

The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis


{kind=link}
{kind=link}
Abstract
1. Introduction
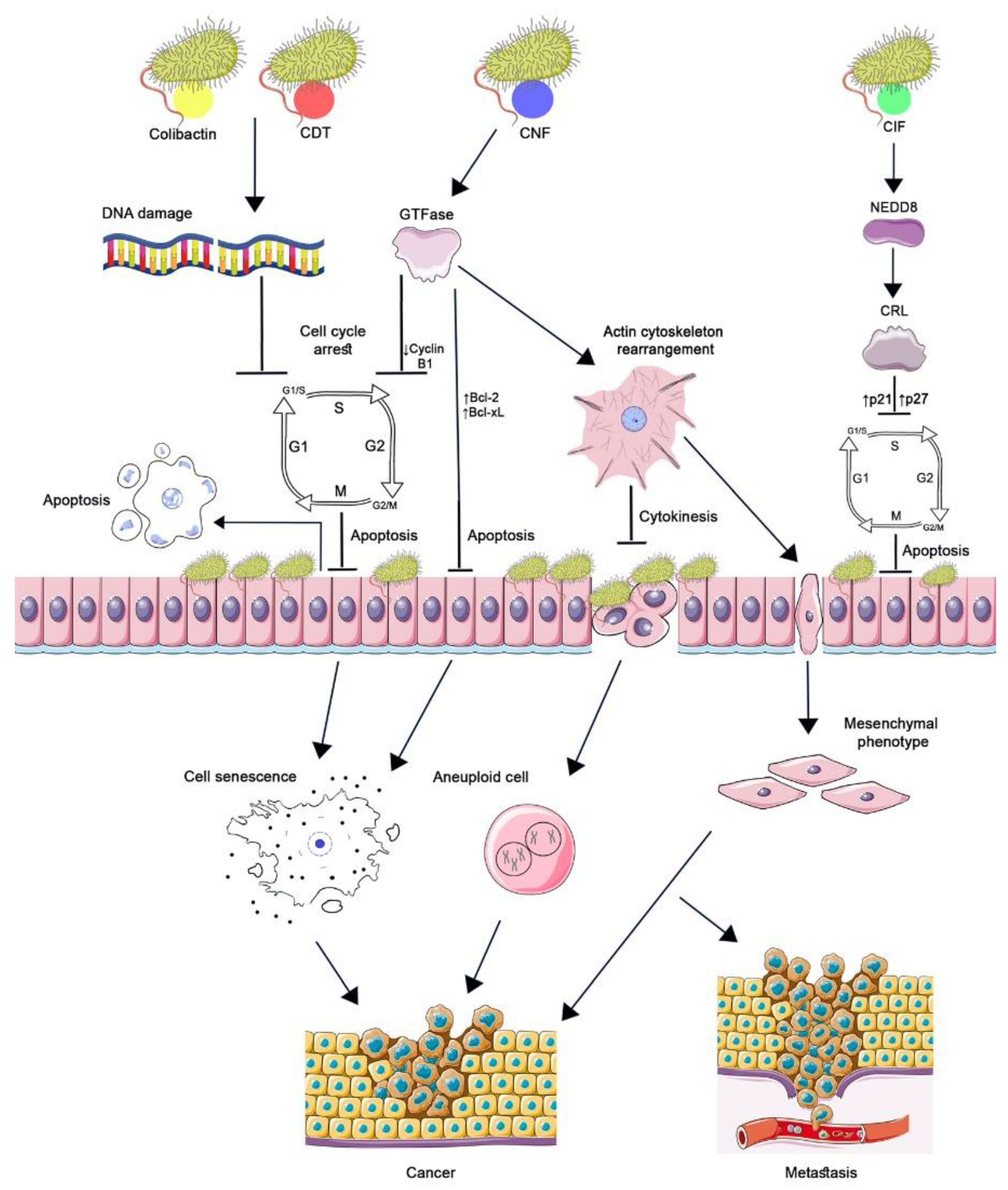
2. Cyclomodulins—CNFs
3. Cyclomodulins—CIFs
4. Cyclomodulins—Genotoxins
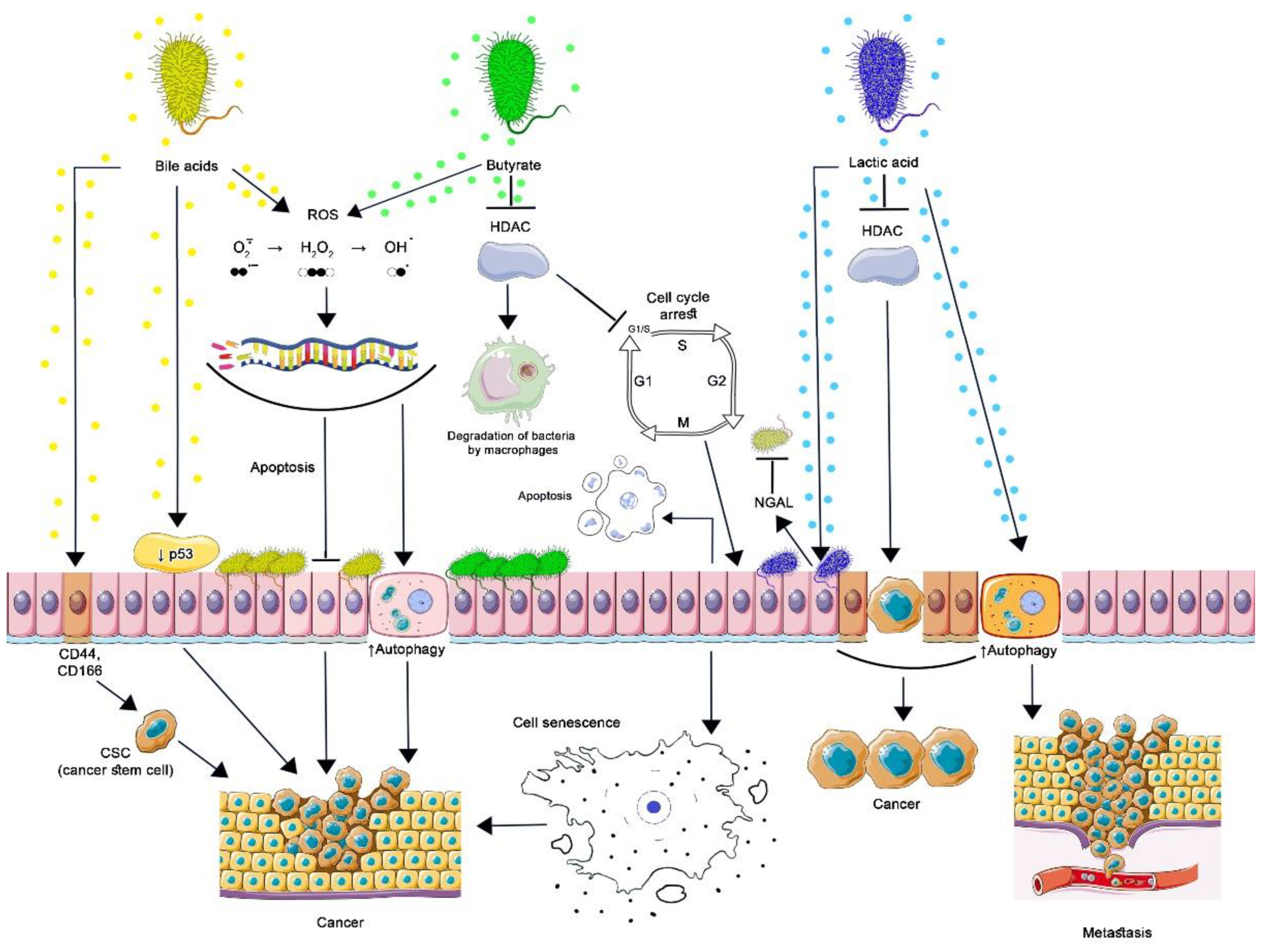
5. Specific Microbial Metabolites
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Elsland, D.; Neefjes, J. Bacterial infections and cancer. EMBO Rep. 2018, 19, e46632. [Google Scholar] [CrossRef]
- Silva-García, O.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M. Wnt/β-catenin signaling as a molecular target by pathogenic bacteria. Front. Immunol. 2019, 10, 2135. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; DePaolo, R.W. “Driver-passenger” bacteria and their metabolites in the pathogenesis of colorectal cancer. Gut Microbes 2021, 13, 1941710. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef]
- El-Aouar Filho, R.A.; Nicolas, A.; De Paula Castro, T.L.; Deplanche, M.; De Carvalho Azevedo, V.A.; Goossens, P.L.; Taieb, F.; Lina, G.; Le Loir, Y.; Berkova, N. Heterogeneous Family of Cyclomodulins: Smart Weapons That Allow Bacteria to Hijack the Eukaryotic Cell Cycle and Promote Infections. Front. Cell Infect. Microbiol. 2017, 23, 208. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef]
- Rosadi, F.; Fiorentini, C.; Fabbri, A. Bacterial protein toxins in human cancers. Pathog. Dis. 2016, 74, ftv105. [Google Scholar] [CrossRef]
- Mezerová, K.; Starý, L.; Zbořil, P.; Klementa, I.; Stašek, M.; Špička, P.; Skalický, P.; Raclavský, V. Cyclomodulins and Hemolysis in E. coli as Potential Low-Cost Non-Invasive Biomarkers for Colorectal Cancer Screening. Life 2021, 11, 1165. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Nougayrède, J.P. The colibactin genotoxin generates DNA interstrand cross-links in infected cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef]
- Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The biology of the cytolethal distending toxins. Toxins 2011, 3, 172–190. [Google Scholar] [CrossRef]
- Zhang, Z.; Aung, K.M.; Uhlin, B.E.; Wai, S.N. Reversible senescence of human colon cancer cells after blockage of mitosis/cytokinesis caused by the CNF1 cyclomodulin from Escherichia coli. Sci. Rep. 2018, 8, 17780. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrède, J.P.; Watrin, C.; Jubelin, G.; Oswald, E.; Taieb, F. Bacterial cyclomodulin Cif blocks the host cell cycle by stabilizing the cyclin-dependent kinase inhibitors p21 and p27. Cell Microbiol. 2008, 10, 2496–2508. [Google Scholar] [CrossRef]
- Barrett, M.; Hand, C.K.; Shanahan, F.; Murphy, T.; O’Toole, P.W. Mutagenesis by microbe: The role of the microbiota in shaping the cancer genome. Trends Cancer 2020, 6, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Duijster, J.W.; Franz, E.; Neefjes, J.; Mughini-Gras, L. Bacterial and Parasitic Pathogens as Risk Factors for Cancers in the Gastrointestinal Tract: A Review of Current Epidemiological Knowledge. Front. Microbiol. 2021, 8, 790256. [Google Scholar] [CrossRef]
- de Savornin Lohman, E.; Duijster, J.; Groot Koerkamp, B.; van der Post, R.; Franz, E.; Mughini Gras, L.; de Reuver, P. Severe Salmonella spp. or Campylobacter spp. Infection and the Risk of Biliary Tract Cancer: A Population-Based Study. Cancers 2020, 12, 3348. [Google Scholar] [CrossRef]
- Iwasaki, M.; Kanehara, R.; Yamaji, T.; Katagiri, R.; Mutoh, M.; Tsunematsu, Y.; Sato, M.; Watanabe, K.; Hosomi, K.; Kakugawa, Y.; et al. Association of Escherichia coli containing polyketide synthase in the gut microbiota with colorectal neoplasia in Japan. Cancer Sci. 2022, 113, 277–286. [Google Scholar] [CrossRef]
- Grasso, F.; Frisan, T. Bacterial genotoxins: Merging the DNA damage response into infection biology. Biomolecules 2015, 5, 1762–1782. [Google Scholar] [CrossRef]
- Chaoprasid, P.; Lukat, P.; Mühlen, S.; Heidler, T.; Gazdag, E.M.; Dong, S.; Blankenfeldt, W. Crystal structure of bacterial cytotoxic necrotizing factor CNFY reveals molecular building blocks for intoxication. EMBO J. 2021, 40, e105202. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Mettouchi, A.; Wilson, B.A.; Lemichez, E. CNF1-like deamidase domains: Common Lego bricks among cancer-promoting immunomodulatory bacterial virulence factors. Pathog. Dis. 2018, 76, fty045. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Ballan, G.; Loizzo, S.; Fiorentini, C. The cytotoxic necrotizing factor 1 from E. coli: A janus toxin playing with cancer regulators. Toxins 2013, 5, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K. Bacterial protein toxins that modify host regulatory GTPases. Nat. Rev. Microbiol. 2011, 9, 487–498. [Google Scholar] [CrossRef]
- Garcia, T.A.; Ventura, C.L.; Smith, M.A.; Merrell, D.S.; O’Brien, A.D. Cytotoxic necrotizing factor 1 and hemolysin from uropathogenic Escherichia coli elicit different host responses in the murine bladder. Infect. Immun. 2013, 81, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Carlini, F.; Maroccia, Z.; Fiorentini, C.; Travaglione, S.; Fabbri, A. Effects of the Escherichia coli Bacterial Toxin Cytotoxic Necrotizing Factor 1 on Different Human and Animal Cells: A Systematic Review. Int. J. Mol. Sci. 2021, 22, 12610. [Google Scholar] [CrossRef] [PubMed]
- Falzano, L.; Filippini, P.; Travaglione, S.; Miraglia, A.G.; Fabbri, A.; Fiorentini, C. Escherichia coli cytotoxic necrotizing factor 1 blocks cell cycle G2/M transition in uroepithelial cells. Infect. Immun. 2006, 74, 3765–3772. [Google Scholar] [CrossRef]
- Fabbri, A.; Travaglione, S.; Rosadi, F.; Ballan, G.; Maroccia, Z.; Giambenedetti, M.; Fiorentini, C. The Escherichia coli protein toxin cytotoxic necrotizing factor 1 induces epithelial mesenchymal transition. Cell. Microbiol. 2020, 22, e13138. [Google Scholar] [CrossRef]
- De Rycke, J.E.A.N.; Mazars, P.; Nougayrede, J.P.; Tasca, C.; Boury, M.; Herault, F.; Oswald, E. Mitotic block and delayed lethality in HeLa epithelial cells exposed to Escherichia coli BM2-1 producing cytotoxic necrotizing factor type 1. Infect. Immun. 1996, 64, 1694–1705. [Google Scholar] [CrossRef]
- Tantillo, E.; Colistra, A.; Vannini, E.; Cerri, C.; Pancrazi, L.; Baroncelli, L.; Caleo, M. Bacterial toxins and targeted brain therapy: New insights from cytotoxic necrotizing factor 1 (CNF1). Int. J. Mol. Sci. 2018, 19, 1632. [Google Scholar] [CrossRef]
- Vannini, E.; Mori, E.; Tantillo, E.; Schmidt, G.; Caleo, M.; Costa, M. CTX-CNF1 Recombinant Protein Selectively Targets Glioma Cells In Vivo. Toxins 2021, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Jubelin, G.; Chavez, C.V.; Taieb, F.; Banfield, M.J.; Samba-Louaka, A.; Nobe, R.; Nougayrède, J.P.; Zumbihl, R.; Givaudan, A.; Escoubas, J.M.; et al. Cycle inhibiting factors (CIFs) are a growing family of functional cyclomodulins present in invertebrate and mammal bacterial pathogens. PLoS ONE 2009, 4, e4855. [Google Scholar] [CrossRef] [PubMed]
- Samba-Louaka, A.; Nougayrède, J.P.; Watrin, C.; Oswald, E.; Taieb, F. The enteropathogenic Escherichia coli effector Cif induces delayed apoptosis in epithelial cells. Infect. Immun. 2009, 77, 5471–5477. [Google Scholar] [CrossRef] [PubMed]
- Loukiadis, E.; Nobe, R.; Herold, S.; Tramuta, C.; Ogura, Y.; Ooka, T.; Morabito, S.; Kérourédan, M.; Brugère, H.; Schmidt, H.; et al. Distribution, functional expression, and genetic organization of Cif, a phage-encoded type III-secreted effector from enteropathogenic and enterohemorrhagic Escherichia coli. J. Bacteriol. 2008, 190, 275–285. [Google Scholar] [CrossRef]
- Taieb, F.; Nougayrède, J.P.; Oswald, E. Cycle inhibiting factors (cifs): Cyclomodulins that usurp the ubiquitin-dependent degradation pathway of host cells. Toxins 2011, 3, 356–368. [Google Scholar] [CrossRef]
- Cui, J.; Yao, Q.; Li, S.; Ding, X.; Lu, Q.; Mao, H.; Liu, L.; Zheng, N.; Chen, S.; Shao, F. Glutamine deamidation and dysfunction of Ubiquitin/NEDD8 by a bacterial effector family. Science 2010, 329, 1215–1218. [Google Scholar] [CrossRef]
- Piciocchi, A.; Germinario, E.A.P.; Garcia Etxebarria, K.; Rossi, S.; Sanchez-Mete, L.; Porowska, B.; Stigliano, V.; Trentino, P.; Oddi, A.; Accarpio, F.; et al. Association of Polygenic Risk Score and Bacterial Toxins at Screening Colonoscopy with Colorectal Cancer Progression: A Multicenter Case-Control Study. Toxins 2021, 13, 569. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12, 63. [Google Scholar] [CrossRef]
- Pokkunuri, V.; Pimentel, M.; Morales, W.; Jee, S.R.; Alpern, J.; Weitsman, S.; Chang, C. Role of cytolethal distending toxin in altered stool form and bowel phenotypes in a rat model of post-infectious irritable bowel syndrome. Neurogastroenterol. Motil. 2012, 18, 434. [Google Scholar] [CrossRef]
- De Rycke, J.; Oswald, E. Cytolethal distending toxin (CDT): A bacterial weapon to control host cell proliferation? FEMS Microbiol. Lett. 2001, 203, 141–148. [Google Scholar] [CrossRef]
- Lai, Y.R.; Chang, Y.F.; Ma, J.; Chiu, C.H.; Kuo, M.L.; Lai, C.H. From DNA Damage to Cancer Progression: Potential Effects of Cytolethal Distending Toxin. Front. Immunol. 2021, 12, 760451. [Google Scholar] [CrossRef] [PubMed]
- Wami, H.; Wallenstein, A.; Sauer, D.; Stoll, M.; von Bünau, R.; Oswald, E.; Dobrindt, U. Insights into evolution and coexistence of the colibactin-and yersiniabactin secondary metabolite determinants in enterobacterial populations. Microb. Genom. 2021, 7, 000577. [Google Scholar] [CrossRef] [PubMed]
- Nougayrède, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J. Clin. Microbiol. 2008, 46, 3906–3911. [Google Scholar] [CrossRef] [PubMed]
- Strakova, N.; Korena, K.; Karpiskova, R. Klebsiella pneumoniae producing bacterial toxin colibactin as a risk of colorectal cancer development-systematic review. Toxicon 2021, 197, 126–135. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Martin, P.; Cloup, E.; Stabler, R.A.; Oswald, E.; Taylor, P.W. The genotoxin colibactin is a determinant of virulence in Escherichia coli K1 experimental neonatal systemic infection. Infect. Immun. 2015, 83, 3704–3711. [Google Scholar] [CrossRef]
- Lu, M.C.; Chen, Y.T.; Chiang, M.K.; Wang, Y.C.; Hsiao, P.Y.; Huang, Y.J.; Lai, Y.C. Colibactin contributes to the hypervirulence of pks+ K1 CC23 Klebsiella pneumoniae in mouse meningitis infections. Front. Cell. Infect. Microbiol. 2017, 7, 103. [Google Scholar] [CrossRef]
- Le Gall, T.; Clermont, O.; Gouriou, S.; Picard, B.; Nassif, X.; Denamur, E.; Tenaillon, O. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol. Biol. Evol. 2007, 24, 2373–2384. [Google Scholar] [CrossRef]
- Nougayrède, J.P.; Chagneau, C.V.; Motta, J.P.; Bossuet-Greif, N.; Belloy, M.; Taieb, F.; Gratadoux, J.J.; Thomas, M.; Langella, P.; Oswald, E.A. Toxic Friend: Genotoxic and Mutagenic Activity of the Probiotic Strain Escherichia coli Nissle 1917. mSphere 2021, 6, e0062421. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More than a new bacterial toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef]
- Tronnet, S.; Floch, P.; Lucarelli, L.; Gaillard, D.; Martin, P.; Serino, M.; Oswald, E. The genotoxin colibactin shapes gut microbiota in mice. mSphere 2020, 5, e00589-20. [Google Scholar] [CrossRef] [PubMed]
- Faïs, T.; Cougnoux, A.; Dalmasso, G.; Laurent, F.; Delmas, J.; Bonnet, R. Antibiotic activity of Escherichia coli against multiresistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 6986–6988. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Balskus, E.P. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Kurnick, S.A.; Mannion, A.J.; Feng, Y.; Madden, C.M.; Chamberlain, P.; Fox, J.G. Genotoxic Escherichia coli strains encoding Colibactin, Cytolethal Distending Toxin, and Cytotoxic necrotizing factor in laboratory rats. Comp. Med. 2019, 69, 103–113. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef]
- Reuter, C.; Alzheimer, M.; Walles, H.; Oelschlaeger, T.A. An adherent mucus layer attenuates the genotoxic effect of colibactin. Cell. Microbiol. 2018, 20, e12812. [Google Scholar] [CrossRef] [PubMed]
- Melhem, H.; Regan-Komito, D.; Niess, J.H. Mucins Dynamics in Physiological and Pathological Conditions. Int. J. Mol. Sci. 2021, 22, 13642. [Google Scholar] [CrossRef] [PubMed]
- Josenhans, C.; Müthing, J.; Elling, L.; Bartfeld, S.; Schmidt, H. How bacterial pathogens of the gastrointestinal tract use the mucosal glyco-code to harness mucus and microbiota: New ways to study an ancient bag of tricks. Int. J. Med. Microbiol. 2020, 310, 151392. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Gagnière, V.; Bonnin, A.S.; Jarrousse, E.; Cardamone, A.; Agus, N.; Uhrhammer, P.; Sauvanet, P.; Déchelotte, N.; Barnich, R.; Bonnet, D.; et al. Bonnet Interactions between microsatellite instability and human gut colonization by Escherichia coli in colorectal cancer. Clin. Sci. 2017, 131, 471–485. [Google Scholar] [CrossRef]
- Taieb, F.; Petit, C.; Nougayrede, J.P.; Oswald, E. The Enterobacterial Genotoxins: Cytolethal Distending Toxin and Colibactin. EcoSal Plus 2016, 7(1), 21. [Google Scholar] [CrossRef] [PubMed]
- Iyadorai, T.; Mariappan, V.; Vellasamy, K.M.; Wanyiri, J.W.; Roslani, A.C.; Lee, G.K.; Vadivelu, J. Prevalence and association of pks+ Escherichia coli with colorectal cancer in patients at the University Malaya Medical Centre, Malaysia. PLoS ONE 2020, 15, e0228217. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Darfeuille-Michaud, A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, S.H.; Hanum, N.; Grotenhuis, A.J.; Castano-Vinyals, G.; Van Der Heijden, A.G.; Aben, K.K.; Kiemeney, L.A. Recurrent urinary tract infection and risk of bladder cancer in the Nijmegen bladder cancer study. Br. J. Cancer 2015, 112, 594–600. [Google Scholar] [CrossRef]
- Faïs, T.; Delmas, J.; Serres, A.; Bonnet, R.; Dalmasso, G. Impact of CDT Toxin on Human Diseases. Toxins 2016, 8, 220. [Google Scholar] [CrossRef]
- Azzi-Martin, L.; He, W.; Péré-Védrenne, C.; Korolik, V.; Alix, C.; Prochazkova-Carlotti, M.; Morel, J.L.; Le Roux-Goglin, E.; Lehours, P.; Djavaheri-Mergny, M.; et al. Cytolethal distending toxin induces the formation of transient messenger-rich ribonucleoprotein nuclear invaginations in surviving cells. PLoS Pathog. 2019, 15, e1007921. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. Response of Chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23. [Google Scholar] [CrossRef]
- Javadi, M.; Bouzari, S.; Oloomi, M. Horizontal gene transfer and the diversity of Escherichia coli. In Escherichia coli—Recent Advances on Physiology, Pathogenesis and Biotechnological Applications; Amidou, S., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef]
- Scott, D.A.; Kaper, J.B. Cloning and sequencing of the genes encoding Escherichia coli cytolethal distending toxin. Infect. Immun. 1994, 62, 244–251. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Karlsson, C.; Lagergård, T.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar] [CrossRef]
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 157 Pt 7, 1851–1875. [Google Scholar] [CrossRef]
- Mathiasen, S.L.; Gall-Mas, L.; Pateras, I.S.; Theodorou, S.D.P.; Namini, M.R.J.; Hansen, M.B.; Martin, O.C.B.; Vadivel, C.K.; Ntostoglou, K.; Butter, D.; et al. Bacterial genotoxins induce T cell senescence. Cell Rep. 2021, 35, 109220. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.; Park, S.C.; Morales, W.; Marsh, E.; Lembo, A.; Kim, J.H.; Pimentel, M. Assessment of anti-vinculin and anti-cytolethal distending toxin B antibodies in subtypes of irritable bowel syndrome. Dig. Dis. Sci. 2017, 62, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gharaibeh, R.Z.; Newsome, R.C.; Pope, J.L.; Dougherty, M.W.; Tomkovich, S.; Pons, B.; Mirey, G.; Vignard, J.; Hendrixson, D.R.; et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2019, 68, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Péré-Védrenne, C.; He, W.; Azzi-Martin, L.; Prouzet-Mauléon, V.; Buissonnière, A.; Cardinaud, B.; Ménard, A. The nuclear remodeling induced by Helicobacter cytolethal distending toxin involves MAFB oncoprotein. Toxins 2020, 12, 174. [Google Scholar] [CrossRef]
- Lai, C.H.; Chang, C.S.; Liu, H.H.; Tsai, Y.S.; Hsu, F.M.; Yu, Y.L.; Lai, C.K.; Gandee, L.; Pong, R.C.; Hsu, H.W.; et al. Sensitization of radio-resistant prostate cancer cells with a unique cytolethal distending toxin. Oncotarget 2014, 5, 5523–5534. [Google Scholar] [CrossRef]
- Tarashi, S.; Siadat, S.D.; Ahmadi Badi, S.; Zali, M.; Biassoni, R.; Ponzoni, M.; Moshiri, A. Gut Bacteria and their Metabolites: Which One Is the Defendant for Colorectal Cancer? Microorganisms 2019, 7, 561. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Hermansen, K. Impact of diet-modulated butyrate production on intestinal barrier function and inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; Van de Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef]
- Schauber, J.; Svanholm, C.; Termen, S.; Iffland, K.; Menzel, T.; Scheppach, W.; Melcher, R.; Agerberth, B.; Lührs, H.; Gudmundsson, G. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: Relevance of signalling pathways. Gut 2003, 52, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Powrie, F. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 2019, 50, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell. 2012, 48, 612–626. [Google Scholar] [CrossRef]
- Davie, J.R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 2003, 133, 2485S–2493S. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Konishi, Y.; Narukawa, M.; Sugiura, Y.; Yoshimoto, S.; Arai, Y.; Sato, S.; Yoshida, Y.; Tsuji, S.; Uemura, K.; et al. Gut bacteria identified in colorectal cancer patients promote tumourigenesis via butyrate secretion. Nat. Commun. 2021, 12, 5674. [Google Scholar] [CrossRef]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Nittayaboon, K.; Leetanaporn, K.; Sangkhathat, S.; Roytrakul, S.; Navakanitworakul, R. Characterization of Butyrate-Resistant Colorectal Cancer Cell Lines and the Cytotoxicity of Anticancer Drugs against These Cells. BioMed Res. Int. 2022. [Google Scholar] [CrossRef]
- Tester, R.; Al-Ghazzewi, F.H. Intrinsic and extrinsic carbohydrates in the vagina: A short review on vaginal glycogen. Int. J. Biol. Macromol. 2018, 112, 203–206. [Google Scholar] [CrossRef]
- Masood, M.I.; Qadir, M.I.; Shirazi, J.H.; Khan, I.U. Beneficial effects of lactic acid bacteria on human beings. Crit. Rev. Microbiol. 2011, 37, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Aldunate, M.; Bradshaw, C.S.; Cone, R.A. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 2017, 168, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S. Lactic acid alleviates stress: Good for female genital tract homeostasis, bad for protection against malignancy. Cell Stress Chaperones 2018, 23, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG 2017, 124, 606–611. [Google Scholar] [CrossRef]
- Eskelinen, E.L. Autophagy: Supporting cellular and organismal homeostasis by self-eating. Int. J. Biochem. Cell Biol. 2019, 111, 1–10. [Google Scholar] [CrossRef]
- Usman, R.M.; Razzaq, F.; Akbar, A.; Farooqui, A.A.; Iftikhar, A.; Latif, A.; Anwer, F. Role and mechanism of autophagyregulating factors in tumorigenesis and drug resistance. Asia Pac. J. Clin. Oncol. 2021, 17, 193–208. [Google Scholar] [CrossRef]
- Nasioudis, D.; Witkin, S.S. Neutrophil gelatinase-associated lipocalin and innate immune responses to bacterial infections. Med. Microbiol. Immunol. 2015, 204, 471–479. [Google Scholar] [CrossRef]
- Latham, T.; Mackay, L.; Sproul, D.; Karim, M.; Culley, J.; Harrison, D.J.; Hayward, L.; Langridge-Smith, P.; Gilbert, N.; Ramsahoye, B.H. Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acid Res. 2012, 40, 4794–4803. [Google Scholar] [CrossRef]
- La Rosa, G.R.M.; Gattuso, G.; Pedullà, E.; Rapisarda, E.; Nicolosi, D.; Salmeri, M. Association of oral dysbiosis with oral cancer development. Oncol. Lett. 2020, 19, 3045–3058. [Google Scholar] [CrossRef]
- Sami, A.; Elimairi, I.; Stanton, C.; Ross, R.P.; Ryan, C.A. The Role of the Microbiome in Oral Squamous Cell Carcinoma with Insight into the Microbiome–Treatment Axis. Int. J. Mol. Sci. 2020, 21, 8061. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ung, T.T.; Kim, N.H.; Jung, Y.D. Role of bile acids in colon carcinogenesis. World J. Clin. Cases 2018, 6, 577. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Fujisawa, T. Analysis of Clostridium cluster XI bacteria in human feces. Biosci. Microbiota Food Health 2019, 38, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Alrubaye, B.; Abraha, M.; Almansour, A.; Bansal, M.; Wang, H.; Kwon, Y.M.; Sun, X. Microbial metabolite deoxycholic acid shapes microbiota against Campylobacter jejuni chicken colonization. PLoS ONE 2019, 14, e0214705. [Google Scholar] [CrossRef] [PubMed]
- Doden, H.L.; Ridlon, J.M. Microbial Hydroxysteroid Dehydrogenases: From Alpha to Omega. Microorganisms 2021, 9, 469. [Google Scholar] [CrossRef]
- Kurdi, P.; Kawanishi, K.; Mizutani, K.; Yokota, A. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J. Bacteriol. 2006, 188, 1979–1986. [Google Scholar] [CrossRef]
- Doden, H.; Sallam, L.A.; Devendran, S.; Ly, L.; Doden, G.; Daniel, S.L.; Ridlon, J.M. Metabolism of oxo-bile acids and characterization of recombinant 12α-hydroxysteroid dehydrogenases from bile acid 7α-dehydroxylating human gut bacteria. Appl. Environ. Microbiol. 2018, 84, e00235-18. [Google Scholar] [CrossRef]
- Kang, J.D.; Myers, C.J.; Harris, S.C.; Kakiyama, G.; Lee, I.K.; Yun, B.S.; Hylemon, P.B. Bile acid 7α-dehydroxylating gut bacteria secrete antibiotics that inhibit Clostridium difficile: Role of secondary bile acids. Cell Chem. Biol. 2019, 26, 27–34. [Google Scholar] [CrossRef]
- Thanissery, R.; Winston, J.A.; Theriot, C.M. Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 2017, 45, 86–100. [Google Scholar] [CrossRef]
- Hegyi, P.; Maléth, J.; Walters, J.R.; Hofmann, A.F.; Keely, S.J. Guts and gall: Bile acids in regulation of intestinal epithelial function in health and disease. Physiol. Rev. 2018, 98, 1983–2023. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Gaitonde, S.V.; Qi, W.; Martinez, J.D. Deoxycholic acid suppresses p53 by stimulating proteasome-mediated p53 protein degradation. Carcinogenesis 2001, 22, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Tortora, K.; Vitali, F.; De Filippo, C.; Caderni, G.; Giovannelli, L. DNA damage in colon mucosa of Pirc rats, an Apc-driven model of colon tumorigenesis. Toxicol. Lett. 2020, 324, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef]
- Farhana, L.; Nangia-Makker, P.; Arbit, E.; Shango, K.; Sarkar, S.; Mahmud, H.; Majumdar, A.P. Bile acid: A potential inducer of colon cancer stem cells. Stem. Cell Res. Ther. 2016, 7, 181. [Google Scholar] [CrossRef]
- Kong, Y.; Bai, P.S.; Sun, H.; Nan, K.J.; Chen, N.Z.; Qi, X.G. The deoxycholic acid targets miRNA-dependent CAC1 gene expression in multidrug resistance of human colorectal cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2321–2332. [Google Scholar] [CrossRef]
- Guz, M.; Jeleniewicz, W.; Malm, A.; Korona-Glowniak, I. A Crosstalk between Diet, Microbiome and microRNA in Epigenetic Regulation of Colorectal Cancer. Nutrients 2021, 13, 2428. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markelova, N.N.; Semenova, E.F.; Sineva, O.N.; Sadykova, V.S. The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis. Int. J. Mol. Sci. 2022, 23, 11706. https://doi.org/10.3390/ijms231911706
Markelova NN, Semenova EF, Sineva ON, Sadykova VS. The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis. International Journal of Molecular Sciences. 2022; 23(19):11706. https://doi.org/10.3390/ijms231911706
Chicago/Turabian StyleMarkelova, Natalia N., Elena F. Semenova, Olga N. Sineva, and Vera S. Sadykova. 2022. "The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis" International Journal of Molecular Sciences 23, no. 19: 11706. https://doi.org/10.3390/ijms231911706
APA StyleMarkelova, N. N., Semenova, E. F., Sineva, O. N., & Sadykova, V. S. (2022). The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis. International Journal of Molecular Sciences, 23(19), 11706. https://doi.org/10.3390/ijms231911706