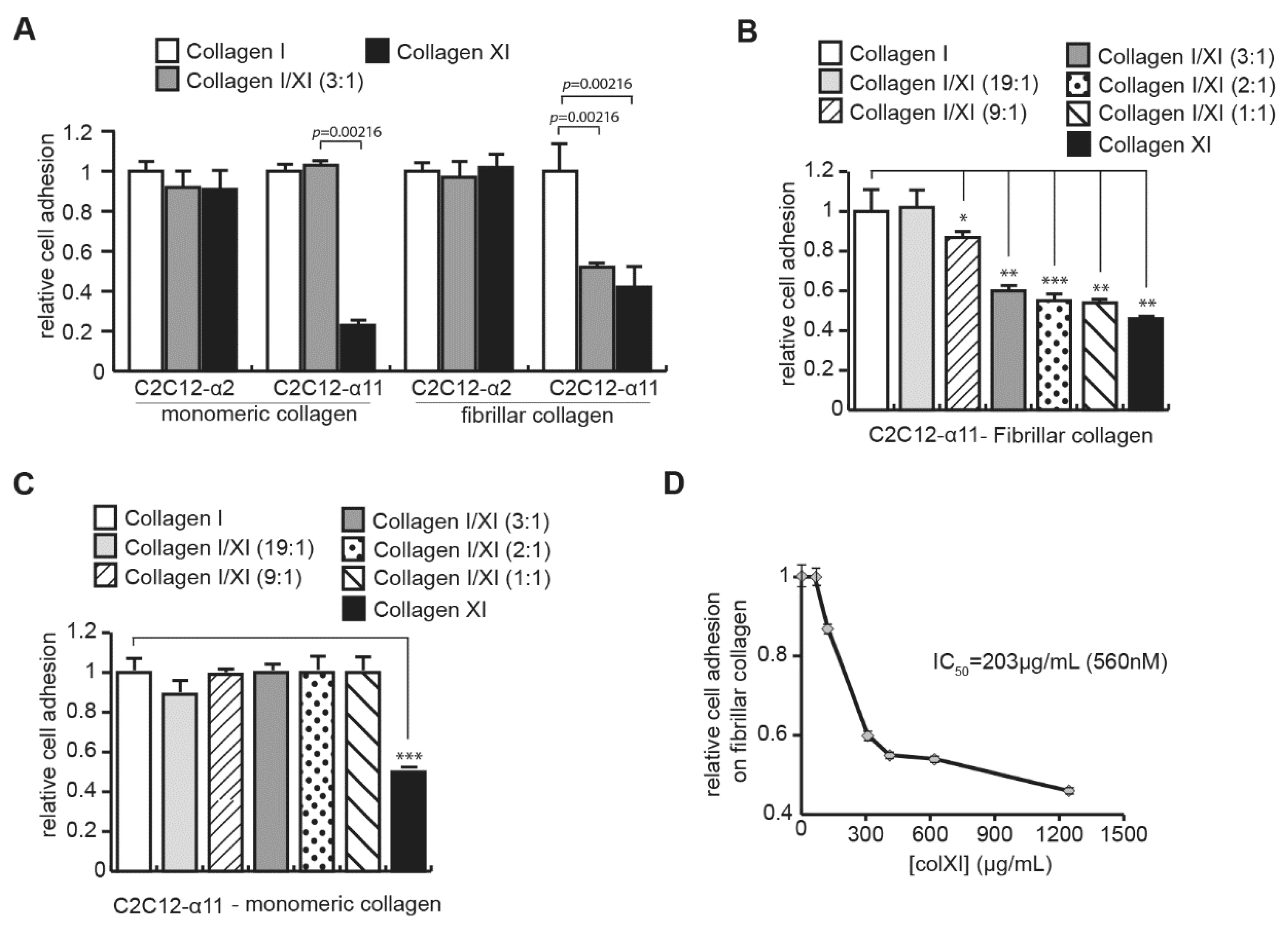
2.3. Collagen Type XI Restrains Collagen Type I Availability for Cells
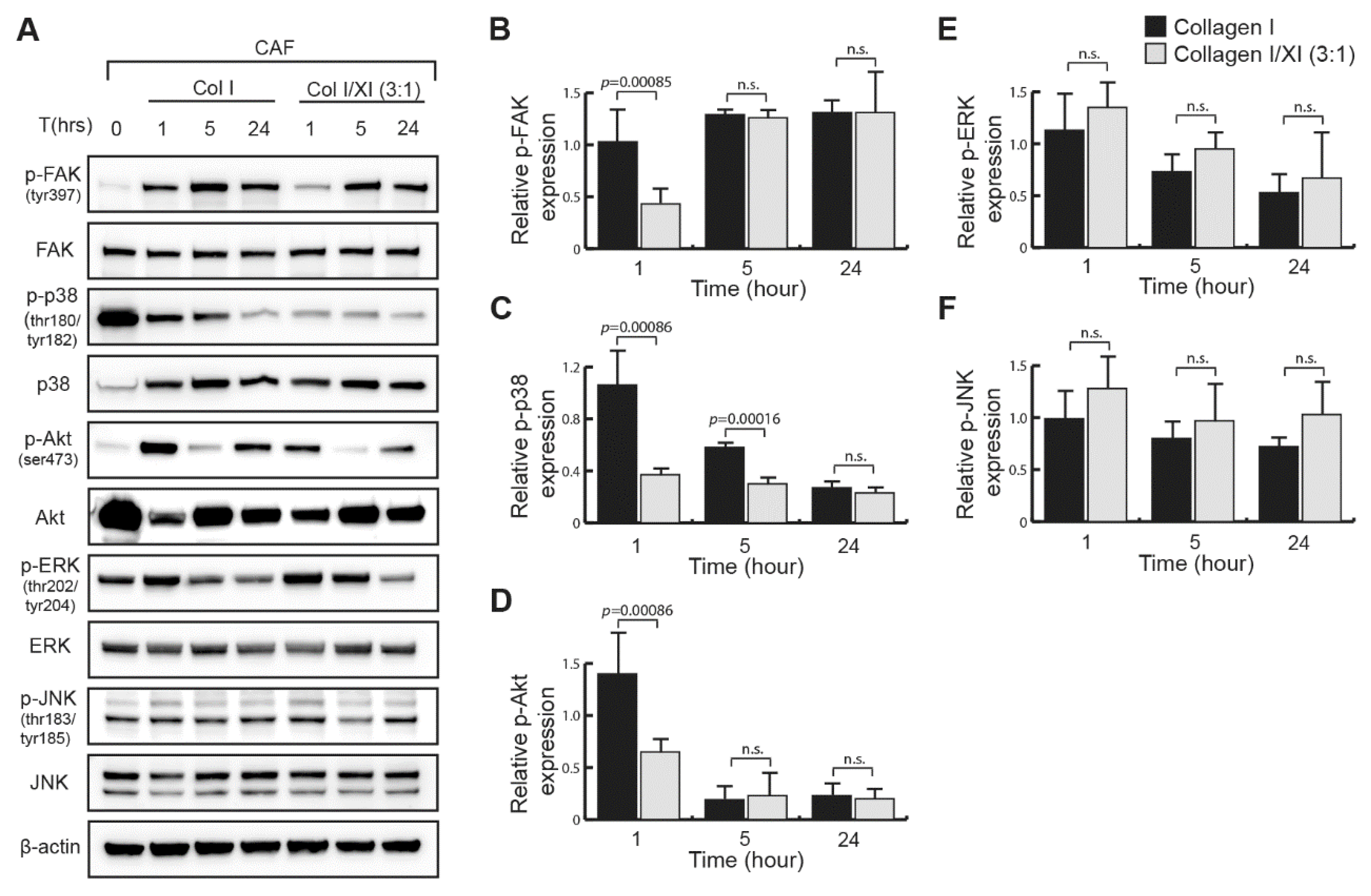
2.4. Defect in FAK, p38 and Akt Activation in Presence of Collagen Type XI
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Adhesion on Collagen Matrices
4.3. Collagen Gel Contraction Assay
4.4. 3D-Matrix Invasion Assay
4.5. Reverse-Transcriptase/Quantitative PCR (RT-qPCR) Expression Profiling
4.6. Solid Phase Assay
4.7. Western Blot Analysis
4.8. Measurement of IC50 Value
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Hao, J.; Zeltz, C.; Pintilie, M.; Li, Q.; Sakashita, S.; Wang, T.; Cabanero, M.; Martins-Filho, S.N.; Wang, D.Y.; Pasko, E.; et al. Characterization of Distinct Populations of Carcinoma-Associated Fibroblasts from Non-Small Cell Lung Carcinoma Reveals a Role for ST8SIA2 in Cancer Cell Invasion. Neoplasia 2019, 21, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; To, C.; Pasko, E.; Kim, K.S.; Park, C.J.; Hai, J.; Liu, J.; Jonkman, J.; Barczyk, M.; et al. Integrin Alpha11beta1 Regulates Cancer Stromal Stiffness and Promotes Tumorigenicity and Metastasis in Non-Small Cell Lung Cancer. Oncogene 2016, 35, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Pasko, E.; Cox, T.R.; Navab, R.; Tsao, M.S. LOXL1 Is Regulated by Integrin Alpha11 and Promotes Non-Small Cell Lung Cancer Tumorigenicity. Cancers 2019, 11, 705. [Google Scholar] [CrossRef]
- Griffith, A.J.; Sprunger, L.K.; Sirko-Osadsa, D.A.; Tiller, G.E.; Meisler, M.H.; Warman, M.L. Marshall Syndrome Associated with a Splicing Defect at the COL11A1 Locus. Am. J. Hum. Genet. 1998, 62, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Vijzelaar, R.; Waller, S.; Errami, A.; Donaldson, A.; Lourenco, T.; Rodrigues, M.; McConnell, V.; Fincham, G.; Snead, M.; Richards, A. Deletions within COL11A1 in Type 2 Stickler Syndrome Detected by Multiplex Ligation-Dependent Probe Amplification (MLPA). BMC Med. Genet. 2013, 14, 48. [Google Scholar] [CrossRef][Green Version]
- Yoshioka, H.; Iyama, K.; Inoguchi, K.; Khaleduzzaman, M.; Ninomiya, Y.; Ramirez, F. Developmental Pattern of Expression of the Mouse Alpha 1 (XI) Collagen Gene (Col11a1). Dev. Dyn. 1995, 204, 41–47. [Google Scholar] [CrossRef]
- Blaschke, U.K.; Eikenberry, E.F.; Hulmes, D.J.; Galla, H.J.; Bruckner, P. Collagen XI Nucleates Self-Assembly and Limits Lateral Growth of Cartilage Fibrils. J. Biol. Chem. 2000, 275, 10370–10378. [Google Scholar] [CrossRef]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Colorectal Carcinogenesis Is Associated with Stromal Expression of COL11A1 and COL5A2. Carcinogenesis 2001, 22, 875–878. [Google Scholar] [CrossRef]
- Toss, M.S.; Miligy, I.M.; Gorringe, K.L.; Aleskandarany, M.A.; Alkawaz, A.; Mittal, K.; Aneja, R.; Ellis, I.O.; Green, A.R.; Rakha, E.A. Collagen (XI) Alpha-1 Chain Is an Independent Prognostic Factor in Breast Ductal Carcinoma in Situ. Mod. Pathol. 2019, 32, 1460–1472. [Google Scholar] [CrossRef]
- Raglow, Z.; Thomas, S.M. Tumor Matrix Protein Collagen XIalpha1 in Cancer. Cancer Lett. 2015, 357, 448–453. [Google Scholar] [CrossRef]
- Liu, Z.; Lai, J.; Jiang, H.; Ma, C.; Huang, H. Collagen XI Alpha 1 Chain, a Potential Therapeutic Target for Cancer. FASEB J. 2021, 35, e21603. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Watkinson, J.; Varadan, V.; Anastassiou, D. Multi-Cancer Computational Analysis Reveals Invasion-Associated Variant of Desmoplastic Reaction Involving INHBA, THBS2 and COL11A1. BMC Med. Genom. 2010, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Liu, Z.; Deng, N.; Tan, T.Z.; Huang, R.Y.; Taylor-Harding, B.; Cheon, D.J.; Lawrenson, K.; Wiedemeyer, W.R.; Walts, A.E.; et al. A COL11A1-Correlated Pan-Cancer Gene Signature of Activated Fibroblasts for the Prioritization of Therapeutic Targets. Cancer Lett. 2016, 382, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Cheon, D.J.; Tong, Y.; Sim, M.S.; Dering, J.; Berel, D.; Cui, X.; Lester, J.; Beach, J.A.; Tighiouart, M.; Walts, A.E.; et al. A Collagen-Remodeling Gene Signature Regulated by TGF-Beta Signaling Is Associated with Metastasis and Poor Survival in Serous Ovarian Cancer. Clin. Cancer Res. 2014, 20, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic Gene-Expression Signature of Carcinoma-Associated Fibroblasts in Non-Small Cell Lung Cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef]
- Wu, Y.H.; Chang, T.H.; Huang, Y.F.; Huang, H.D.; Chou, C.Y. COL11A1 Promotes Tumor Progression and Predicts Poor Clinical Outcome in Ovarian Cancer. Oncogene 2014, 33, 3432–3440. [Google Scholar] [CrossRef]
- Sok, J.C.; Lee, J.A.; Dasari, S.; Joyce, S.; Contrucci, S.C.; Egloff, A.M.; Trevelline, B.K.; Joshi, R.; Kumari, N.; Grandis, J.R.; et al. Collagen Type XI Alpha1 Facilitates Head and Neck Squamous Cell Cancer Growth and Invasion. Br. J. Cancer 2013, 109, 3049–3056. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chen, C.C.; Chou, C.Y. Akt Inhibitor SC66 Promotes Cell Sensitivity to Cisplatin in Chemoresistant Ovarian Cancer Cells through Inhibition of COL11A1 Expression. Cell Death Dis. 2019, 10, 322. [Google Scholar] [CrossRef]
- Wu, Y.H.; Huang, Y.F.; Chang, T.H.; Chou, C.Y. Activation of TWIST1 by COL11A1 Promotes Chemoresistance and Inhibits Apoptosis in Ovarian Cancer Cells by Modulating NF-KappaB-Mediated IKKbeta Expression. Int. J. Cancer 2017, 141, 2305–2317. [Google Scholar] [CrossRef]
- Wang, K.K.; Liu, N.; Radulovich, N.; Wigle, D.A.; Johnston, M.R.; Shepherd, F.A.; Minden, M.D.; Tsao, M.S. Novel Candidate Tumor Marker Genes for Lung Adenocarcinoma. Oncogene 2002, 21, 7598–7604. [Google Scholar] [CrossRef][Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-Led Collective Invasion of Carcinoma Cells with Differing Roles for RhoGTPases in Leading and Following Cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Tiger, C.F.; Fougerousse, F.; Grundstrom, G.; Velling, T.; Gullberg, D. Alpha11beta1 Integrin Is a Receptor for Interstitial Collagens Involved in Cell Migration and Collagen Reorganization on Mesenchymal Nonmuscle Cells. Dev. Biol. 2001, 237, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Yamato, M.; Aoyagi, M.; Yamamoto, K. Identification of Integrins Involved in Cell Adhesion to Native and Denatured Type I Collagens and the Phenotypic Transition of Rabbit Arterial Smooth Muscle Cells. Exp. Cell Res. 1995, 219, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Tulla, M.; Pentikainen, O.T.; Viitasalo, T.; Kapyla, J.; Impola, U.; Nykvist, P.; Nissinen, L.; Johnson, M.S.; Heino, J. Selective Binding of Collagen Subtypes by Integrin Alpha 1I, Alpha 2I, and Alpha 10I Domains. J. Biol. Chem. 2001, 276, 48206–48212. [Google Scholar] [CrossRef]
- Erusappan, P.; Alam, J.; Lu, N.; Zeltz, C.; Gullberg, D. Integrin Alpha11 Cytoplasmic Tail Is Required for FAK Activation to Initiate 3D Cell Invasion and ERK-Mediated Cell Proliferation. Sci. Rep. 2019, 9, 15283. [Google Scholar] [CrossRef]
- Zhang, K.; Corsa, C.A.; Ponik, S.M.; Prior, J.L.; Piwnica-Worms, D.; Eliceiri, K.W.; Keely, P.J.; Longmore, G.D. The Collagen Receptor Discoidin Domain Receptor 2 Stabilizes SNAIL1 to Facilitate Breast Cancer Metastasis. Nat. Cell Biol. 2013, 15, 677–687. [Google Scholar] [CrossRef]
- Rada, M.; Nallanthighal, S.; Cha, J.; Ryan, K.; Sage, J.; Eldred, C.; Ullo, M.; Orsulic, S.; Cheon, D.J. Inhibitor of Apoptosis Proteins (IAPs) Mediate Collagen Type XI Alpha 1-Driven Cisplatin Resistance in Ovarian Cancer. Oncogene 2018, 37, 4809–4820. [Google Scholar] [CrossRef]
- Ivaska, J.; Reunanen, H.; Westermarck, J.; Koivisto, L.; Kähäri, V.M.; Heino, J. Integrin Alpha2beta1 Mediates Isoform-Specific Activation of P38 and Upregulation of Collagen Gene Transcription by a Mechanism Involving the Alpha2 Cytoplasmic Tail. J. Cell Biol. 1999, 147, 401–416. [Google Scholar] [CrossRef]
- Schulz, J.-N.; Zeltz, C.; Sørensen, I.W.; Barczyk, M.; Carracedo, S.; Hallinger, R.; Niehoff, A.; Eckes, B.; Gullberg, D. Reduced Granulation Tissue and Wound Strength in the Absence of A11β1 Integrin. J. Investig. Dermatol. 2015, 135, 1435–1444. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.X.; Müller, S.; Keerthivasan, S.; Koeppen, H.; Hung, J.; Gierke, S.; Breart, B.; Foreman, O.; Bainbridge, T.W.; Castiglioni, A.; et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15 + Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020, 10, 232–253. [Google Scholar] [CrossRef]
- Zeltz, C.; Alam, J.; Liu, H.; Erusappan, P.M.; Hoschuetzky, H.; Molven, A.; Parajuli, H.; Cukierman, E.; Costea, D.E.; Lu, N.; et al. Alpha11beta1 Integrin Is Induced in a Subset of Cancer-Associated Fibroblasts in Desmoplastic Tumor Stroma and Mediates In Vitro Cell Migration. Cancers 2019, 11, 765. [Google Scholar] [CrossRef]
- Girotti, M.R.; Fernandez, M.; Lopez, J.A.; Camafeita, E.; Fernandez, E.A.; Albar, J.P.; Benedetti, L.G.; Valacco, M.P.; Brekken, R.A.; Podhajcer, O.L.; et al. SPARC Promotes Cathepsin B-Mediated Melanoma Invasiveness through a Collagen I/Alpha2beta1 Integrin Axis. J. Investig. Dermatol. 2011, 131, 2438–2447. [Google Scholar] [CrossRef]
- Hall, C.L.; Dubyk, C.W.; Riesenberger, T.A.; Shein, D.; Keller, E.T.; van Golen, K.L. Type I Collagen Receptor (Alpha2beta1) Signaling Promotes Prostate Cancer Invasion through RhoC GTPase. Neoplasia 2008, 10, 797–803. [Google Scholar] [CrossRef]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z.; Shenoy, V.B.; McCulloch, C.A.; Hinz, B. Dynamic Fibroblast Contractions Attract Remote Macrophages in Fibrillar Collagen Matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef]
- Jokinen, J.; Dadu, E.; Nykvist, P.; Kapyla, J.; White, D.J.; Ivaska, J.; Vehvilainen, P.; Reunanen, H.; Larjava, H.; Hakkinen, L.; et al. Integrin-Mediated Cell Adhesion to Type I Collagen Fibrils. J. Biol. Chem. 2004, 279, 31956–31963. [Google Scholar] [CrossRef]
- Mercier, I.; Lechaire, J.P.; Desmouliere, A.; Gaill, F.; Aumailley, M. Interactions of Human Skin Fibroblasts with Monomeric or Fibrillar Collagens Induce Different Organization of the Cytoskeleton. Exp. Cell Res. 1996, 225, 245–256. [Google Scholar] [CrossRef]
- Woltersdorf, C.; Bonk, M.; Leitinger, B.; Huhtala, M.; Kapyla, J.; Heino, J.; Gil Girol, C.; Niland, S.; Eble, J.A.; Bruckner, P.; et al. The Binding Capacity of Alpha1beta1-, Alpha2beta1- and Alpha10beta1-Integrins Depends on Non-Collagenous Surface Macromolecules Rather than the Collagens in Cartilage Fibrils. Matrix Biol. 2017, 63, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Orgel, J.; Gullberg, D. Molecular Composition and Function of Integrin-Based Collagen Glues-Introducing COLINBRIs. Biochim. Biophys. Acta 2014, 1840, 2533–2548. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, M.M.; Olsen, L.H.; da Franca, P.; Loos, B.G.; Mustafa, K.; Gullberg, D.; Bolstad, A.I. A Role for Alpha11beta1 Integrin in the Human Periodontal Ligament. J. Dent. Res. 2009, 88, 621–626. [Google Scholar] [CrossRef]
- Wu, X.; Cai, J.; Zuo, Z.; Li, J. Collagen Facilitates the Colorectal Cancer Stemness and Metastasis through an Integrin/PI3K/AKT/Snail Signaling Pathway. Biomed. Pharmacother. 2019, 114, 108708. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. Pp125FAK a Structurally Distinctive Protein-Tyrosine Kinase Associated with Focal Adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef]
- Higuchi, M.; Kihara, R.; Okazaki, T.; Aoki, I.; Suetsugu, S.; Gotoh, Y. Akt1 Promotes Focal Adhesion Disassembly and Cell Motility through Phosphorylation of FAK in Growth Factor-Stimulated Cells. J. Cell Sci. 2013, 126, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Herrera, M.L.; Quezada-Calvillo, R. DDR2 Plays a Role in Fibroblast Migration Independent of Adhesion Ligand and Collagen Activated DDR2 Tyrosine Kinase. Biochem. Biophys. Res. Commun. 2012, 429, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.A.; Jarai, G. Discoidin Domain Receptors Regulate the Migration of Primary Human Lung Fibroblasts through Collagen Matrices. Fibrogenesis Tissue Repair 2012, 5, 3. [Google Scholar] [CrossRef]
- Jia, S.; Agarwal, M.; Yang, J.; Horowitz, J.C.; White, E.S.; Kim, K.K. Discoidin Domain Receptor 2 Signaling Regulates Fibroblast Apoptosis through PDK1/Akt. Am. J. Respir. Cell Mol. Biol. 2018, 59, 295–305. [Google Scholar] [CrossRef]
- Harikrishnan, V.; Titus, A.S.; Cowling, R.T.; Kailasam, S. Collagen Receptor Cross-Talk Determines Alpha-Smooth Muscle Actin-Dependent Collagen Gene Expression in Angiotensin II-Stimulated Cardiac Fibroblasts. J. Biol. Chem. 2019, 294, 19723–19739. [Google Scholar] [CrossRef]
- Bayer, S.V.; Grither, W.R.; Brenot, A.; Hwang, P.Y.; Barcus, C.E.; Ernst, M.; Pence, P.; Walter, C.; Pathak, A.; Longmore, G.D. DDR2 Controls Breast Tumor Stiffness and Metastasis by Regulating Integrin Mediated Mechanotransduction in CAFs. eLife 2019, 8, e45508. [Google Scholar] [CrossRef] [PubMed]
- Nykvist, P.; Tu, H.; Ivaska, J.; Kapyla, J.; Pihlajaniemi, T.; Heino, J. Distinct Recognition of Collagen Subtypes by Alpha(1)Beta(1) and Alpha(2)Beta(1) Integrins. Alpha(1)Beta(1) Mediates Cell Adhesion to Type XIII Collagen. J. Biol. Chem. 2000, 275, 8255–8261. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Smoking History | Sex | Clinical Stage | Histology | |
|---|---|---|---|---|
| CAF 094 | Unknown | F | 3A | ADC |
| CAF 448 | Ex-Smoker | M | 1B | ADC |
| CAF 453 | Smoker | M | 1A | ADC |
| CAF 455 | Never | F | 2A | ADC |
| CAF 458 | Never | M | 3 | SqCC |
| CAF 462 | Smoker | F | 1B | SqCC |
| CAF 466 | Ex-Smoker | M | 1B | ADC |
| CAF 472 | Never | F | 1A | ADC |
| CAF 474 | Unknown | F | 1B | ADC |
| CAF 476 | Ex-Smoker | F | 3A | ADC |
| CAF 480 | Ex-Smoker | M | 1B | ADC |
| CAF 481 | Never | F | 1B | ADC |
| CAF 482 | Ex-Smoker | M | 2B | SqCC |
| CAF 487 | Ex-Smoker | M | 3A | SqCC |
| (Collagen Type I) mg/mL | (Collagen Type XI) mg/mL | (Collagen Type I) µM | (Collagen Type XI) µM | |
|---|---|---|---|---|
| Coll I | 1.24 | 0 | 4.1 | 0 |
| Coll I/XI (19:1) | 1.178 | 0.062 | 3.93 | 0.17 |
| Coll I/XI (9:1) | 1.116 | 0.124 | 3.72 | 0.34 |
| Coll I/XI (3:1) | 0.93 | 0.31 | 3.1 | 0.85 |
| Coll I/XI (2:1) | 0.83 | 0.41 | 2.75 | 1.14 |
| Coll I/XI (1:1) | 0.62 | 0.62 | 2.07 | 1.7 |
| Coll XI | 0 | 1.24 | 0 | 3.4 |
| Gene | Primer Sequence |
|---|---|
| ITGA1 | Forward 5’-CACTGTTGTTCTACGCTGCT-3’ Reverse 5’-ACGGAGAACCAATAAGCAC-3’ |
| ITGA2 | Forward 5’-CTAGAAGCCCATCCTGTGCC-3’ Reverse 5’-GGTTACTGCCAGTCAGCCAA-3’ |
| ITGA10 | Forward 5’-CATCACCCACGCCTATTCCC-3’ Reverse 5’-TGCCTCCCCTTTGTTAGCAC-3’ |
| ITGA11 | Forward 5’-GCCTCCAGTATTTTGGCTGC-3’ Reverse 5’-GCTCAAAGTGGAGGCTGGC-3’ |
| DDR1 | Forward 5’-GATCTCGACTCCGCTTCAAG-3’ Reverse 5’-CAAAGGGTGTCCCTTACGC-3’ |
| DDR2 | Forward 5’-AACGAGAGTGCCACCAAT-3’ Reverse 5’-ACTCACTGGCTTCAGAGCG-3’ |
| RPS13 | Forward 5’-GTTGCTGTTCGAAAGCATCTTG-3’ Reverse 5’-AATATCGAGCCAAACGGTGAA-3’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeltz, C.; Khalil, M.; Navab, R.; Tsao, M.-S. Collagen Type XI Inhibits Lung Cancer-Associated Fibroblast Functions and Restrains the Integrin Binding Site Availability on Collagen Type I Matrix. Int. J. Mol. Sci. 2022, 23, 11722. https://doi.org/10.3390/ijms231911722
Zeltz C, Khalil M, Navab R, Tsao M-S. Collagen Type XI Inhibits Lung Cancer-Associated Fibroblast Functions and Restrains the Integrin Binding Site Availability on Collagen Type I Matrix. International Journal of Molecular Sciences. 2022; 23(19):11722. https://doi.org/10.3390/ijms231911722
Chicago/Turabian StyleZeltz, Cédric, Maryam Khalil, Roya Navab, and Ming-Sound Tsao. 2022. "Collagen Type XI Inhibits Lung Cancer-Associated Fibroblast Functions and Restrains the Integrin Binding Site Availability on Collagen Type I Matrix" International Journal of Molecular Sciences 23, no. 19: 11722. https://doi.org/10.3390/ijms231911722
APA StyleZeltz, C., Khalil, M., Navab, R., & Tsao, M.-S. (2022). Collagen Type XI Inhibits Lung Cancer-Associated Fibroblast Functions and Restrains the Integrin Binding Site Availability on Collagen Type I Matrix. International Journal of Molecular Sciences, 23(19), 11722. https://doi.org/10.3390/ijms231911722